
A Comparative Genomic Analysis of Epstein–Barr Virus Strains with a Focus on EBV2 Variability

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
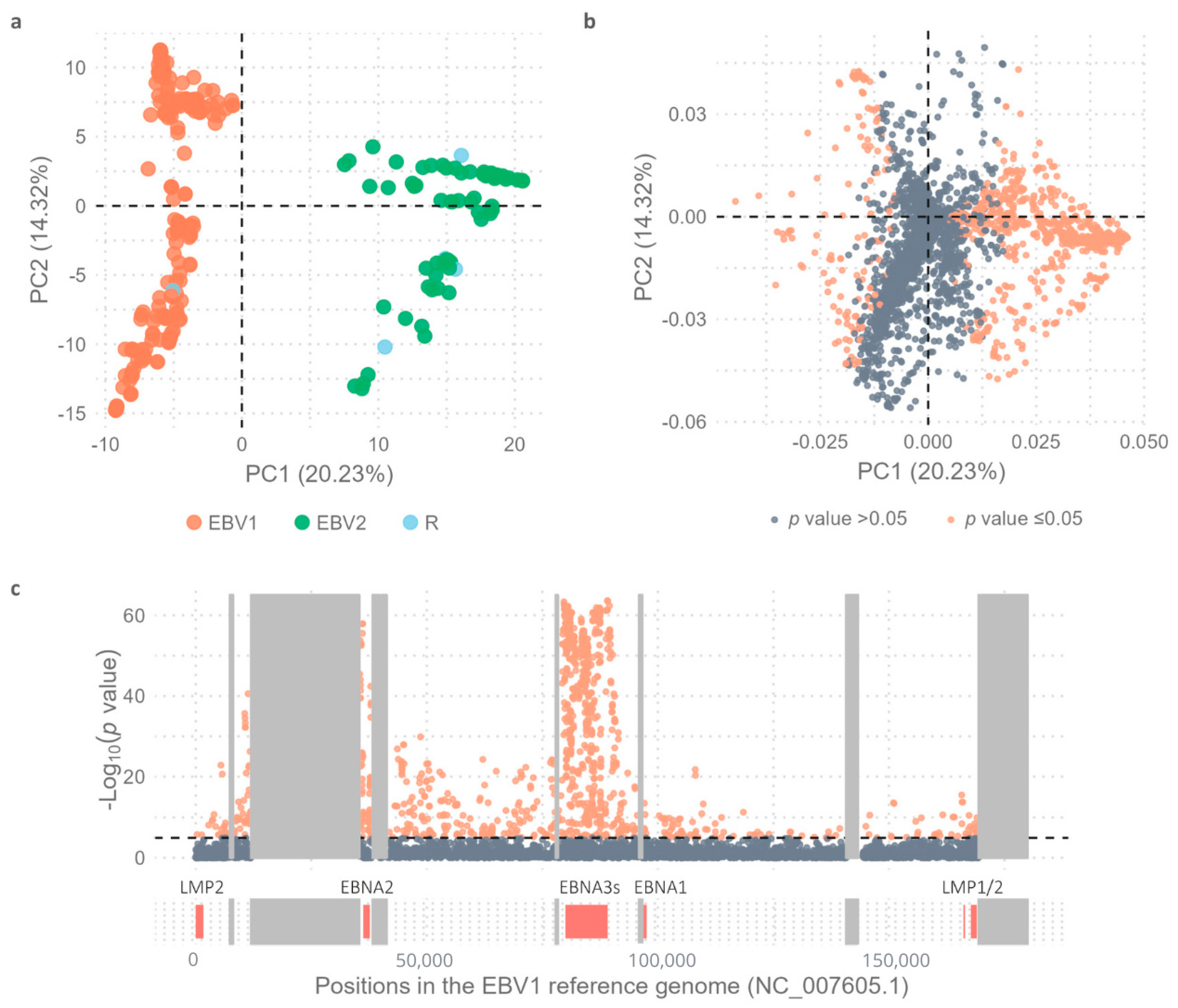
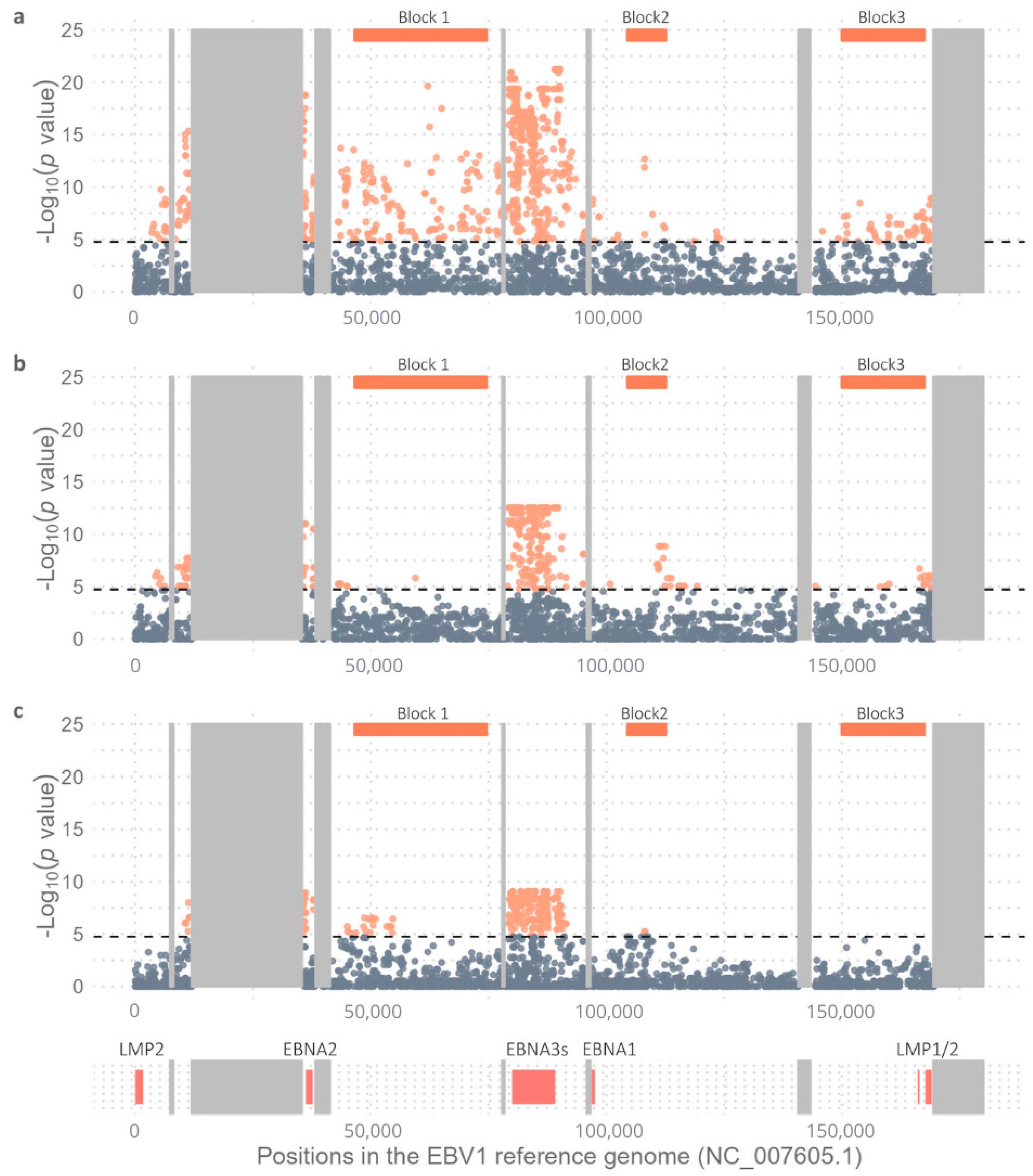
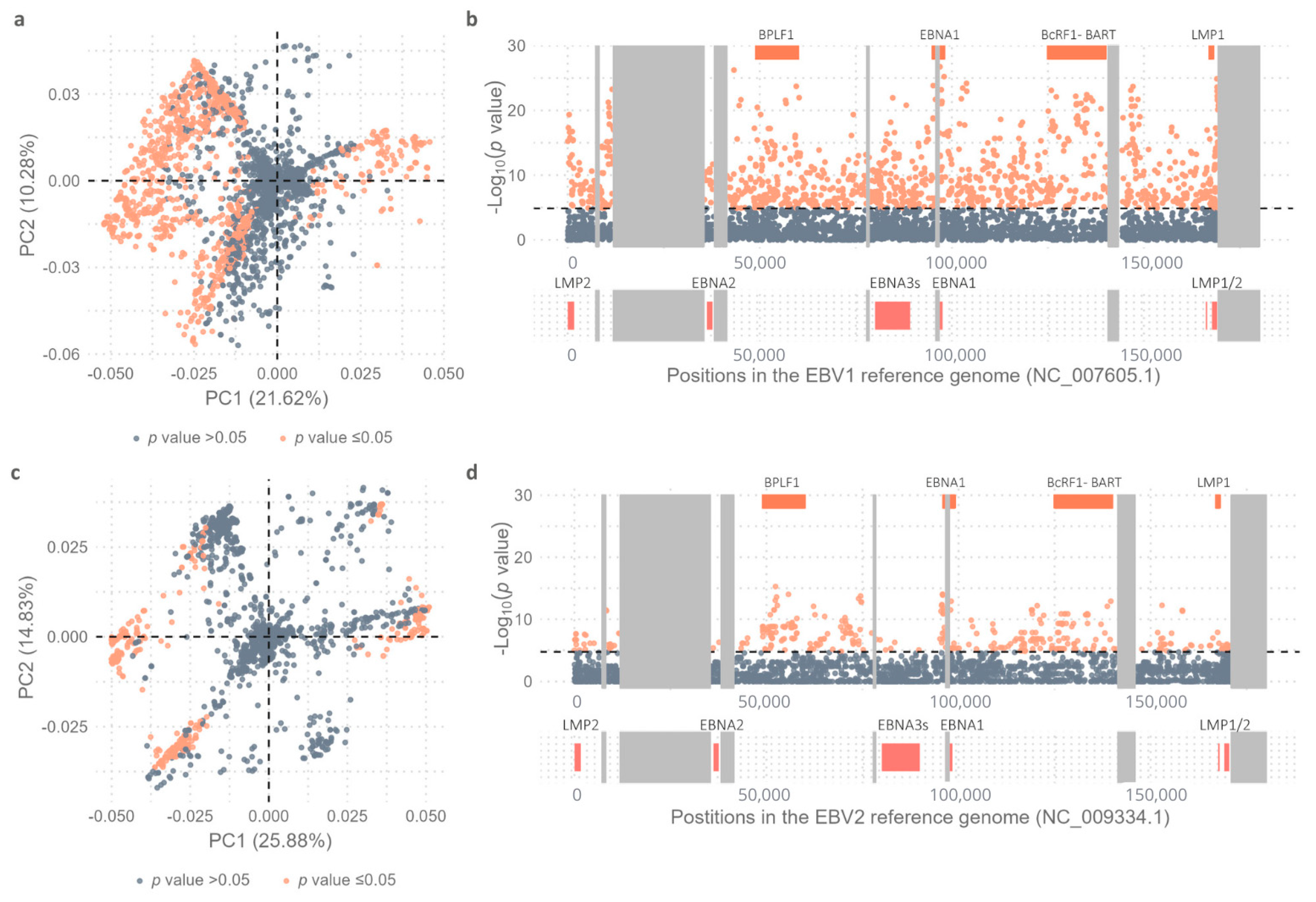
2.1. Variability Between EBV1 and EBV2 Genomes
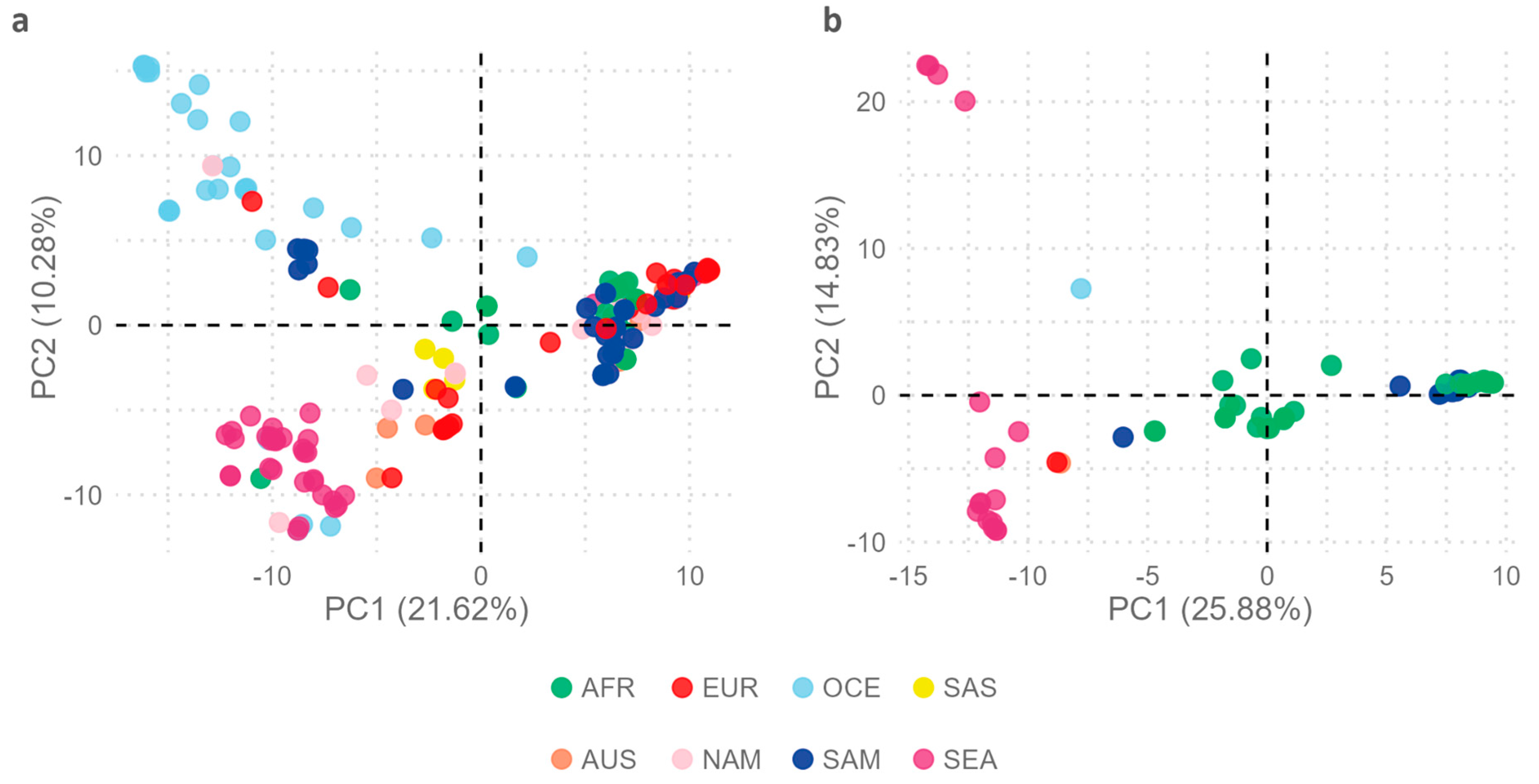
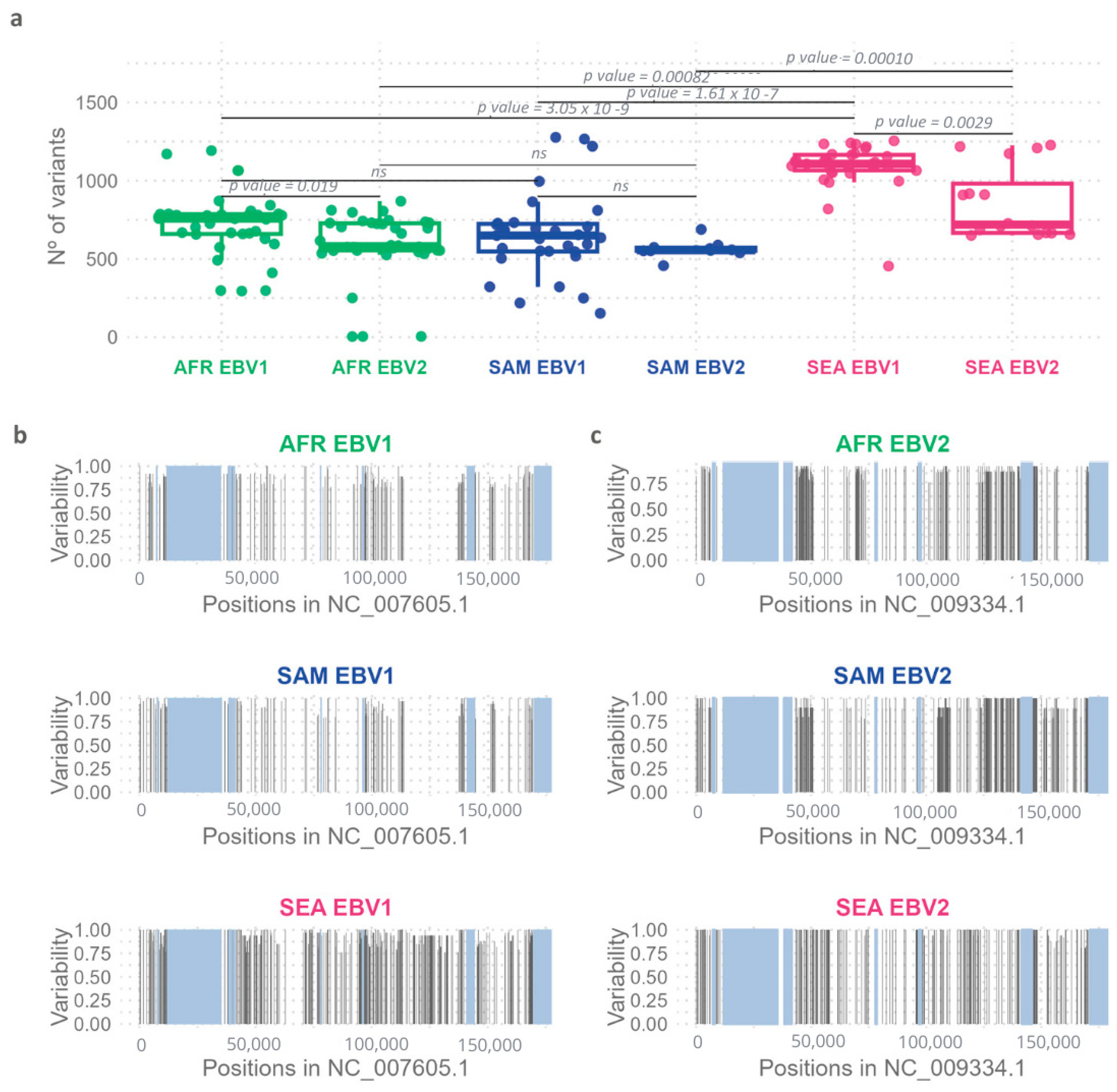
2.2. Comparative Geographic Variability Analysis in Each EBV Type
3. Discussion
4. Methods
4.1. Ethic Statement
4.2. Patients and Samples
4.3. DNA Extraction and Viral Load Analysis
4.4. Library Preparation and NGS Sequencing on the Illumina Platform
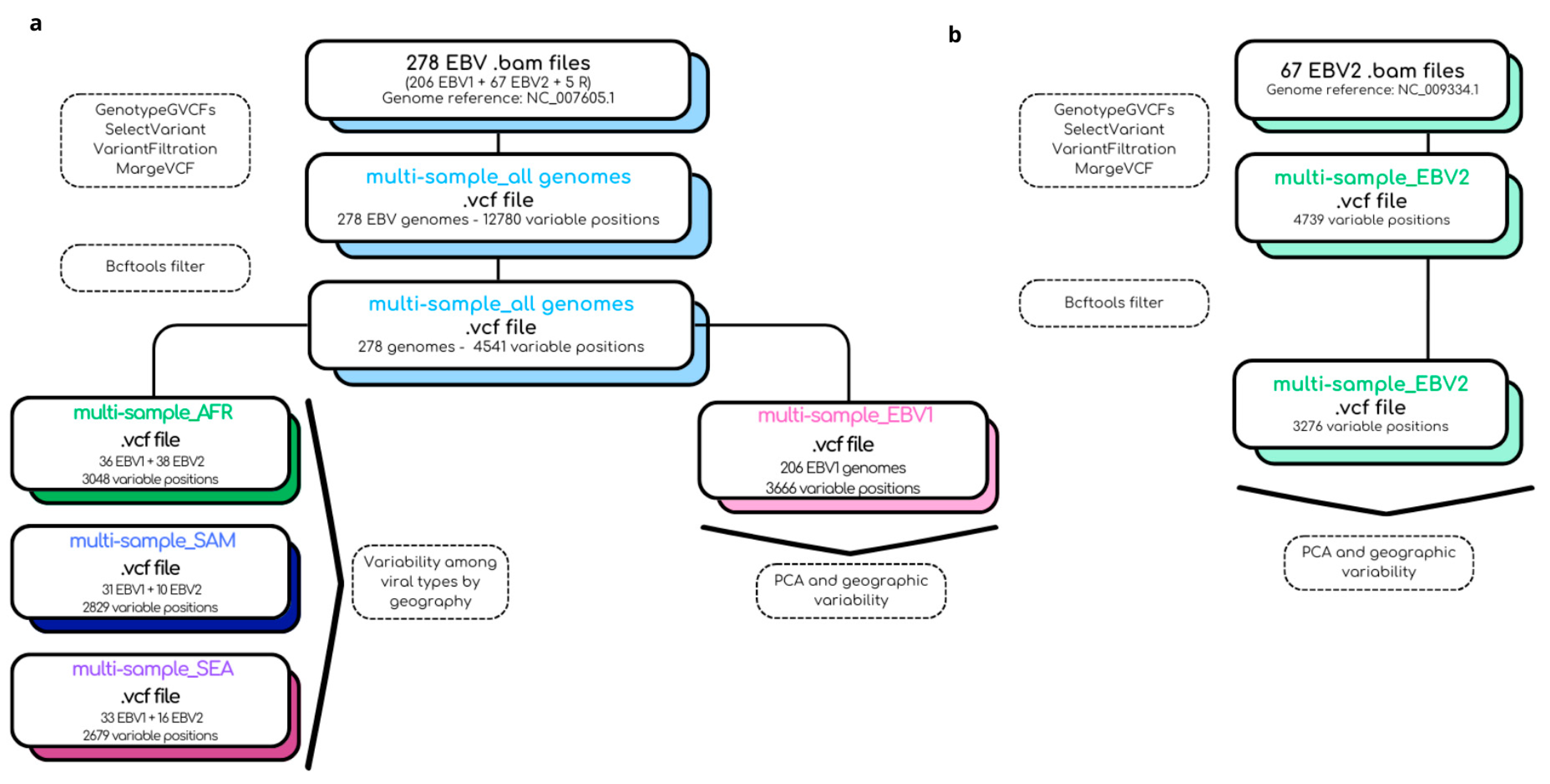
4.5. Bioinformatics, Viral Typing and Recombination Analysis Between Viral Types
4.6. Bioinformatic Pipeline for the Construction of Multi-Sample .vcf Files
4.7. Principal Component Analysis
4.8. Variant Frequency
4.9. Statistical Analysis
4.10. Data Availability
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yu, H.; Robertson, E.S. Epstein-Barr Virus History and Pathogenesis. Viruses 2023, 15, 714. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.L.; Glaser, S.L. Epstein-Barr Virus-Associated Malignancies: Epidemiologic Patterns and Etiologic Implications. Crit. Rev. Oncol. Hematol. 2000, 34, 27–53. [Google Scholar] [CrossRef] [PubMed]
- Chabay, P.A.; Preciado, M.V. EBV Primary Infection in Childhood and Its Relation to B-Cell Lymphoma Development: A Mini-Review from a Developing Region. Int. J. Cancer 2013, 133, 1286–1292. [Google Scholar] [CrossRef] [PubMed]
- Tzellos, S.; Farrell, P.J. Epstein-Barr Virus Sequence Variation-Biology and Disease. Pathogens 2012, 1, 156–174. [Google Scholar] [CrossRef]
- Ward, B.J.H.; Schaal, D.L.; Nkadi, E.H.; Scott, R.S. EBV Association with Lymphomas and Carcinomas in the Oral Compartment. Viruses 2022, 14, 2700. [Google Scholar] [CrossRef]
