
Drug Discovery and Development for Heart Failure Using Multi-Omics Approaches


Abstract
1. Introduction
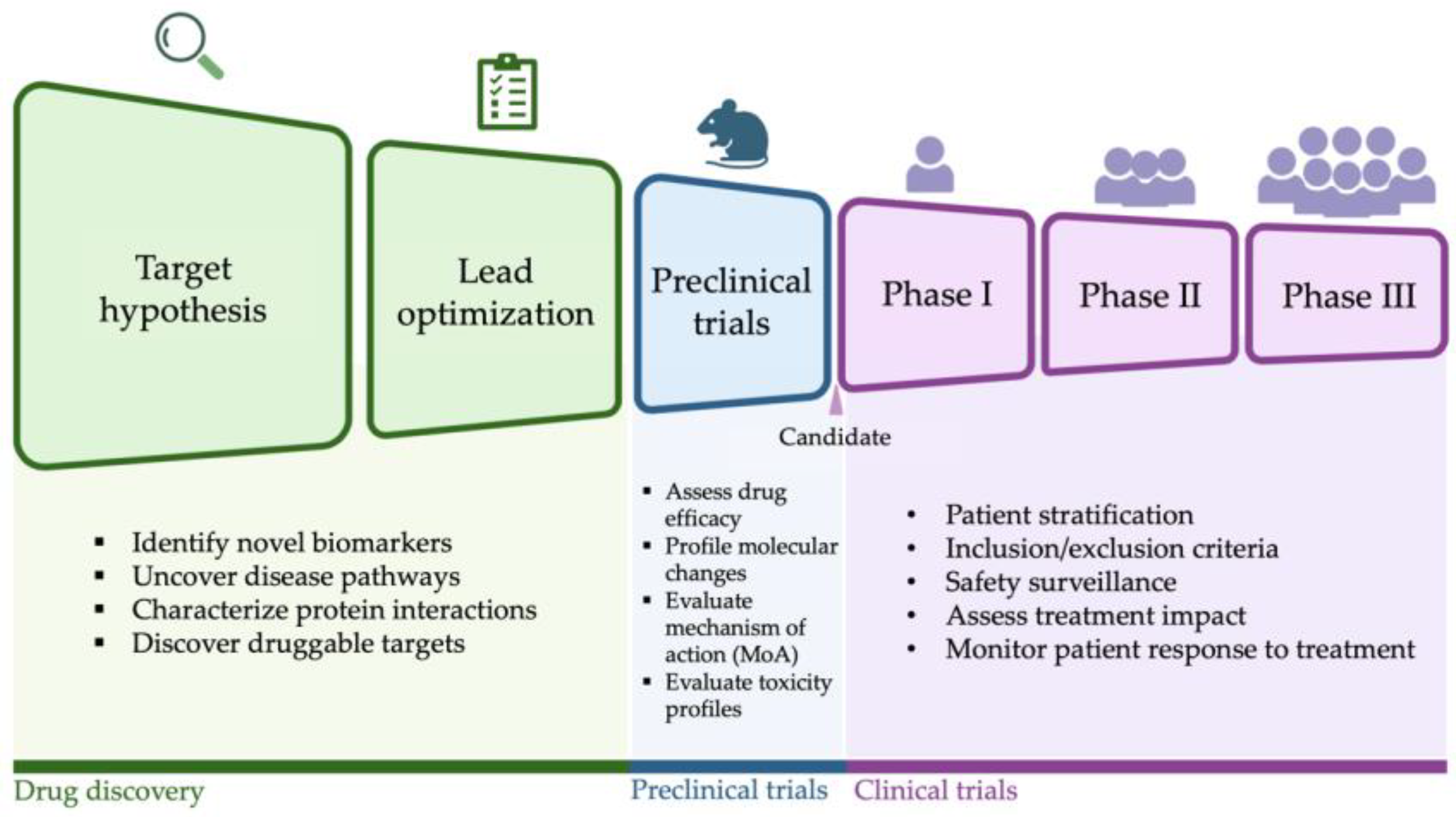
2. Target Discovery and Lead Identification
Multi-Omics Approaches for Therapeutic Target Identification
3. Multi-Omics Approaches in Preclinical Studies for Heart Failure
3.1. Evaluating Drug Mechanism of Action
3.2. Assessing Drug Safety in Preclinical Models
4. Multi-Omics Approaches in Clinical Trials for Heart Failure
4.1. Enhancing Patient Stratification
4.2. Monitoring Treatment Response and Disease Progression
4.3. Accelerating Drug Development Through Early Endpoint Detection
5. Current Limitations and Future Perspectives
5.1. Translational Challenges in Advancing Omics from Bench to Bedside
5.2. Future Perspectives
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Savarese, G.; Becher, P.M.; Lund, L.H.; Seferovic, P.; Rosano, G.M.C.; Coats, A.J.S. Global burden of heart failure: A comprehensive and updated review of epidemiology. Cardiovasc. Res. 2022, 118, 3272–3287. [Google Scholar] [CrossRef] [PubMed]
- Sayed, A.; Abramov, D.; Fonarow, G.C.; Mamas, M.A.; Kobo, O.; Butler, J.; Fudim, M. Reversals in the Decline of Heart Failure Mortality in the US, 1999 to 2021. JAMA Cardiol. 2024, 9, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Heidenreich, P.A.; Bozkurt, B.; Aguilar, D.; Allen, L.A.; Byun, J.J.; Colvin, M.M.; Deswal, A.; Drazner, M.H.; Dunlay, S.M.; Evers, L.R.; et al. 2022 AHA/ACC/HFSA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 2022, 145, e895–e1032. [Google Scholar] [CrossRef] [PubMed]
- Carnicelli, A.P.; Clare, R.; Hofmann, P.; Chiswell, K.; DeVore, A.D.; Vemulapalli, S.; Felker, G.M.; Sarocco, P.; Mentz, R.J. Characteristics and Outcomes of Patients with Heart Failure with Reduced Ejection Fraction After a Recent Worsening Heart Failure Event. J. Am. Heart Assoc. 2021, 10, e021276. [Google Scholar] [CrossRef]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2023 Focused Update of the 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2023, 44, 3627–3639. [Google Scholar] [CrossRef]
- Ferrari, R.; Böhm, M.; Cleland, J.G.F.; Paulus, W.J.S.; Pieske, B.; Rapezzi, C.; Tavazzi, L. Heart failure with preserved ejection fraction: Uncertainties and dilemmas. Eur. J. Heart Fail. 2015, 17, 665–671. [Google Scholar] [CrossRef]
- Raufaste-Cazavieille, V.; Santiago, R.; Droit, A. Multi-omics analysis: Paving the path toward achieving precision medicine in cancer treatment and immuno-oncology. Front. Mol. Biosci. 2022, 9, 962743. [Google Scholar] [CrossRef]
- Crowther, L.M.; Poms, M.; Plecko, B. Multiomics tools for the diagnosis and treatment of rare neurological disease. J. Inherit. Metab. Dis. 2018, 41, 425–434. [Google Scholar] [CrossRef]
- Ward, R.A.; Aghaeepour, N.; Bhattacharyya, R.P.; Clish, C.B.; Gaudillière, B.; Hacohen, N.; Mansour, M.K.; Mudd, P.A.; Pasupneti, S.; Presti, R.M.; et al. Harnessing the potential of multiomics studies for precision medicine in infectious disease. Open Forum Infect. Dis. 2021, 8, ofab483. [Google Scholar] [CrossRef]
- DiMasi, J.A.; Grabowski, H.G.; Hansen, R.W. Innovation in the pharmaceutical industry: New estimates of R&D costs. J. Health Econ. 2016, 47, 20–33. [Google Scholar]
- Sun, D.; Gao, W.; Hu, H.; Zhou, S. Why 90% of clinical drug development fails and how to improve it? Acta Pharm. Sin. B 2022, 12, 3049–3062. [Google Scholar] [CrossRef] [PubMed]
- Rasooly, D.; Peloso, G.M.; Pereira, A.C.; Dashti, H.; Giambartolomei, C.; Wheeler, E.; Aung, N.; Ferolito, B.R.; Pietzner, M.; Farber-Eger, E.H.; et al. Genome-wide association analysis and Mendelian randomization proteomics identify drug targets for heart failure. Nat. Commun. 2023, 14, 3826. [Google Scholar] [CrossRef] [PubMed]
