
Methamphetamine and Methamphetamine-Induced Neuronal Exosomes Modulate the Activity of Rab7a via PTEN to Exert an Influence on the Disordered Autophagic Flux Induced in Neurons

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results
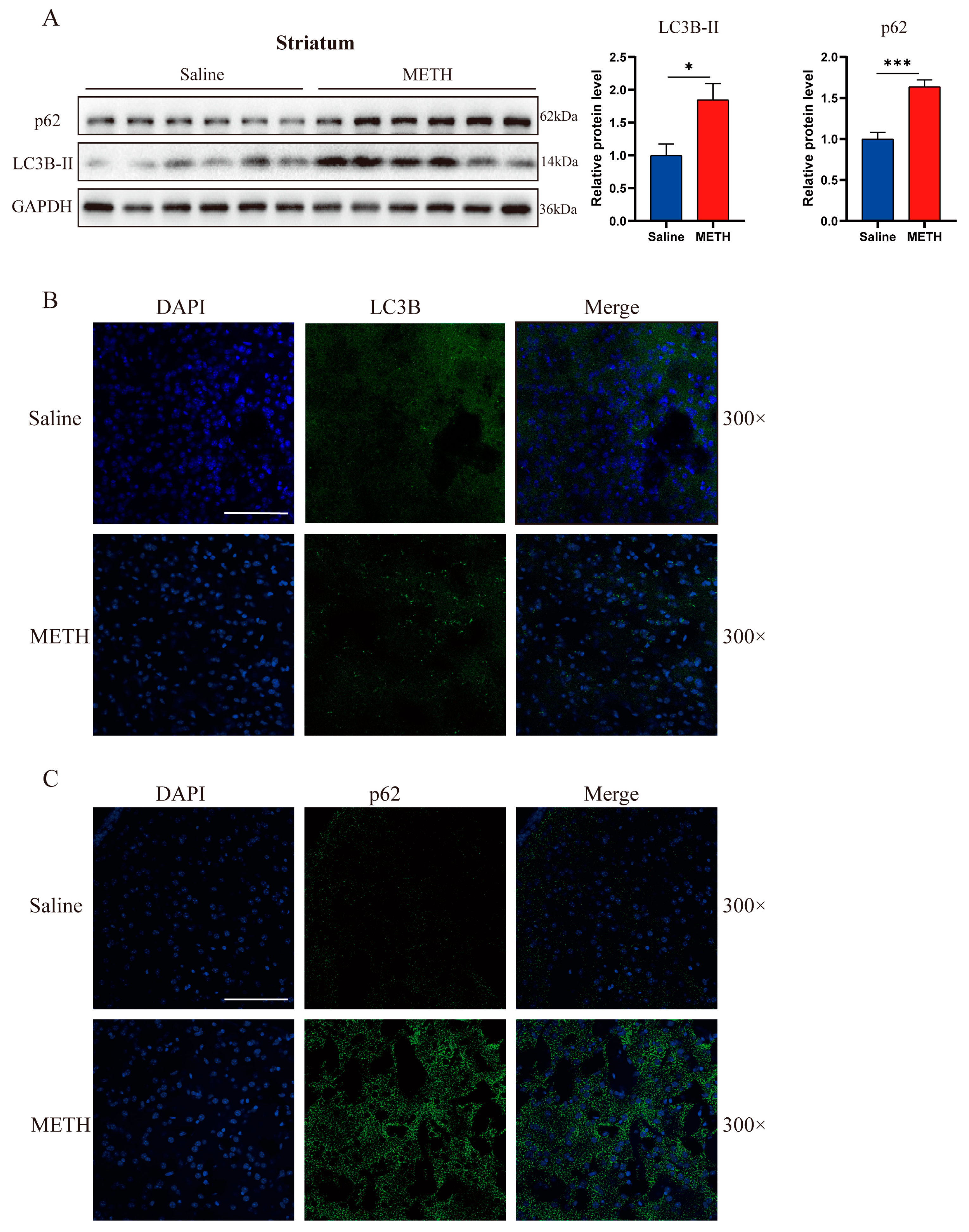
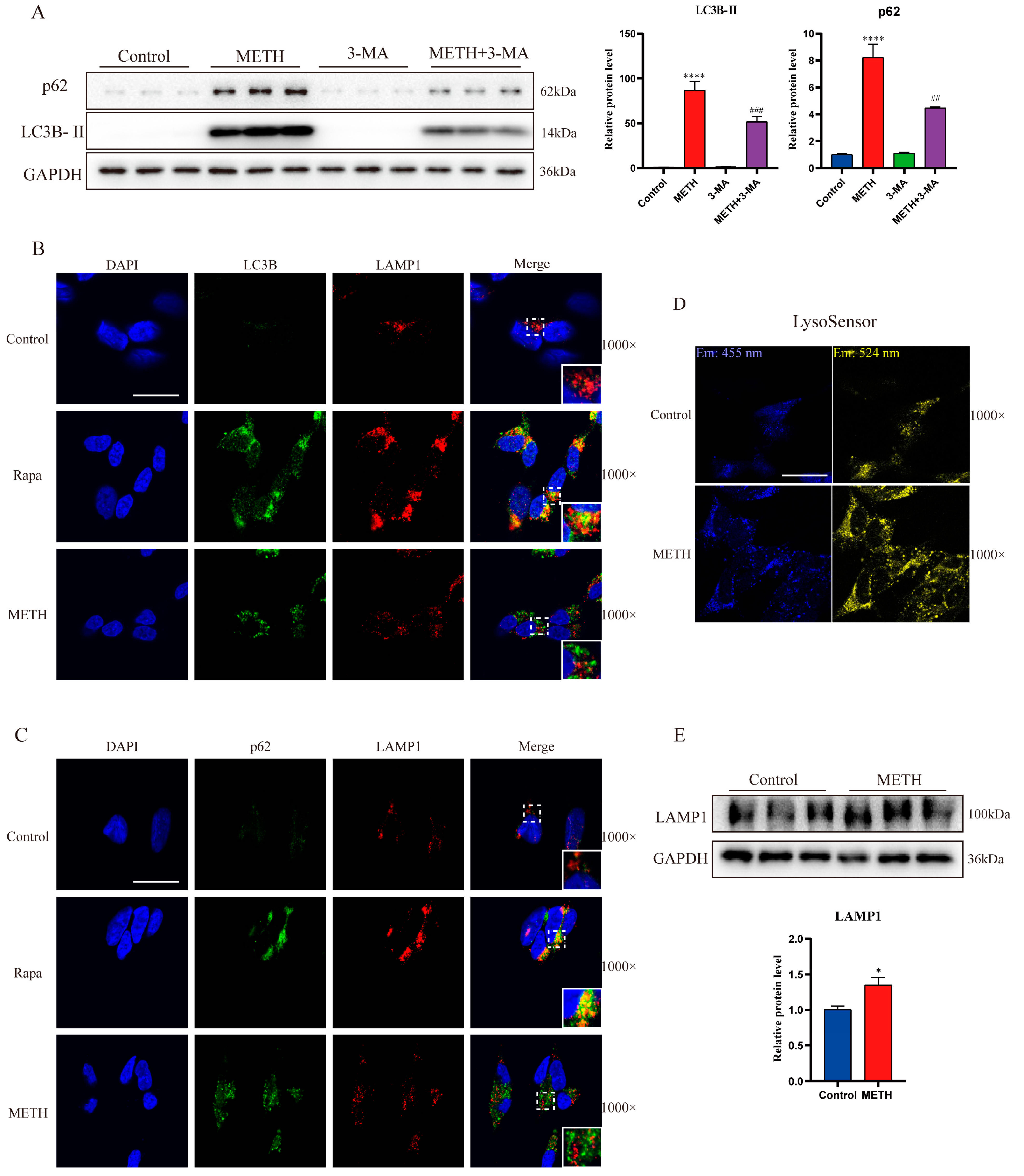
2.1. Exposure to Methamphetamine Induces Autophagosome Accumulation Both In Vivo and In Vitro
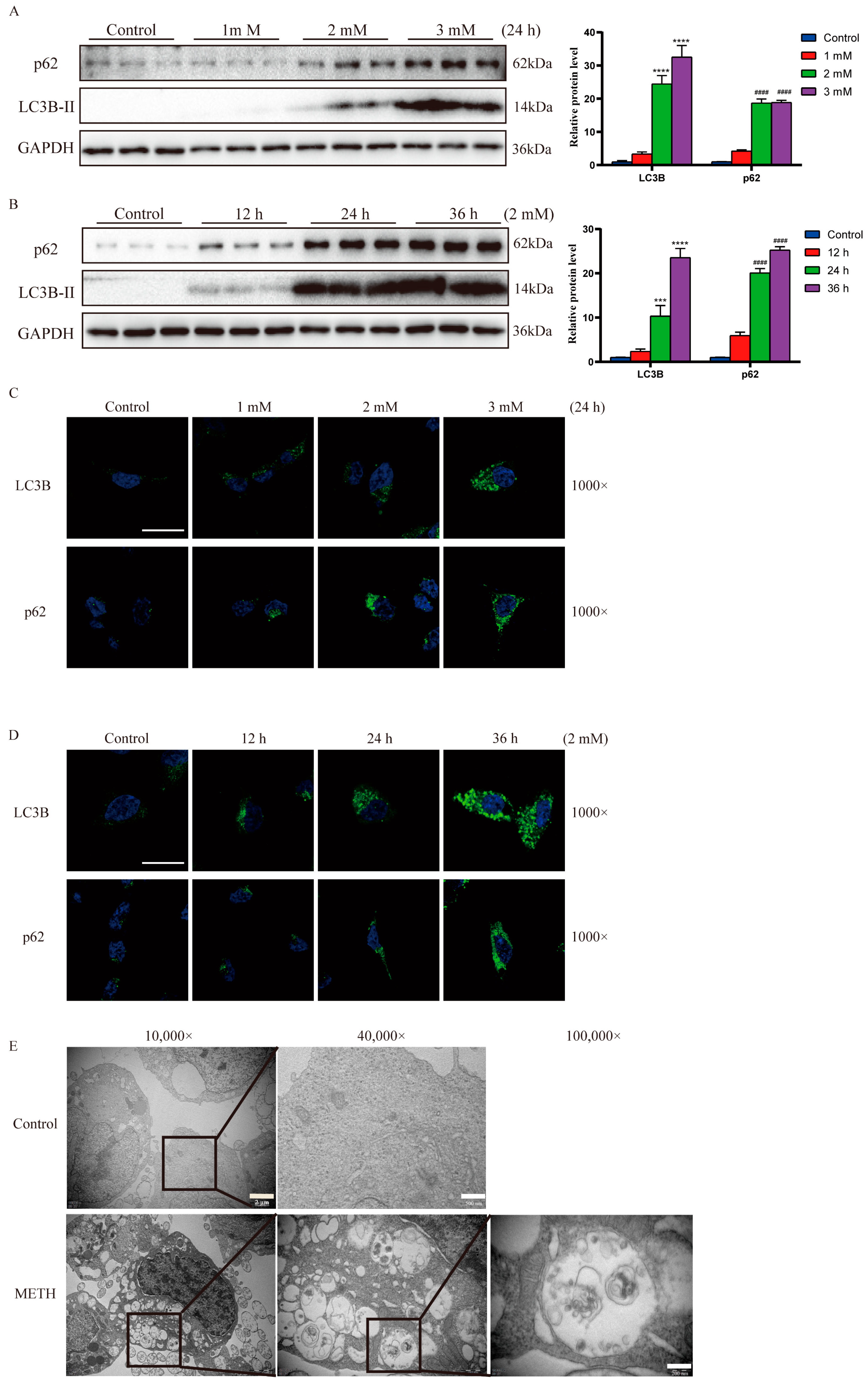
2.2. Methamphetamine Exposure Induces an Upregulation of Autophagosome Formation
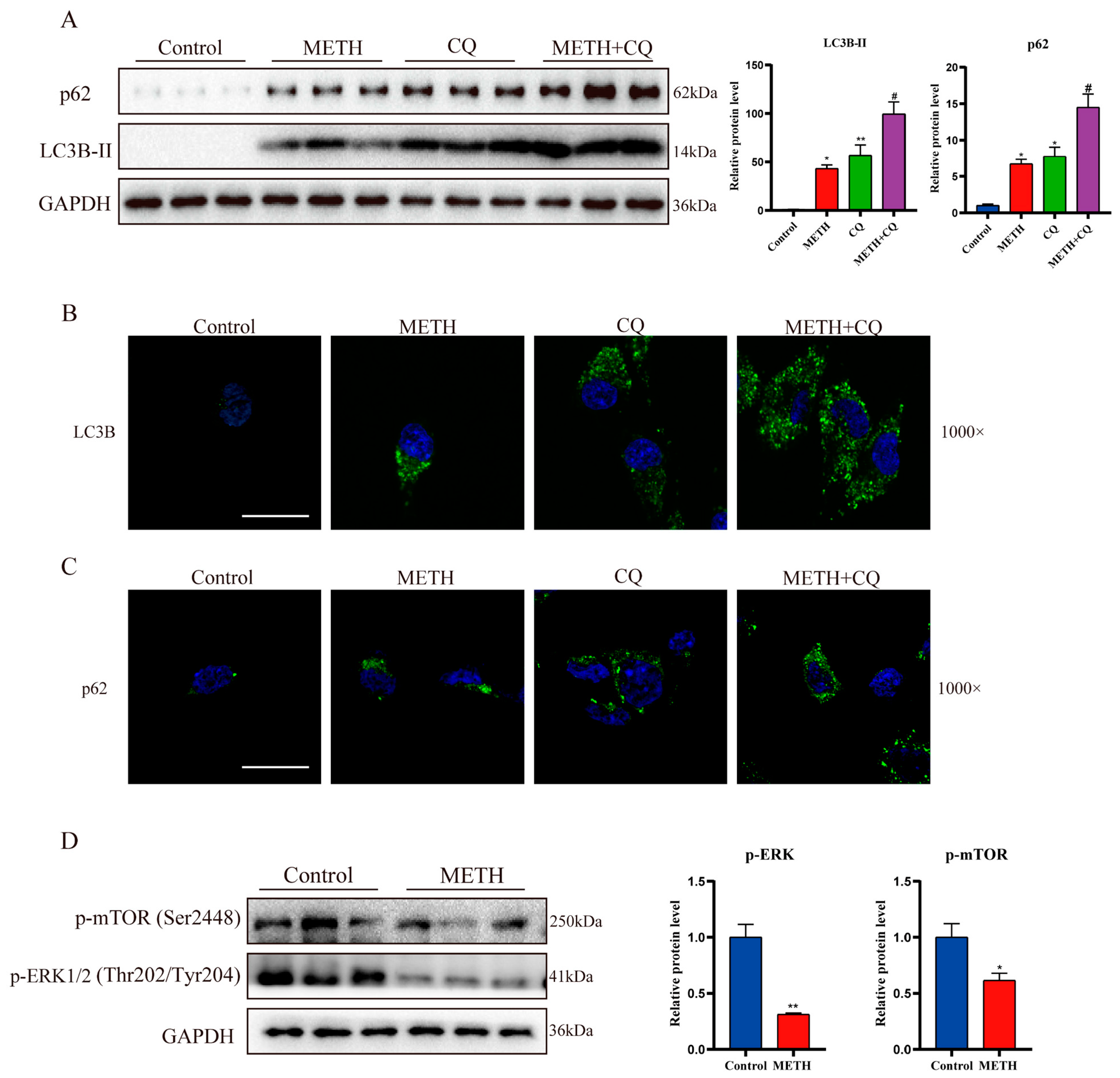
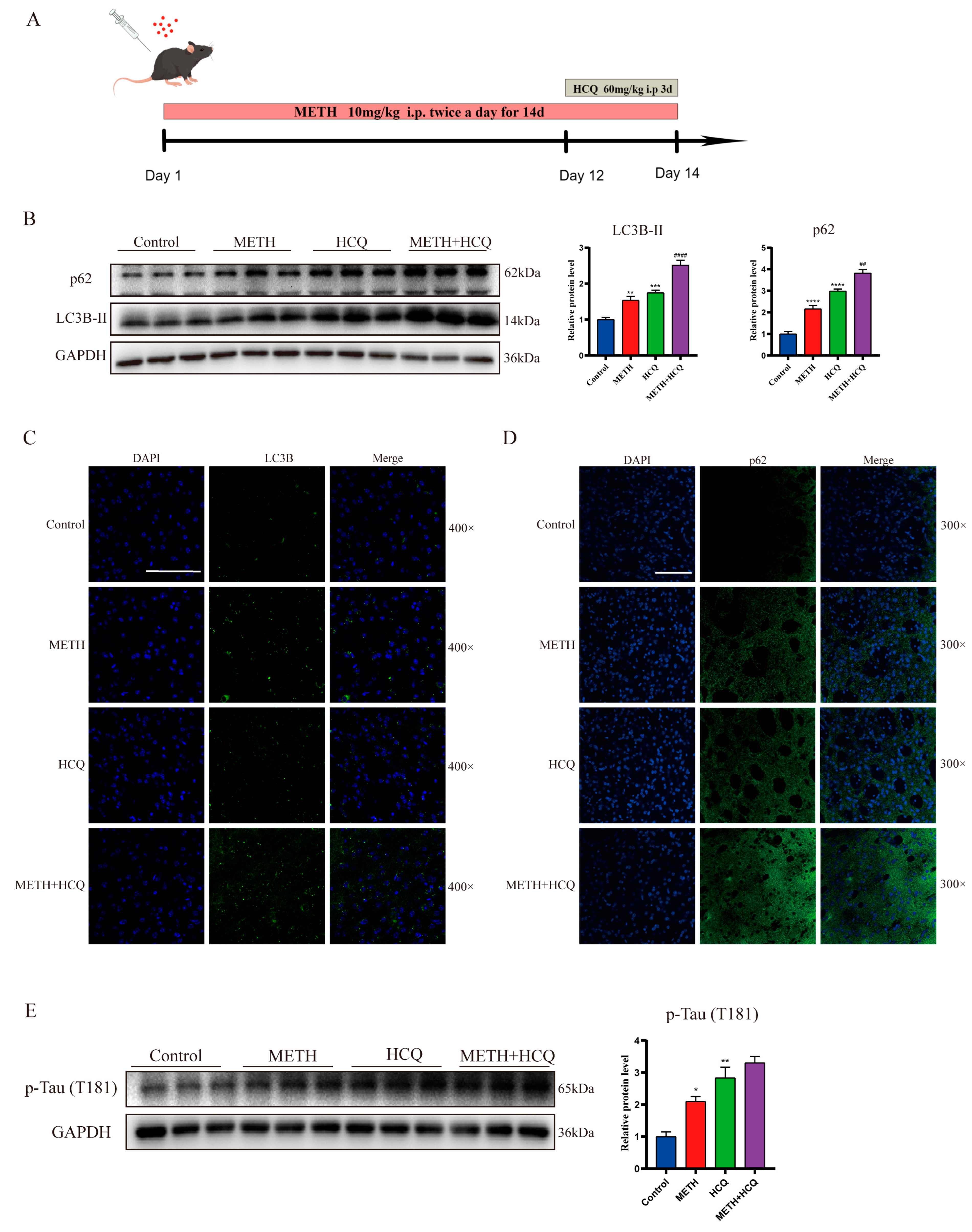
2.3. Methamphetamine Exposure Leads to a Decrease in Autophagosome Degradation In Vitro
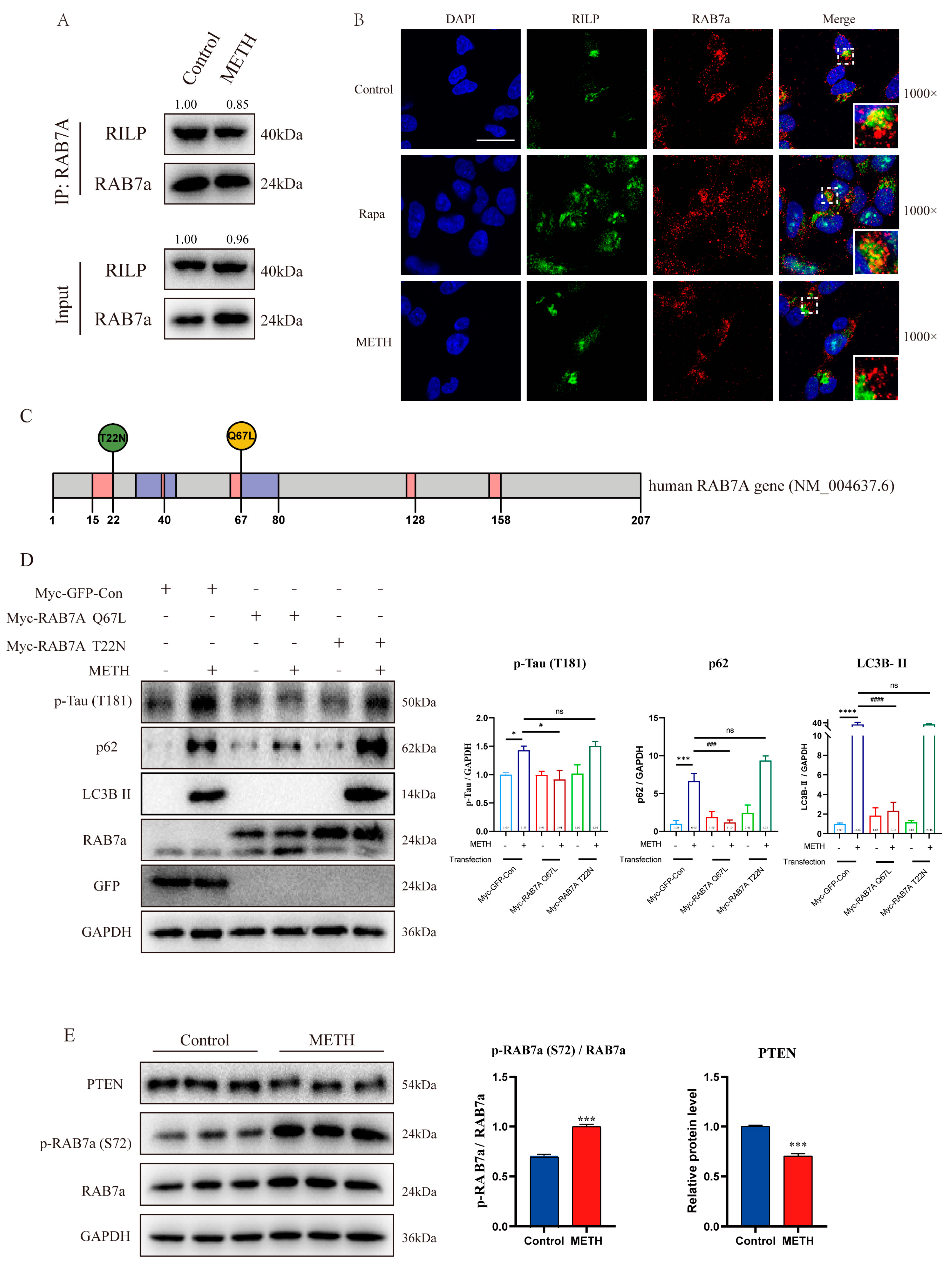
2.4. METH Inhibits the Transport and Fusion of Autophagosome by Inhibiting Rab7a Activity
2.5. METH Can Induce Neurons to Release Exosomes Containing Differential Express miRNAs (DE-miRNAs) and Lead to a Decrease in the Level of PTEN in Neurons, Thereby Affecting the Phosphorylation Level of Rab7a
3. Discussion
4. Materials and Methods
4.1. Animal Protocol
4.2. Cell Culture and Transfection
4.3. Immunofluorescence Labelling
4.4. Electron Microscopy
4.5. Lysosomal Acidity Analysis
4.6. Western Blotting
4.7. Co-Immunoprecipitation
4.8. Extraction and Application of Exosomes
4.9. Identification of Exosomes
4.10. MiRNA Array Analysis
4.11. Prediction of Differentially Expressed miRNA Target Genes and Bioinformatics Analysis
4.12. Quantitative Real-Time PCR Validation of miRNA Expression
4.13. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- UNODC. World Drug Report 2023 (United Nations Publication, 2023). Available online: www.unodc.org/unodc/en/data-and-analysis/world-drug-report-2023.html (accessed on 15 March 2024).
- Xu, X.; Huang, E.; Luo, B.; Cai, D.; Zhao, X.; Luo, Q.; Jin, Y.; Chen, L.; Wang, Q.; Liu, C.; et al. Methamphetamine exposure triggers apoptosis and autophagy in neuronal cells by activating the C/EBPbeta-related signaling pathway. FASEB. J. 2018, 32, 6737–6759. [Google Scholar] [CrossRef]