- Chabay, P.; Lens, D.; Hassan, R.; Rodríguez Pinilla, S.M.; Valvert Gamboa, F.; Rivera, I.; Huamán Garaicoa, F.; Ranuncolo, S.M.; Barrionuevo, C.; Morales Sánchez, A.; et al. Lymphotropic Viruses EBV, KSHV and HTLV in Latin America: Epidemiology and Associated Malignancies. A Literature-Based Study by the RIAL-CYTED. Cancers 2020, 12, 2166. [Google Scholar] [CrossRef]
- Farrell, P.J.; White, R.E. Do Epstein-Barr Virus Mutations and Natural Genome Sequence Variations Contribute to Disease? Biomolecules 2021, 12, 17. [Google Scholar] [CrossRef]
- Monteiro, T.A.F.; Costa, I.B.; Costa, I.B.; Corrêa, T.L.D.S.; Coelho, B.M.R.; Silva, A.E.S.; de Paula Ramos, F.L.; Filho, A.J.M.; Monteiro, J.L.F.; Siqueira, J.A.M.; et al. Genotypes of Epstein-Barr Virus (EBV1/EBV2) in Individuals with Infectious Mononucleosis in the Metropolitan Area of Belém, Brazil, between 2005 and 2016. Braz. J. Infect. Dis. 2020, 24, 322–329. [Google Scholar] [CrossRef]
- Neves, M.; Marinho-Dias, J.; Ribeiro, J.; Sousa, H. Epstein-Barr Virus Strains and Variations: Geographic or Disease-Specific Variants? J. Med. Virol. 2017, 89, 373–387. [Google Scholar] [CrossRef]
- Tzellos, S.; Correia, P.B.; Karstegl, C.E.; Cancian, L.; Cano-Flanagan, J.; McClellan, M.J.; West, M.J.; Farrell, P.J. A Single Amino Acid in EBNA-2 Determines Superior B Lymphoblastoid Cell Line Growth Maintenance by Epstein-Barr Virus Type 1 EBNA-2. J. Virol. 2014, 88, 8743–8753. [Google Scholar] [CrossRef]
- Rickinson, A.B.; Young, L.S.; Rowe, M. Influence of the Epstein-Barr Virus Nuclear Antigen EBNA 2 on the Growth Phenotype of Virus-Transformed B Cells. J. Virol. 1987, 61, 1310–1317. [Google Scholar] [CrossRef] [PubMed]
- Romero-Masters, J.C.; Huebner, S.M.; Ohashi, M.; Bristol, J.A.; Benner, B.E.; Barlow, E.A.; Turk, G.L.; Nelson, S.E.; Baiu, D.C.; Van Sciver, N.; et al. B Cells Infected with Type 2 Epstein-Barr Virus (EBV) Have Increased NFATc1/NFATc2 Activity and Enhanced Lytic Gene Expression in Comparison to Type 1 EBV Infection. PLoS Pathog. 2020, 16, e1008365. [Google Scholar] [CrossRef]