- Minikel, E.V.; Painter, J.L.; Dong, C.C.; Nelson, M.R. Refining the impact of genetic evidence on clinical success. Nature 2024, 629, 624–629. [Google Scholar] [CrossRef] [PubMed]
- Ochoa, D.; Karim, M.; Ghoussaini, M.; Hulcoop, D.G.; McDonagh, E.M.; Dunham, I. Human genetics evidence supports two-thirds of the 2021 FDA-approved drugs. Nat. Rev. Drug Discov. 2022, 21, 551. [Google Scholar] [CrossRef]
- Levin, M.G.; Tsao, N.L.; Singhal, P.; Liu, C.; Vy, H.M.T.; Paranjpe, I.; Backman, J.D.; Bellomo, T.R.; Bone, W.P.; Biddinger, K.J.; et al. Genome-wide association and multi-trait analyses characterize the common genetic architecture of heart failure. Nat. Commun. 2022, 13, 6914. [Google Scholar] [CrossRef]
- Shah, S.; Henry, A.; Roselli, C.; Lin, H.; Sveinbjörnsson, G.; Fatemifar, G.; Hedman, Å.K.; Wilk, J.B.; Morley, M.P.; Chaffin, M.D.; et al. Genome-wide association and Mendelian randomisation analysis provide insights into the pathogenesis of heart failure. Nat. Commun. 2020, 11, 163. [Google Scholar] [CrossRef]
- Gaziano, J.M.; Concato, J.; Brophy, M.; Fiore, L.; Pyarajan, S.; Breeling, J.; Whitbourne, S.; Deen, J.; Shannon, C.; Humphries, D.; et al. Million Veteran Program: A mega-biobank to study genetic influences on health and disease. J. Clin. Epidemiol. 2016, 70, 214–223. [Google Scholar] [CrossRef]
- Joseph, J.; Liu, C.; Hui, Q.; Aragam, K.; Wang, Z.; Charest, B.; Huffman, J.E.; Keaton, J.M.; Edwards, T.L.; Demissie, S.; et al. Genetic architecture of heart failure with preserved versus reduced ejection fraction. Nat. Commun. 2022, 13, 7753. [Google Scholar] [CrossRef]
- Rasooly, D.; Giambartolomei, C.; Peloso, G.M.; Dashti, H.; Ferolito, B.R.; Golden, D.; Horimoto, A.R.V.R.; Pietzner, M.; Farber-Eger, E.H.; Wells, Q.S.; et al. Large-scale multi-omics identifies drug targets for heart failure with reduced and preserved ejection fraction. Nat. Cardiovasc. Res. 2025, 1–19. [Google Scholar] [CrossRef]
- Bozkurt, B.; Coats, A.J.; Tsutsui, H.; Abdelhamid, M.; Adamopoulos, S.; Albert, N.; Anker, S.D.; Atherton, J.; Böhm, M.; Butler, J.; et al. Universal Definition and Classification of Heart Failure: A Report of the Heart Failure Society of America, Heart Failure Association of the European Society of Cardiology, Japanese Heart Failure Society and Writing Committee of the Universal Definition of Heart Failure. J. Card. Fail. 2021, 27, 387–413. [Google Scholar] [CrossRef]
- Rasooly, D.; Patel, C.J. Conducting a Reproducible Mendelian Randomization Analysis Using the R Analytic Statistical Environment. Curr. Protoc. Hum. Genet. 2019, 101, e82. [Google Scholar] [CrossRef] [PubMed]
- Rasooly, D.; Peloso, G.M. Two-Sample Multivariable Mendelian Randomization Analysis Using R. Curr. Protoc. 2021, 1, e335. [Google Scholar] [CrossRef] [PubMed]
- Zuber, V.; Grinberg, N.F.; Gill, D.; Manipur, I.; Slob, E.A.W.; Patel, A.; Wallace, C.; Burgess, S. Combining evidence from Mendelian randomization and colocalization: Review and comparison of approaches. Am. J. Hum. Genet. 2022, 109, 767–782. [Google Scholar] [CrossRef] [PubMed]
- Rasooly, D.; Peloso, G.M.; Giambartolomei, C. Bayesian Genetic Colocalization Test of Two Traits Using coloc. Curr. Protoc. 2022, 2, e627. [Google Scholar] [CrossRef]
- Beauverger, P.; Ozoux, M.-L.; Bégis, G.; Glénat, V.; Briand, V.; Philippo, M.-C.; Daveu, C.; Tavares, G.; Roy, S.; Corbier, A.; et al. Reversion of cardiac dysfunction by a novel orally available calcium/calmodulin-dependent protein kinase II inhibitor, RA306, in a genetic model of dilated cardiomyopathy. Cardiovasc. Res. 2020, 116, 329–338. [Google Scholar] [CrossRef]