- Tan, X.H.; Zhang, K.K.; Xu, J.T.; Qu, D.; Chen, L.J.; Li, J.H.; Wang, Q.; Wang, H.J.; Xie, X.L. Luteolin alleviates methamphetamine-induced neurotoxicity by suppressing PI3K/Akt pathway-modulated apoptosis and autophagy in rats. Food. Chem. Toxicol. 2020, 137, 111179. [Google Scholar] [CrossRef]
- Yang, X.; Wang, Y.; Li, Q.; Zhong, Y.; Chen, L.; Du, Y.; He, J.; Liao, L.; Xiong, K.; Yi, C.X.; et al. The Main Molecular Mechanisms Underlying Methamphetamine- Induced Neurotoxicity and Implications for Pharmacological Treatment. Front. Molec. Neurosci. 2018, 11, 186. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Huang, E.; Tai, Y.; Zhao, X.; Chen, X.; Chen, C.; Chen, R.; Liu, C.; Lin, Z.; Wang, H.; et al. Nupr1 Modulates Methamphetamine-Induced Dopaminergic Neuronal Apoptosis and Autophagy through CHOP-Trib3-Mediated Endoplasmic Reticulum Stress Signaling Pathway. Front. Molec. Neurosci. 2017, 10, 203. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Chen, R.; Chen, L.; Qiu, P.; Ai, X.; Huang, E.; Huang, W.; Chen, C.; Liu, C.; Lin, Z.; et al. Effects of DDIT4 in Methamphetamine-Induced Autophagy and Apoptosis in Dopaminergic Neurons. Mol. Neurobiol. 2017, 54, 1642–1660. [Google Scholar] [CrossRef]
- Larsen, K.E.; Fon, E.A.; Hastings, T.G.; Edwards, R.H.; Sulzer, D. Methamphetamine-induced degeneration of dopaminergic neurons involves autophagy and upregulation of dopamine synthesis. J. Neurosci. 2002, 22, 8951–8960. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Chandramani-Shivalingappa, P.; Jin, H.; Ghosh, A.; Anantharam, V.; Ali, S.; Kanthasamy, A.G.; Kanthasamy, A. Methamphetamine-induced neurotoxicity linked to ubiquitin-proteasome system dysfunction and autophagy-related changes that can be modulated by protein kinase C delta in dopaminergic neuronal cells. Neuroscience 2012, 210, 308–332. [Google Scholar] [CrossRef]
- Nopparat, C.; Porter, J.E.; Ebadi, M.; Govitrapong, P. The mechanism for the neuroprotective effect of melatonin against methamphetamine-induced autophagy. J. Pineal Res. 2010, 49, 382–389. [Google Scholar] [CrossRef]