- Bristol, J.A.; Brand, J.; Ohashi, M.; Eichelberg, M.R.; Casco, A.; Nelson, S.E.; Hayes, M.; Romero-Masters, J.C.; Baiu, D.C.; Gumperz, J.E.; et al. Reduced IRF4 Expression Promotes Lytic Phenotype in Type 2 EBV-Infected B Cells. PLoS Pathog. 2022, 18, e1010453. [Google Scholar] [CrossRef] [PubMed]
- Bristol, J.A.; Djavadian, R.; Albright, E.R.; Coleman, C.B.; Ohashi, M.; Hayes, M.; Romero-Masters, J.C.; Barlow, E.A.; Farrell, P.J.; Rochford, R.; et al. A Cancer-Associated Epstein-Barr Virus BZLF1 Promoter Variant Enhances Lytic Infection. PLoS Pathog. 2018, 14, e1007179. [Google Scholar] [CrossRef]
- Correia, S.; Palser, A.; Elgueta Karstegl, C.; Middeldorp, J.M.; Ramayanti, O.; Cohen, J.I.; Hildesheim, A.; Fellner, M.D.; Wiels, J.; White, R.E.; et al. Natural Variation of Epstein-Barr Virus Genes, Proteins, and Primary MicroRNA. J. Virol. 2017, 91, e00375-17. [Google Scholar] [CrossRef]
- Correia, S.; Bridges, R.; Wegner, F.; Venturini, C.; Palser, A.; Middeldorp, J.M.; Cohen, J.I.; Lorenzetti, M.A.; Bassano, I.; White, R.E.; et al. Sequence Variation of Epstein-Barr Virus: Viral Types, Geography, Codon Usage, and Diseases. J. Virol. 2018, 92, e01132-18. [Google Scholar] [CrossRef]
- Palser, A.L.; Grayson, N.E.; White, R.E.; Corton, C.; Correia, S.; Ba Abdullah, M.M.; Watson, S.J.; Cotten, M.; Arrand, J.R.; Murray, P.G.; et al. Genome Diversity of Epstein-Barr Virus from Multiple Tumor Types and Normal Infection. J. Virol. 2015, 89, 5222–5237. [Google Scholar] [CrossRef]
- Bridges, R.; Correia, S.; Wegner, F.; Venturini, C.; Palser, A.; White, R.E.; Kellam, P.; Breuer, J.; Farrell, P.J. Essential Role of Inverted Repeat in Epstein-Barr Virus IR-1 in B Cell Transformation; Geographical Variation of the Viral Genome. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2019, 374, 20180299. [Google Scholar] [CrossRef]