- Alexander, V.J.; Xia, S.; Hurh, E.; Hughes, S.G.; O’Dea, L.; Geary, R.S.; Witztum, J.L.; Tsimikas, S. N-acetyl galactosamine-conjugated antisense drug to APOC3 mRNA, triglycerides and atherogenic lipoprotein levels. Eur. Heart J. 2019, 40, 2785–2796. [Google Scholar] [CrossRef]
- Ritchie, S.C. Discovery of drug targets for heart failure with preserved and reduced ejection fraction. Nat. Cardiovasc. Res. 2025, 1–2. [Google Scholar] [CrossRef]
- Tsimikas, S.; Moriarty, P.M.; Stroes, E.S. Emerging RNA therapeutics to lower blood levels of LP(a): JACC focus seminar 2/4. J. Am. Coll. Cardiol. 2021, 77, 1576–1589. [Google Scholar] [CrossRef]
- Connelly, C.M.; Moon, M.H.; Schneekloth, J.S., Jr. The Emerging Role of RNA as a Therapeutic Target for Small Molecules. Cell Chem. Biol. 2016, 23, 1077–1090. [Google Scholar] [CrossRef]
- Franco-Serrano, L.; Hernández, S.; Calvo, A.; Severi, M.A.; Ferragut, G.; Pérez-Pons, J.; Piñol, J.; Pich, Ò.; Mozo-Villarias, Á.; Amela, I.; et al. MultitaskProtDB-II: An update of a database of multitasking/moonlighting proteins. Nucleic Acids Res. 2018, 46, D645–D648. [Google Scholar] [CrossRef]
- Aebersold, R.; Mann, M. Mass-spectrometric exploration of proteome structure and function. Nature 2016, 537, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Cao, T.H.; Jones, D.J.L.; Voors, A.A.; Quinn, P.A.; Sandhu, J.K.; Chan, D.C.S.; Parry, H.M.; Mohan, M.; Mordi, I.R.; Sama, I.E.; et al. Plasma proteomic approach in patients with heart failure: Insights into pathogenesis of disease progression and potential novel treatment targets. Eur. J. Heart Fail. 2020, 22, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Voors, A.A.; Anker, S.D.; Cleland, J.G.; Dickstein, K.; Filippatos, G.; van der Harst, P.; Hillege, H.L.; Lang, C.C.; Ter Maaten, J.M.; Ng, L.; et al. A systems BIOlogy Study to TAilored Treatment in Chronic Heart Failure: Rationale, design, and baseline characteristics of BIOSTAT-CHF. Eur. J. Heart Fail. 2016, 18, 716–726. [Google Scholar] [CrossRef] [PubMed]
- Adamy, C.; Mulder, P.; Khouzami, L.; Andrieu-abadie, N.; Defer, N.; Candiani, G.; Pavoine, C.; Caramelle, P.; Souktani, R.; Le Corvoisier, P.; et al. Neutral sphingomyelinase inhibition participates to the benefits of N-acetylcysteine treatment in post-myocardial infarction failing heart rats. J. Mol. Cell Cardiol. 2007, 43, 344–353. [Google Scholar] [CrossRef]
- Smith, J.G.; Gerszten, R.E. Emerging Affinity-Based Proteomic Technologies for Large-Scale Plasma Profiling in Cardiovascular Disease. Circulation 2017, 135, 1651–1664. [Google Scholar] [CrossRef]
- McDermott, J.E.; Wang, J.; Mitchell, H.; Webb-Robertson, B.-J.; Hafen, R.; Ramey, J.; Rodland, K.D. Challenges in Biomarker Discovery: Combining Expert Insights with Statistical Analysis of Complex Omics Data. Expert. Opin. Med. Diagn. 2013, 7, 37–51. [Google Scholar] [CrossRef]
- Egerstedt, A.; Berntsson, J.; Smith, M.L.; Gidlöf, O.; Nilsson, R.; Benson, M.; Wells, Q.S.; Celik, S.; Lejonberg, C.; Farrell, L.; et al. Profiling of the plasma proteome across different stages of human heart failure. Nat. Commun. 2019, 10, 5830. [Google Scholar] [CrossRef]
- Nayor, M.; Short, M.I.; Rasheed, H.; Lin, H.; Jonasson, C.; Yang, Q.; Hveem, K.; Felix, J.F.; Morrison, A.C.; Wild, P.S.; et al. Aptamer-Based Proteomic Platform Identifies Novel Protein Predictors of Incident Heart Failure and Echocardiographic Traits. Circ. Heart Fail. 2020, 13, e006749. [Google Scholar] [CrossRef]
- Dieden, A.; Girerd, N.; Ottosson, F.; Molvin, J.; Pareek, M.; Melander, O.; Bachus, E.; Råstam, L.; Lindblad, U.; Daka, B.; et al. Proteomic biomarkers and pathway analysis for progression to heart failure in three epidemiological representative cohorts. Eur. J. Heart Fail. 2024. [Google Scholar] [CrossRef]