- Subu, R.; Jayanthi, S.; Cadet, J.L. Compulsive methamphetamine taking induces autophagic and apoptotic markers in the rat dorsal striatum. Arch. Toxicol. 2020, 94, 3515–3526. [Google Scholar] [CrossRef]
- Ryskalin, L.; Puglisi-Allegra, S.; Lazzeri, G.; Biagioni, F.; Busceti, C.L.; Balestrini, L.; Fornasiero, A.; Leone, S.; Pompili, E.; Ferrucci, M.; et al. Neuroprotective Effects of Curcumin in Methamphetamine-Induced Toxicity. Molecules 2021, 26, 2493. [Google Scholar] [CrossRef]
- Xie, X.L.; He, J.T.; Wang, Z.T.; Xiao, H.Q.; Zhou, W.T.; Du, S.H.; Xue, Y.; Wang, Q. Lactulose attenuates METH-induced neurotoxicity by alleviating the impaired autophagy, stabilizing the perturbed antioxidant system and suppressing apoptosis in rat striatum. Toxicol. Lett. 2018, 289, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 2021, 17, 1797280. [Google Scholar] [CrossRef]
- Tian, X.; Teng, J.; Chen, J. New insights regarding SNARE proteins in autophagosome-lysosome fusion. Autophagy 2021, 17, 2680–2688. [Google Scholar] [CrossRef] [PubMed]
- Tan, E.; Tang, B.L. Rab7a and Mitophagosome Formation. Cells 2019, 8, 224. [Google Scholar] [CrossRef]
- Jin, X.; Wang, K.; Wang, L.; Liu, W.; Zhang, C.; Qiu, Y.; Liu, W.; Zhang, H.; Zhang, D.; Yang, Z.; et al. RAB7 activity is required for the regulation of mitophagy in oocyte meiosis and oocyte quality control during ovarian aging. Autophagy 2022, 18, 643–660. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Luong, G.; Plastikwala, F.; Sun, Y. Control of Rab7a activity and localization through endosomal type Igamma PIP 5-kinase is required for endosome maturation and lysosome function. FASEB. J. 2020, 34, 2730–2748. [Google Scholar] [CrossRef]
- Gutierrez, M.G.; Munafo, D.B.; Beron, W.; Colombo, M.I. Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J. Cell Sci. 2004, 117, 2687–2697. [Google Scholar] [CrossRef]
- Shinde, S.R.; Maddika, S. PTEN modulates EGFR late endocytic trafficking and degradation by dephosphorylating Rab7. Nat. Commun. 2016, 7, 10689. [Google Scholar] [CrossRef]