- Blazquez, A.C.; Berenstein, A.J.; Torres, C.; Izquierdo, A.; Lezama, C.; Moscatelli, G.; De Matteo, E.N.; Lorenzetti, M.A.; Preciado, M.V. Comprehensive Evolutionary Analysis of Complete Epstein-Barr Virus Genomes from Argentina and Other Geographies. Viruses 2021, 13, 1172. [Google Scholar] [CrossRef]
- Reyes, M.E.; Zanella, L.; Riquelme, I.; Buchegger, K.; Mora-Lagos, B.; Guzmán, P.; García, P.; Roa, J.C.; Ili, C.G.; Brebi, P. Exploring the Genetic Diversity of Epstein-Barr Virus among Patients with Gastric Cancer in Southern Chile. Int. J. Mol. Sci. 2023, 24, 11276. [Google Scholar] [CrossRef]
- Hui, K.F.; Chan, T.F.; Yang, W.; Shen, J.J.; Lam, K.P.; Kwok, H.; Sham, P.C.; Tsao, S.W.; Kwong, D.L.; Lung, M.L.; et al. High Risk Epstein-Barr Virus Variants Characterized by Distinct Polymorphisms in the EBER Locus Are Strongly Associated with Nasopharyngeal Carcinoma. Int. J. Cancer 2019, 144, 3031–3042. [Google Scholar] [CrossRef] [PubMed]
- Kaymaz, Y.; Oduor, C.I.; Aydemir, O.; Luftig, M.A.; Otieno, J.A.; Ong’echa, J.M.; Bailey, J.A.; Moormann, A.M. Epstein-Barr Virus Genomes Reveal Population Structure and Type 1 Association with Endemic Burkitt Lymphoma. J. Virol. 2020, 94, e02007-19. [Google Scholar] [CrossRef]
- Correa, R.M.; Fellner, M.D.; Alonio, L.V.; Durand, K.; Teyssié, A.R.; Picconi, M.A. Epstein-Barr Virus (EBV) in Healthy Carriers: Distribution of Genotypes and 30 Bp Deletion in Latent Membrane Protein-1 (LMP-1) Oncogene. J. Med. Virol. 2004, 73, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Lorenzetti, M.A.; Gantuz, M.; Altcheh, J.; De Matteo, E.; Chabay, P.A.; Preciado, M.V. Epstein-Barr Virus BZLF1 Gene Polymorphisms: Malignancy Related or Geographically Distributed Variants? Clin. Microbiol. Infect. 2014, 20, O861–O869. [Google Scholar] [CrossRef] [PubMed]
- Alves, P.; Larrate, M.; Garcia-Costa, A.; Rohan, P.; Gama, B.E.; Abdelhay, E.; Delatorre, E.; Hassan, R. Spatial Dispersal of Epstein-Barr Virus in South America Reveals an African American Variant in Brazilian Lymphomas. Viruses 2022, 14, 1762. [Google Scholar] [CrossRef] [PubMed]