- Qin, H.; Tromp, J.; Ter Maaten, J.M.; Voordes, G.H.D.; van Essen, B.J.; André de la Rambelje, M.; van der Hoef, C.C.S.; Santema, B.T.; Lam, C.S.P.; Voors, A.A. Clinical and Proteomic Risk Profiles of New-Onset Heart Failure in Men and Women. JACC Heart Fail. 2024, 13, 435–449. [Google Scholar] [CrossRef]
- Nath, M.; Romaine, S.P.R.; Koekemoer, A.; Hamby, S.; Webb, T.R.; Nelson, C.P.; Castellanos-Uribe, M.; Papakonstantinou, M.; Anker, S.D.; Lang, C.C.; et al. Whole blood transcriptomic profiling identifies molecular pathways related to cardiovascular mortality in heart failure. Eur. J. Heart Fail. 2022, 24, 1009–1019. [Google Scholar] [CrossRef] [PubMed]
- Jovanovich, A.; Ix, J.H.; Gottdiener, J.; McFann, K.; Katz, R.; Kestenbaum, B.; de Boer, I.H.; Sarnak, M.; Shlipak, M.G.; Mukamal, K.J.; et al. Fibroblast growth factor 23, left ventricular mass, and left ventricular hypertrophy in community-dwelling older adults. Atherosclerosis 2013, 231, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Ix, J.H.; Katz, R.; Kestenbaum, B.R.; de Boer, I.H.; Chonchol, M.; Mukamal, K.J.; Rifkin, D.; Siscovick, D.S.; Sarnak, M.J.; Shlipak, M.G. Fibroblast growth factor-23 and death, heart failure, and cardiovascular events in community-living individuals: CHS (Cardiovascular Health Study). J. Am. Coll. Cardiol. 2012, 60, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Joseph, J.; Chonchol, M.; Kaufman, J.S.; Cheung, A.K.; Rafeq, Z.; Smits, G.; Kendrick, J.; HOST Investigators. Higher fibroblast growth factor-23 concentrations associate with left ventricular systolic dysfunction in dialysis patients. Clin. Nephrol. 2013, 80, 313–321. [Google Scholar] [CrossRef]
- Wagner, A.; Cohen, N.; Kelder, T.; Amit, U.; Liebman, E.; Steinberg, D.M.; Radonjic, M.; Ruppin, E. Drugs that reverse disease transcriptomic signatures are more effective in a mouse model of dyslipidemia. Mol. Syst. Biol. 2015, 11, 791. [Google Scholar] [CrossRef]
- Ahmad, T.; Kelly, J.P.; McGarrah, R.W.; Hellkamp, A.S.; Fiuzat, M.; Testani, J.M.; Wang, T.S.; Verma, A.; Samsky, M.D.; Donahue, M.P.; et al. Prognostic Implications of Long-Chain Acylcarnitines in Heart Failure and Reversibility with Mechanical Circulatory Support. J. Am. Coll. Cardiol. 2016, 67, 291–299. [Google Scholar] [CrossRef]
- Perry, A.S.; Amancherla, K.; Huang, X.; Lance, M.L.; Farber-Eger, E.; Gajjar, P.; Amrute, J.; Stolze, L.; Zhao, S.; Sheng, Q.; et al. Clinical-transcriptional prioritization of the circulating proteome in human heart failure. Cell Rep. Med. 2024, 5, 101704. [Google Scholar] [CrossRef]
- Paul, S.M.; Mytelka, D.S.; Dunwiddie, C.T.; Persinger, C.C.; Munos, B.H.; Lindborg, S.R.; Schacht, A.L. How to improve R&D productivity: The pharmaceutical industry’s grand challenge. Nat. Rev. Drug Discov. 2010, 9, 203–214. [Google Scholar]
- Nguyen, N.; Jennen, D.; Kleinjans, J. Omics technologies to understand drug toxicity mechanisms. Drug Discov. Today 2022, 27, 103348. [Google Scholar] [CrossRef]
- Schirle, M.; Bantscheff, M.; Kuster, B. Mass spectrometry-based proteomics in preclinical drug discovery. Chem. Biol. 2012, 19, 72–84. [Google Scholar] [CrossRef]
- Selvaraj, S.; Fu, Z.; Jones, P.; Kwee, L.C.; Windsor, S.L.; Ilkayeva, O.; Newgard, C.B.; Margulies, K.B.; Husain, M.; Inzucchi, S.E.; et al. Metabolomic Profiling of the Effects of Dapagliflozin in Heart Failure with Reduced Ejection Fraction: DEFINE-HF. Circulation 2022, 146, 808–818. [Google Scholar] [CrossRef] [PubMed]