- Deng, P.; Fan, T.; Gao, P.; Peng, Y.; Li, M.; Li, J.; Qin, M.; Hao, R.; Wang, L.; Li, M.; et al. SIRT5-Mediated Desuccinylation of RAB7A Protects Against Cadmium-Induced Alzheimer’s Disease-Like Pathology by Restoring Autophagic Flux. Adv. Sci. 2024, 11, e2402030. [Google Scholar] [CrossRef]
- Modica, G.; Skorobogata, O.; Sauvageau, E.; Vissa, A.; Yip, C.M.; Kim, P.K.; Wurtele, H.; Lefrancois, S. Rab7 palmitoylation is required for efficient endosome-to-TGN trafficking. J. Cell Sci. 2017, 130, 2579–2590. [Google Scholar] [CrossRef]
- Rak, A.; Pylypenko, O.; Niculae, A.; Pyatkov, K.; Goody, R.S.; Alexandrov, K. Structure of the Rab7:REP-1 complex: Insights into the mechanism of Rab prenylation and choroideremia disease. Cell 2004, 117, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Shinde, S.R.; Maddika, S. A modification switch on a molecular switch: Phosphoregulation of Rab7 during endosome maturation. Small GTPases 2016, 7, 164–167. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367, aau6977. [Google Scholar] [CrossRef]
- Cheng, L.; Hill, A.F. Therapeutically harnessing extracellular vesicles. Nat. Rev. Drug Discov. 2022, 21, 379–399. [Google Scholar] [CrossRef] [PubMed]
- Budnik, V.; Ruiz-Canada, C.; Wendler, F. Extracellular vesicles round off communication in the nervous system. Nat. Rev. Neurosci. 2016, 17, 160–172. [Google Scholar] [CrossRef]
- Jiao, Z.; He, Z.; Liu, N.; Lai, Y.; Zhong, T. Multiple roles of neuronal extracellular vesicles in neurological disorders. Front. Cell. Neurosci. 2022, 16, 979856. [Google Scholar] [CrossRef]
- Thompson, A.G.; Gray, E.; Heman-Ackah, S.M.; Mager, I.; Talbot, K.; Andaloussi, S.E.; Wood, M.J.; Turner, M.R. Extracellular vesicles in neurodegenerative disease—Pathogenesis to biomarkers. Nat. Rev. Neurol. 2016, 12, 346–357. [Google Scholar] [CrossRef]
- Xia, X.; Wang, Y.; Zheng, J.C. Extracellular vesicles, from the pathogenesis to the therapy of neurodegenerative diseases. Transl. Neurodegener. 2022, 11, 53. [Google Scholar] [CrossRef]