- Arturo-Terranova, D.; Giraldo-Ocampo, S.; Castillo, A. Molecular Characterization of Epstein-Barr Virus Variants Detected in the Oral Cavity of Adolescents in Cali, Colombia. Biomedica 2020, 40, 76–88. [Google Scholar] [CrossRef]
- Lorenzetti, M.A.; Gutiérrez, M.I.; Altcheh, J.; Moscatelli, G.; Moroni, S.; Chabay, P.A.; Preciado, M.V. Epstein-Barr Virus BZLF1 Gene Promoter Variants in Pediatric Patients with Acute Infectious Mononucleosis: Its Comparison with Pediatric Lymphomas. J. Med. Virol. 2009, 81, 1912–1917. [Google Scholar] [CrossRef]
- Alves, P.D.; Rohan, P.; Hassan, R.; Abdelhay, E. Lytic and Latent Genetic Diversity of the Epstein-Barr Virus Reveals Raji-Related Variants from Southeastern Brazil Associated with Recombination Markers. Int. J. Mol. Sci. 2024, 25, 5002. [Google Scholar] [CrossRef]
- Sathiyamoorthy, K.; Hu, Y.X.; Möhl, B.S.; Chen, J.; Longnecker, R.; Jardetzky, T.S. Structural Basis for Epstein-Barr Virus Host Cell Tropism Mediated by gp42 and gHgL Entry Glycoproteins. Nat. Commun. 2016, 7, 13557. [Google Scholar] [CrossRef]
- Hong, J.; Wei, D.; Wu, Q.; Zhong, L.; Chen, K.; Huang, Y.; Zhang, W.; Chen, J.; Xia, N.; Zhang, X.; et al. Antibody Generation and Immunogenicity Analysis of EBV gp42 N-Terminal Region. Viruses 2021, 13, 2380. [Google Scholar] [CrossRef]
- Jean-Pierre, V.; Lupo, J.; Buisson, M.; Morand, P.; Germi, R. Main Targets of Interest for the Development of a Prophylactic or Therapeutic Epstein-Barr Virus Vaccine. Front. Microbiol. 2021, 12, 701611. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Rabaan, A.A.; Alwashmi, A.S.S.; Albayat, H.; Mashraqi, M.M.; Alshehri, A.A.; Garout, M.; Abduljabbar, W.A.; Yusof, N.Y.; Yean, C.Y. Immunoinformatic Execution and Design of an Anti-Epstein-Barr Virus Vaccine with Multiple Epitopes Triggering Innate and Adaptive Immune Responses. Microorganisms 2023, 11, 2448. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.I. Epstein-Barr Virus Vaccines. Clin. Transl. Immunol. 2015, 4, e32. [Google Scholar] [CrossRef] [PubMed]