- Yurista, S.R.; Silljé, H.H.W.; Oberdorf-Maass, S.U.; Schouten, E.-M.; Pavez Giani, M.G.; Hillebrands, J.-L.; van Goor, H.; van Veldhuisen, D.J.; de Boer, R.A.; Westenbrink, B.D. Sodium-glucose co-transporter 2 inhibition with empagliflozin improves cardiac function in non-diabetic rats with left ventricular dysfunction after myocardial infarction. Eur. J. Heart Fail. 2019, 21, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Scisciola, L.; Taktaz, F.; Fontanella, R.A.; Pesapane, A.; Surina; Cataldo, V.; Ghosh, P.; Franzese, M.; Puocci, A.; Paolisso, P.; et al. Targeting high glucose-induced epigenetic modifications at cardiac level: The role of SGLT2 and SGLT2 inhibitors. Cardiovasc. Diabetol. 2023, 22, 24. [Google Scholar] [CrossRef]
- Harrington, J.; Fonarow, G.C.; Khan, M.S.; Hernandez, A.; Anker, S.; Böhm, M.; Greene, S.J.; Felker, G.M.; Vaduganathan, M.; Butler, J. Medication-Attributable Adverse Events in Heart Failure Trials. JACC Heart Fail. 2023, 11, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.; Xiong, Y.; Siddiq, M.M.; Dhanan, P.; Hu, B.; Shewale, B.; Yadaw, A.S.; Jayaraman, G.; Tolentino, R.E.; Chen, Y.; et al. Multiscale mapping of transcriptomic signatures for cardiotoxic drugs. Nat. Commun. 2024, 15, 7968. [Google Scholar] [CrossRef]
- Sharma, A.; Burridge, P.W.; McKeithan, W.L.; Serrano, R.; Shukla, P.; Sayed, N.; Churko, J.M.; Kitani, T.; Wu, H.; Holmström, A.; et al. High-throughput screening of tyrosine kinase inhibitor cardiotoxicity with human induced pluripotent stem cells. Sci. Transl. Med. 2017, 9, eaaf2584. [Google Scholar] [CrossRef]
- van Hasselt, J.G.C.; Rahman, R.; Hansen, J.; Stern, A.; Shim, J.V.; Xiong, Y.; Pickard, A.; Jayaraman, G.; Hu, B.; Mahajan, M.; et al. Transcriptomic profiling of human cardiac cells predicts protein kinase inhibitor-associated cardiotoxicity. Nat. Commun. 2020, 11, 4809. [Google Scholar] [CrossRef]
- Al Sultan, A.; Rattray, Z.; Rattray NJ, W. Integrative analysis of toxicometabolomics and toxicoproteomics data: New molecular insights into thiazolidinedione-induced cardiotoxicity. Metabolomics 2024, 21, 1. [Google Scholar] [CrossRef]
- Iorio, A.; Pozzi, A.; Senni, M. Addressing the Heterogeneity of Heart Failure in Future Randomized Trials. Curr. Heart Fail. Rep. 2017, 14, 197–202. [Google Scholar] [CrossRef]
- Zielinski, J.M.; Luke, J.J.; Guglietta, S.; Krieg, C. High Throughput Multi-Omics Approaches for Clinical Trial Evaluation and Drug Discovery. Front. Immunol. 2021, 12, 590742. [Google Scholar] [CrossRef]
- Rushton, C.A.; Satchithananda, D.K.; Jones, P.W.; Kadam, U.T. Non-cardiovascular comorbidity, severity and prognosis in non-selected heart failure populations: A systematic review and meta-analysis. Int. J. Cardiol. 2015, 196, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Bozkurt, B.; Aguilar, D.; Deswal, A.; Dunbar, S.B.; Francis, G.S.; Horwich, T.; Jessup, M.; Kosiborod, M.; Pritchett, A.M.; Ramasubbu, K.; et al. Contributory Risk and Management of Comorbidities of Hypertension, Obesity, Diabetes Mellitus, Hyperlipidemia, and Metabolic Syndrome in Chronic Heart Failure: A Scientific Statement From the American Heart Association. Circulation 2016, 134, e535–e578. [Google Scholar] [CrossRef] [PubMed]
- Antman, E.M.; Loscalzo, J. Precision medicine in cardiology. Nat. Rev. Cardiol. 2016, 13, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Lavalle, C.; Mariani, M.V.; Severino, P.; Palombi, M.; Trivigno, S.; D’Amato, A.; Silvetti, G.; Pierucci, N.; Di Lullo, L.; Chimenti, C.; et al. Efficacy of modern therapies for heart failure with reduced ejection fraction in specific population subgroups: A systematic review and network meta-analysis. Cardiorenal Med. 2024, 14, 570–580. [Google Scholar] [CrossRef]
- Center for Drug Evaluation & Research. Treatment for Heart Failure: Endpoints for Drug Development Guidance for Industry. U.S. Food and Drug Administration. 2020. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/treatment-heart-failure-endpoints-drug-development-guidance-industry (accessed on 1 February 2025).