- Jia, L.; Zhu, M.; Yang, J.; Pang, Y.; Wang, Q.; Li, T.; Li, F.; Wang, Q.; Li, Y.; Wei, Y. Exosomal MicroRNA-Based Predictive Model for Preclinical Alzheimer’s Disease: A Multicenter Study. Biol. Psychiatry 2022, 92, 44–53. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdalla, F.C.; Abeliovich, H.; Abraham, R.T.; Arozena, A.A.; Adeli, K.; Agholme, L.; Agnello, M.; Agostinis, P.; Aguirre-Ghiso, J.A. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012, 8, 445–544. [Google Scholar] [CrossRef]
- Bucci, C.; Thomsen, P.; Nicoziani, P.; McCarthy, J.; van Deurs, B. Rab7: A key to lysosome biogenesis. Mol. Biol. Cell. 2000, 11, 467–480. [Google Scholar] [CrossRef]
- Seaman, M.; Mukadam, A.S.; Breusegem, S.Y. Inhibition of TBC1D5 activates Rab7a and can enhance the function of the retromer cargo-selective complex. J. Cell Sci. 2018, 131, 217398. [Google Scholar] [CrossRef] [PubMed]
- Cantalupo, G.; Alifano, P.; Roberti, V.; Bruni, C.B.; Bucci, C. Rab-interacting lysosomal protein (RILP): The Rab7 effector required for transport to lysosomes. EMBO. J. 2001, 20, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Romero, R.K.; Peralta, E.R.; Guenther, G.G.; Wong, S.Y.; Edinger, A.L. Rab7 activation by growth factor withdrawal contributes to the induction of apoptosis. Mol. Biol. Cell. 2009, 20, 2831–2840. [Google Scholar] [CrossRef] [PubMed]
- Hanafusa, H.; Yagi, T.; Ikeda, H.; Hisamoto, N.; Nishioka, T.; Kaibuchi, K.; Shirakabe, K.; Matsumoto, K. LRRK1 phosphorylation of Rab7 at S72 links trafficking of EGFR-containing endosomes to its effector RILP. J. Cell Sci. 2019, 132, 228809. [Google Scholar] [CrossRef]
- Xu, H.; Zhu, Y.; Chen, X.; Yang, T.; Wang, X.; Song, X.; Xie, X.; Hu, M.; Jiang, L.; Cheng, J.; et al. Mystery of methamphetamine-induced autophagosome accumulation in hippocampal neurons: Loss of syntaxin 17 in defects of dynein-dynactin driving and autophagosome-late endosome/lysosome fusion. Arch. Toxicol. 2021, 95, 3263–3284. [Google Scholar] [CrossRef]