- Sokal, E.M.; Hoppenbrouwers, K.; Vandermeulen, C.; Moutschen, M.; Léonard, P.; Moreels, A.; Haumont, M.; Bollen, A.; Smets, F.; Denis, M. Recombinant gp350 Vaccine for Infectious Mononucleosis: A Phase 2, Randomized, Double-Blind, Placebo-Controlled Trial to Evaluate the Safety, Immunogenicity, and Efficacy of an Epstein-Barr Virus Vaccine in Healthy Young Adults. J. Infect. Dis. 2007, 196, 1749–1753. [Google Scholar] [CrossRef] [PubMed]
- Weiss, E.R.; Alter, G.; Ogembo, J.G.; Henderson, J.L.; Tabak, B.; Bakiş, Y.; Somasundaran, M.; Garber, M.; Selin, L.; Luzuriaga, K. High Epstein-Barr Virus Load and Genomic Diversity Are Associated with Generation of gp350-Specific Neutralizing Antibodies Following Acute Infectious Mononucleosis. J. Virol. 2017, 91, e01562. [Google Scholar] [CrossRef]
- Luo, B.; Liu, M.; Chao, Y.; Wang, Y.; Jing, Y.; Sun, Z. Characterization of Epstein-Barr Virus gp350/220 Gene Variants in Virus Isolates from Gastric Carcinoma and Nasopharyngeal Carcinoma. Arch. Virol. 2012, 157, 207–216. [Google Scholar] [CrossRef]
- Avena, S.; Via, M.; Ziv, E.; Pérez-Stable, E.J.; Gignoux, C.R.; Dejean, C.; Huntsman, S.; Torres-Mejía, G.; Dutil, J.; Matta, J.L.; et al. Heterogeneity in Genetic Admixture across Different Regions of Argentina. PLoS ONE 2012, 7, e34695. [Google Scholar] [CrossRef]
- Sun, C.; Chen, X.-C.; Kang, Y.-F.; Zeng, M.-S. The Status and Prospects of Epstein-Barr Virus Prophylactic Vaccine Development. Front. Immunol. 2021, 12, 677027. [Google Scholar] [CrossRef]
- Rühl, J.; Leung, C.S.; Münz, C. Vaccination against the Epstein-Barr Virus. Cell Mol. Life Sci. 2020, 77, 4315–4324. [Google Scholar] [CrossRef]