- Using machine learning approaches for multi-omics data analysis: A review. Biotechnol. Adv. 2021, 49, 107739.
- Piccini, J.P.; Abraham, W.T.; Dufton, C.; Carroll, I.A.; Healey, J.S.; van Veldhuisen, D.J.; Sauer, W.H.; Anand, I.S.; White, M.; Wilton, S.B.; et al. Bucindolol for the Maintenance of Sinus Rhythm in a Genotype-Defined HF Population: The GENETIC-AF Trial. JACC Heart Fail. 2019, 7, 586–598. [Google Scholar] [CrossRef]
- Ibrahim, N.E.; Burnett, J.C., Jr.; Butler, J.; Camacho, A.; Felker, G.M.; Fiuzat, M.; O’Connor, C.; Solomon, S.D.; Vaduganathan, M.; Zile, M.R.; et al. Natriuretic Peptides as Inclusion Criteria in Clinical Trials: A JACC: Heart Failure Position Paper. JACC Heart Fail. 2020, 8, 347–358. [Google Scholar] [CrossRef]
- Solomon, S.D.; McMurray, J.J.V.; Anand, I.S.; Ge, J.; Lam, C.S.P.; Maggioni, A.P.; Martinez, F.; Packer, M.; Pfeffer, M.A.; Pieske, B.; et al. Angiotensin-Neprilysin Inhibition in Heart Failure with Preserved Ejection Fraction. N. Engl. J. Med. 2019, 381, 1609–1620. [Google Scholar] [CrossRef]
- Anker, S.D.; Butler, J.; Filippatos, G.; Ferreira, J.P.; Bocchi, E.; Böhm, M.; Brunner-La Rocca, H.-P.; Choi, D.-J.; Chopra, V.; Chuquiure-Valenzuela, E.; et al. Empagliflozin in Heart Failure with a Preserved Ejection Fraction. N. Engl. J. Med. 2021, 385, 1451–1461. [Google Scholar] [CrossRef]
- Pitt, B.; Pfeffer, M.A.; Assmann, S.F.; Boineau, R.; Anand, I.S.; Claggett, B.; Clausell, N.; Desai, A.S.; Diaz, R.; Fleg, J.L.; et al. Spironolactone for heart failure with preserved ejection fraction. N. Engl. J. Med. 2014, 370, 1383–1392. [Google Scholar] [CrossRef]
- Ibrahim, N.E.; Gaggin, H.K.; Konstam, M.A.; Januzzi, J.L., Jr. Established and Emerging Roles of Biomarkers in Heart Failure Clinical Trials. Circ. Heart Fail. 2016, 9, e002528. [Google Scholar] [CrossRef] [PubMed]
- Pabón, M.A.; Cunningham, J.W.; Claggett, B.L.; Packer, M.; Zile, M.; Pfeffer, M.A.; Lefkowitz, M.; Shi, V.; Rizkala, A.; McMurray, J.J.V.; et al. Natriuretic peptide-based inclusion criteria in heart failure with preserved ejection fraction clinical trials: Insights from PARAGON-HF. Eur. J. Heart Fail. 2022, 24, 672–677. [Google Scholar] [CrossRef] [PubMed]
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey, D.E., Jr.; Colvin, M.M.; Drazner, M.H.; Filippatos, G.S.; Fonarow, G.C.; Givertz, M.M.; et al. 2017 ACC/AHA/HFSA Focused Update of the 2013 ACCF/AHA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. Circulation 2017, 136, e137–e161. [Google Scholar] [PubMed]
- Packer, M.; Fowler, M.B.; Roecker, E.B.; Coats, A.J.S.; Katus, H.A.; Krum, H.; Mohacsi, P.; Rouleau, J.L.; Tendera, M.; Staiger, C.; et al. Effect of carvedilol on the morbidity of patients with severe chronic heart failure: Results of the carvedilol prospective randomized cumulative survival (COPERNICUS) study. Circulation 2002, 106, 2194–2199. [Google Scholar] [CrossRef]
- Motiwala, S.R.; Szymonifka, J.; Belcher, A.; Weiner, R.B.; Baggish, A.L.; Sluss, P.; Gaggin, H.K.; Bhardwaj, A.; Januzzi, J.L. Serial measurement of galectin-3 in patients with chronic heart failure: Results from the ProBNP Outpatient Tailored Chronic Heart Failure Therapy (PROTECT) study. Eur. J. Heart Fail. 2013, 15, 1157–1163. [Google Scholar] [CrossRef]
- Lanfear, D.E.; Luzum, J.A.; She, R.; Gui, H.; Donahue, M.P.; O’Connor, C.M.; Adams, K.F.; Sanders-van Wijk, S.; Zeld, N.; Maeder, M.T.; et al. Polygenic Score for β-Blocker Survival Benefit in European Ancestry Patients with Reduced Ejection Fraction Heart Failure. Circ. Heart Fail. 2020, 13, e007012. [Google Scholar] [CrossRef]
- Topkara, V.K.; Mann, D.L. Role of microRNAs in cardiac remodeling and heart failure. Cardiovasc. Drugs Ther. 2011, 25, 171–182. [Google Scholar] [CrossRef]
- Montgomery, R.L.; Hullinger, T.G.; Semus, H.M.; Dickinson, B.A.; Seto, A.G.; Lynch, J.M.; Stack, C.; Latimer, P.A.; Olson, E.N.; van Rooij, E. Therapeutic inhibition of miR-208a improves cardiac function and survival during heart failure. Circulation 2011, 124, 1537–1547. [Google Scholar] [CrossRef]