- Grosshans, B.L.; Ortiz, D.; Novick, P. Rabs and their effectors: Achieving specificity in membrane traffic. Proc. Natl. Acad. Sci. USA 2006, 103, 11821–11827. [Google Scholar] [CrossRef]
- Pfeffer, S.; Aivazian, D. Targeting Rab GTPases to distinct membrane compartments. Nat. Rev. Mol. Cell Biol. 2004, 5, 886–896. [Google Scholar] [CrossRef]
- Toyofuku, T.; Morimoto, K.; Sasawatari, S.; Kumanogoh, A. Leucine-Rich Repeat Kinase 1 Regulates Autophagy through Turning On TBC1D2-Dependent Rab7 Inactivation. Mol. Cell. Biol. 2015, 35, 3044–3058. [Google Scholar] [CrossRef]
- Xin, W.; Pan, Y.; Wei, W.; Tatenhorst, L.; Graf, I.; Popa-Wagner, A.; Gerner, S.T.; Huber, S.; Kilic, E.; Hermann, D.M.; et al. Preconditioned extracellular vesicles from hypoxic microglia reduce poststroke AQP4 depolarization, disturbed cerebrospinal fluid flow, astrogliosis, and neuroinflammation. Theranostics 2023, 13, 4197–4216. [Google Scholar] [CrossRef]
- Guo, M.; Wang, J.; Zhao, Y.; Feng, Y.; Han, S.; Dong, Q.; Cui, M.; Tieu, K. Microglial exosomes facilitate alpha-synuclein transmission in Parkinson’s disease. Brain. 2020, 143, 1476–1497. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Zhang, Y.D.; Ma, R.Y.; Chen, Y.J.; Xiang, X.M.; Hou, D.Y.; Li, X.H.; Huang, H.; Li, T.; Duan, C.Y. Activated Drp1 regulates p62-mediated autophagic flux and aggravates inflammation in cerebral ischemia-reperfusion via the ROS-RIP1/RIP3-exosome axis. Mil. Med. Res. 2022, 9, 25. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.X.; Liang, F.C.; Gu, P.; Xu, B.L.; Xu, H.J.; Wang, W.T.; Hou, J.Y.; Xie, D.X.; Chai, X.Q.; An, S.J. Exosomes derived from mesenchymal stem cells repair a Parkinson’s disease model by inducing autophagy. Cell Death Dis. 2020, 11, 288. [Google Scholar] [CrossRef]
- Chen, R.; Huang, P.; Wei, S.; Zhang, C.; Lai, X.; Wang, H.; Tang, J. Methamphetamine exposure increases cardiac microvascular permeability by activating the VEGF-PI3K-Akt-eNOS signaling pathway, reversed by Bevacizumab. Hum. Exp. Toxicol. 2022, 41, 774827261. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Chen, C.; Zhao, X.; Lu, J.; Zhang, M.; Qiu, H.; Yue, X.; Wang, H. New insight into methamphetamine-associated heart failure revealed by transcriptomic analyses: Circadian rhythm disorder. Toxicol. Appl. Pharmacol. 2022, 451, 116172. [Google Scholar] [CrossRef]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef] [PubMed]