- Zhong, L.; Krummenacher, C.; Zhang, W.; Hong, J.; Feng, Q.; Chen, Y.; Zhao, Q.; Zeng, M.-S.; Zeng, Y.-X.; Xu, M.; et al. Urgency and Necessity of Epstein-Barr Virus Prophylactic Vaccines. NPJ Vaccines 2022, 7, 159. [Google Scholar] [CrossRef]
- Tornesello, A.L.; Tagliamonte, M.; Buonaguro, F.M.; Tornesello, M.L.; Buonaguro, L. Virus-like Particles as Preventive and Therapeutic Cancer Vaccines. Vaccines 2022, 10, 227. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Zhao, Q.; Zeng, M.-S.; Zhang, X. Prophylactic Vaccines against Epstein-Barr Virus. Lancet 2024, 404, 845. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.I. Therapeutic Vaccines for Herpesviruses. J. Clin. Investig. 2024, 134, e179483. [Google Scholar] [CrossRef]
- Sample, J.; Young, L.; Martin, B.; Chatman, T.; Kieff, E.; Rickinson, A.; Kieff, E. Epstein-Barr Virus Types 1 and 2 Differ in Their EBNA-3A, EBNA-3B, and EBNA-3C Genes. J. Virol. 1990, 64, 4084–4092. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Assefa, S.; Keane, T.M.; Otto, T.D.; Newbold, C.; Berriman, M. ABACAS: Algorithm-Based Automatic Contiguation of Assembled Sequences. Bioinformatics 2009, 25, 1968–1969. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and Analysis of Recombination Patterns in Virus Genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Schmieder, R.; Edwards, R. Quality Control and Preprocessing of Metagenomic Datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A Flexible Suite of Utilities for Comparing Genomic Features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A Program for Annotating and Predicting the Effects of Single Nucleotide Polymorphisms, SnpEff: SNPs in the Genome of Drosophila Melanogaster Strain w1118; Iso-2; Iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Jombart, T. Adegenet: A R Package for the Multivariate Analysis of Genetic Markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef]