- Bocchi, E.A.; Vilella de Moraes, A.V.; Esteves-Filho, A.; Bacal, F.; Auler, J.O.; Carmona, M.J.; Bellotti, G.; Ramires, A.F. L-arginine reduces heart rate and improves hemodynamics in severe congestive heart failure. Clin. Cardiol. 2000, 23, 205–210. [Google Scholar] [CrossRef]
- Hermann, H.P.; Pieske, B.; Schwarzmüller, E.; Keul, J.; Just, H.; Hasenfuss, G. Haemodynamic effects of intracoronary pyruvate in patients with congestive heart failure: An open study. Lancet 1999, 353, 1321–1323. [Google Scholar] [CrossRef]
- Ouwerkerk, W.; Belo Pereira, J.P.; Maasland, T.; Emmens, J.E.; Figarska, S.M.; Tromp, J.; Koekemoer, A.L.; Nelson, C.P.; Nath, M.; Romaine, S.P.R.; et al. Multiomics Analysis Provides Novel Pathways Related to Progression of Heart Failure. J. Am. Coll. Cardiol. 2023, 82, 1921–1931. [Google Scholar] [CrossRef]
- Paulus, W.J.; Tschöpe, C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Raphael, R.; Purushotham, D.; Gastonguay, C.; Chesnik, M.A.; Kwok, W.-M.; Wu, H.-E.; Shah, S.J.; Mirza, S.P.; Strande, J.L. Combining patient proteomics and in vitro cardiomyocyte phenotype testing to identify potential mediators of heart failure with preserved ejection fraction. J. Transl. Med. 2016, 14, 18. [Google Scholar] [CrossRef] [PubMed]
- Yadalam, A.K.; Gold, M.E.; Patel, K.J.; Liu, C.; Razavi, A.C.; Jain, V.; Vatsa, N.; Gold, D.; Owais, M.; Haroun, N.; et al. Proteomics-Based Soluble Urokinase Plasminogen Activator Receptor Levels Are Associated with Incident Heart Failure Risk. JACC Adv. 2025, 4, 101442. [Google Scholar] [CrossRef] [PubMed]
- Thummel, K.E.; Lin, Y.S. Sources of interindividual variability. Methods Mol. Biol. 2014, 1113, 363–415. [Google Scholar]
- Lanfear, D.E.; Hrobowski, T.N.; Peterson, E.L.; Wells, K.E.; Swadia, T.V.; Spertus, J.A.; Williams, L.K. Association of β-blocker exposure with outcomes in heart failure differs between African American and white patients. Circ. Heart Fail. 2012, 5, 202–208. [Google Scholar] [CrossRef]
- Cohn, J.N.; Archibald, D.G.; Ziesche, S.; Franciosa, J.A.; Harston, W.E.; Tristani, F.E.; Dunkman, W.B.; Jacobs, W.; Francis, G.S.; Flohr, K.H. Effect of vasodilator therapy on mortality in chronic congestive heart failure. Results of a Veterans Administration Cooperative Study. N. Engl. J. Med. 1986, 314, 1547–1552. [Google Scholar] [CrossRef]
- Hjalmarson, A.; Goldstein, S.; Fagerberg, B.; Wedel, H.; Waagstein, F.; Kjekshus, J.; Wikstrand, J.; El Allaf, D.; Vítovec, J.; Aldershvile, J.; et al. Effects of controlled-release metoprolol on total mortality, hospitalizations, and well-being in patients with heart failure: The Metoprolol CR/XL Randomized Intervention Trial in congestive heart failure (MERIT-HF). MERIT-HF Study Group. JAMA 2000, 283, 1295–1302. [Google Scholar] [CrossRef]
- CIBIS II Investigators and Committees. The Cardiac Insufficiency Bisoprolol Study II (CIBIS-II): A randomised trial. Lancet 1999, 353, 9–13. [Google Scholar] [CrossRef]
- Shin, J.; Johnson, J.A. Beta-blocker pharmacogenetics in heart failure. Heart Fail. Rev. 2010, 15, 187–196. [Google Scholar] [CrossRef]
- Ibrahim, N.E.; Januzzi, J.L., Jr. Beyond Natriuretic Peptides for Diagnosis and Management of Heart Failure. Clin. Chem. 2017, 63, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Bikdeli, B.; Punnanithinont, N.; Akram, Y.; Lee, I.; Desai, N.R.; Ross, J.S.; Krumholz, H.M. Two Decades of Cardiovascular Trials With Primary Surrogate Endpoints: 1990–2011. J. Am. Heart Assoc. 2017, 6, e005285. [Google Scholar] [CrossRef]
- Zannad, F.; Garcia, A.A.; Anker, S.D.; Armstrong, P.W.; Calvo, G.; Cleland, J.G.F.; Cohn, J.N.; Dickstein, K.; Domanski, M.J.; Ekman, I.; et al. Clinical outcome endpoints in heart failure trials: A European Society of Cardiology Heart Failure Association consensus document. Eur. J. Heart Fail. 2013, 15, 1082–1094. [Google Scholar] [CrossRef]
- Greene, S.J.; Mentz, R.J.; Fiuzat, M.; Butler, J.; Solomon, S.D.; Ambrosy, A.P.; Mehta, C.; Teerlink, J.R.; Zannad, F.; O’Connor, C.M. Reassessing the Role of Surrogate End Points in Drug Development for Heart Failure. Circulation 2018, 138, 1039–1053. [Google Scholar] [CrossRef] [PubMed]