- McAfee, Q.; Zhang, Z.; Samanta, A.; Amaravadi, R.K. Autophagy inhibitor Lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. Proc. Natl. Acad. Sci. USA 2012, 109, 8253–8258. [Google Scholar] [CrossRef]
- Kartik, S.; Pal, R.; Chaudhary, M.J.; Nath, R.; Kumar, M.; Bawankule, D.U. Neuroprotective role of chloroquine via modulation of autophagy and neuroinflammation in MPTP-induced Parkinson’s disease. Inflammopharmacology 2023, 31, 927–941. [Google Scholar] [CrossRef]
- Garber, J.C. On the care and use of US lab animals. Nature 2011, 476, 152. [Google Scholar] [CrossRef]
- Melbye, M. Research Article The natural history of human T lymphotropic virus-III infection: The cause of AIDS. BMJ 1986, 292, 5–10. [Google Scholar] [CrossRef]
- Tian, Y.; Gong, M.; Hu, Y.; Liu, H.; Zhang, W.; Zhang, M.; Hu, X.; Aubert, D.; Zhu, S.; Wu, L.; et al. Quality and efficiency assessment of six extracellular vesicle isolation methods by nano-flow cytometry. J. Extracell. Vesicles. 2019, 9, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.Y.; Lin, Y.C.; Cui, S.; Huang, Y.; Tang, Y.; Xu, J.; Bao, J.; Li, Y.; Wen, J.; Zuo, H.; et al. miRTarBase update 2022: An informative resource for experimentally validated miRNA-target interactions. Nucleic Acids Res. 2022, 50, D222–D230. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. ClusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2023; Available online: https://www.R-project.org/ (accessed on 25 February 2024).
- Ushey, K.; Allaire, J.; Wickham, H.; Ritchie, G. rstudioapi: Safely Access the RStudio API. 2022. Available online: https://rstudio.github.io/rstudioapi/ (accessed on 25 February 2024).
- Available online: https://github.com/rstudio/rstudioapi (accessed on 25 February 2024).