- Knaus, B.J.; Grünwald, N.J. Vcfr: A Package to Manipulate and Visualize Variant Call Format Data in R. Mol. Ecol. Resour. 2017, 17, 44–53. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The Variant Call Format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Luo, B.; Tang, X.; Jia, Y.; Wang, Y.; Chao, Y.; Zhao, C. Sequence Variation of Epstein-Barr Virus (EBV) BZLF1 Gene in EBV-Associated Gastric Carcinomas and Nasopharyngeal Carcinomas in Northern China. Microbes Infect. 2011, 13, 776–782. [Google Scholar] [CrossRef]
- Gutiérrez, M.I.; Ibrahim, M.M.; Dale, J.K.; Greiner, T.C.; Straus, S.E.; Bhatia, K. Discrete Alterations in the BZLF1 Promoter in Tumor and Non-Tumor-Associated Epstein-Barr Virus. J. Natl. Cancer Inst. 2002, 94, 1757–1763. [Google Scholar] [CrossRef] [PubMed]
- R Development Core Team. The R Reference Manual: Base Package; Network Theory; R Development Core Team: Vienna, Austria, 2003; ISBN 9780954612009. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blazquez, A.C.; Fellner, M.D.; Lorenzetti, M.A.; Preciado, M.V. A Comparative Genomic Analysis of Epstein–Barr Virus Strains with a Focus on EBV2 Variability. Int. J. Mol. Sci. 2025, 26, 2708. https://doi.org/10.3390/ijms26062708
Blazquez AC, Fellner MD, Lorenzetti MA, Preciado MV. A Comparative Genomic Analysis of Epstein–Barr Virus Strains with a Focus on EBV2 Variability. International Journal of Molecular Sciences. 2025; 26(6):2708. https://doi.org/10.3390/ijms26062708
Chicago/Turabian StyleBlazquez, Ana Catalina, María Dolores Fellner, Mario Alejandro Lorenzetti, and María Victoria Preciado. 2025. "A Comparative Genomic Analysis of Epstein–Barr Virus Strains with a Focus on EBV2 Variability" International Journal of Molecular Sciences 26, no. 6: 2708. https://doi.org/10.3390/ijms26062708
APA StyleBlazquez, A. C., Fellner, M. D., Lorenzetti, M. A., & Preciado, M. V. (2025). A Comparative Genomic Analysis of Epstein–Barr Virus Strains with a Focus on EBV2 Variability. International Journal of Molecular Sciences, 26(6), 2708. https://doi.org/10.3390/ijms26062708