- Vaduganathan, M.; Claggett, B.; Packer, M.; McMurray, J.J.V.; Rouleau, J.L.; Zile, M.R.; Swedberg, K.; Solomon, S.D. Natriuretic Peptides as Biomarkers of Treatment Response in Clinical Trials of Heart Failure. JACC Heart Fail. 2018, 6, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Gheorghiade, M.; Böhm, M.; Greene, S.J.; Fonarow, G.C.; Lewis, E.F.; Zannad, F.; Solomon, S.D.; Baschiera, F.; Botha, J.; Hua, T.A.; et al. Effect of aliskiren on postdischarge mortality and heart failure readmissions among patients hospitalized for heart failure: The ASTRONAUT randomized trial. JAMA 2013, 309, 1125–1135. [Google Scholar] [CrossRef]
- Greene, S.J.; Fonarow, G.C.; Solomon, S.D.; Subacius, H.P.; Ambrosy, A.P.; Vaduganathan, M.; Maggioni, A.P.; Böhm, M.; Lewis, E.F.; Zannad, F.; et al. Influence of atrial fibrillation on post-discharge natriuretic peptide trajectory and clinical outcomes among patients hospitalized for heart failure: Insights from the ASTRONAUT trial. Eur. J. Heart Fail. 2017, 19, 552–562. [Google Scholar] [CrossRef]
- Savarese, G.; Uijl, A.; Ouwerkerk, W.; Tromp, J.; Anker, S.D.; Dickstein, K.; Hage, C.; Lam, C.S.P.; Lang, C.C.; Metra, M.; et al. Biomarker changes as surrogate endpoints in early-phase trials in heart failure with reduced ejection fraction. ESC Heart Fail. 2022, 9, 2107–2118. [Google Scholar] [CrossRef]
- Packer, M.; McMurray, J.J.V.; Desai, A.S.; Gong, J.; Lefkowitz, M.P.; Rizkala, A.R.; Rouleau, J.L.; Shi, V.C.; Solomon, S.D.; Swedberg, K.; et al. Angiotensin receptor neprilysin inhibition compared with enalapril on the risk of clinical progression in surviving patients with heart failure. Circulation 2015, 131, 54–61. [Google Scholar] [CrossRef]
- Peacock, W.F., 4th; De Marco, T.; Fonarow, G.C.; Diercks, D.; Wynne, J.; Apple, F.S.; Wu, A.H.B.; ADHERE Investigators. Cardiac troponin and outcome in acute heart failure. N. Engl. J. Med. 2008, 358, 2117–2126. [Google Scholar] [CrossRef]
- Felker, G.M.; Mentz, R.J.; Teerlink, J.R.; Voors, A.A.; Pang, P.S.; Ponikowski, P.; Greenberg, B.H.; Filippatos, G.; Davison, B.A.; Cotter, G.; et al. Serial high sensitivity cardiac troponin T measurement in acute heart failure: Insights from the RELAX-AHF study. Eur. J. Heart Fail. 2015, 17, 1262–1270. [Google Scholar] [CrossRef] [PubMed]
- Pang, P.S.; Teerlink, J.R.; Voors, A.A.; Ponikowski, P.; Greenberg, B.H.; Filippatos, G.; Felker, G.M.; Davison, B.A.; Cotter, G.; Kriger, J.; et al. Use of High-Sensitivity Troponin T to Identify Patients with Acute Heart Failure at Lower Risk for Adverse Outcomes: An Exploratory Analysis From the RELAX-AHF Trial. JACC Heart Fail. 2016, 4, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Bayes-Genis, A.; Aimo, A.; Jhund, P.; Richards, M.; de Boer, R.A.; Arfsten, H.; Fabiani, I.; Lupón, J.; Anker, S.D.; González, A.; et al. Biomarkers in heart failure clinical trials. A review from the Biomarkers Working Group of the Heart Failure Association of the European Society of Cardiology. Eur. J. Heart Fail. 2022, 24, 1767–1777. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Omics Type | Advantages | Limitations |
|---|---|---|
| Genomics |
|
|
| Proteomics |
|
|
| Transcriptomics |
|
|
| Metabolomics |
|
|
| Epigenomics |
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rasooly, D.; Pereira, A.C.; Joseph, J. Drug Discovery and Development for Heart Failure Using Multi-Omics Approaches. Int. J. Mol. Sci. 2025, 26, 2703. https://doi.org/10.3390/ijms26062703
Rasooly D, Pereira AC, Joseph J. Drug Discovery and Development for Heart Failure Using Multi-Omics Approaches. International Journal of Molecular Sciences. 2025; 26(6):2703. https://doi.org/10.3390/ijms26062703
Chicago/Turabian StyleRasooly, Danielle, Alexandre C. Pereira, and Jacob Joseph. 2025. "Drug Discovery and Development for Heart Failure Using Multi-Omics Approaches" International Journal of Molecular Sciences 26, no. 6: 2703. https://doi.org/10.3390/ijms26062703
APA StyleRasooly, D., Pereira, A. C., & Joseph, J. (2025). Drug Discovery and Development for Heart Failure Using Multi-Omics Approaches. International Journal of Molecular Sciences, 26(6), 2703. https://doi.org/10.3390/ijms26062703