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Primer Sequence (5′-3′) | Primer Length |
|---|---|---|
| Ecor1-RAB7-F | CCGGAATTCATGACCTCTAGGAAGAAAGTG- | 30 bp |
| Xhol-RAB7-R | CCGCTCGAGTCAGCAACTGCAGCTTTCTG- | 29 bp |
| Primer Name | Primer Sequence (5′-3′) | Primer Length |
|---|---|---|
| hsa-miR-15b-5p-F | TAGCAGCACATCATGGTTTACA | 22 bp |
| hsa-miR-16-5p-F | TAGCAGCACGTAAATATTGGCG | 22 bp |
| hsa-miR-17-5p-F | CAAAGTGCTTACAGTGCAGGTAG | 23 bp |
| hsa-miR-19b-3p-F | TGTGCAAATCCATGCAAAACTGA | 23 bp |
| hsa-miR-106a-5p-F | AAAAGTGCTTACAGTGCAGGTAG | 23 bp |
| hsa-miR-503-5p-F | TAGCAGCGGGAACAGTTCTGCAG | 23 bp |
| hsa-miR-25-3p-F | AACCTCCCATTGCACTTGTCTC | 22 bp |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiu, H.; Zhang, M.; Li, M.; Chen, C.; Wang, H.; Yue, X. Methamphetamine and Methamphetamine-Induced Neuronal Exosomes Modulate the Activity of Rab7a via PTEN to Exert an Influence on the Disordered Autophagic Flux Induced in Neurons. Int. J. Mol. Sci. 2025, 26, 2644. https://doi.org/10.3390/ijms26062644
Qiu H, Zhang M, Li M, Chen C, Wang H, Yue X. Methamphetamine and Methamphetamine-Induced Neuronal Exosomes Modulate the Activity of Rab7a via PTEN to Exert an Influence on the Disordered Autophagic Flux Induced in Neurons. International Journal of Molecular Sciences. 2025; 26(6):2644. https://doi.org/10.3390/ijms26062644
Chicago/Turabian StyleQiu, Hai, Manting Zhang, Minchun Li, Chuanxiang Chen, Huijun Wang, and Xia Yue. 2025. "Methamphetamine and Methamphetamine-Induced Neuronal Exosomes Modulate the Activity of Rab7a via PTEN to Exert an Influence on the Disordered Autophagic Flux Induced in Neurons" International Journal of Molecular Sciences 26, no. 6: 2644. https://doi.org/10.3390/ijms26062644
APA StyleQiu, H., Zhang, M., Li, M., Chen, C., Wang, H., & Yue, X. (2025). Methamphetamine and Methamphetamine-Induced Neuronal Exosomes Modulate the Activity of Rab7a via PTEN to Exert an Influence on the Disordered Autophagic Flux Induced in Neurons. International Journal of Molecular Sciences, 26(6), 2644. https://doi.org/10.3390/ijms26062644