
The Role of the Mitogen-Activated Protein Kinase Pathway in the Development of Laser-Induced Choroidal Neovascularization




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
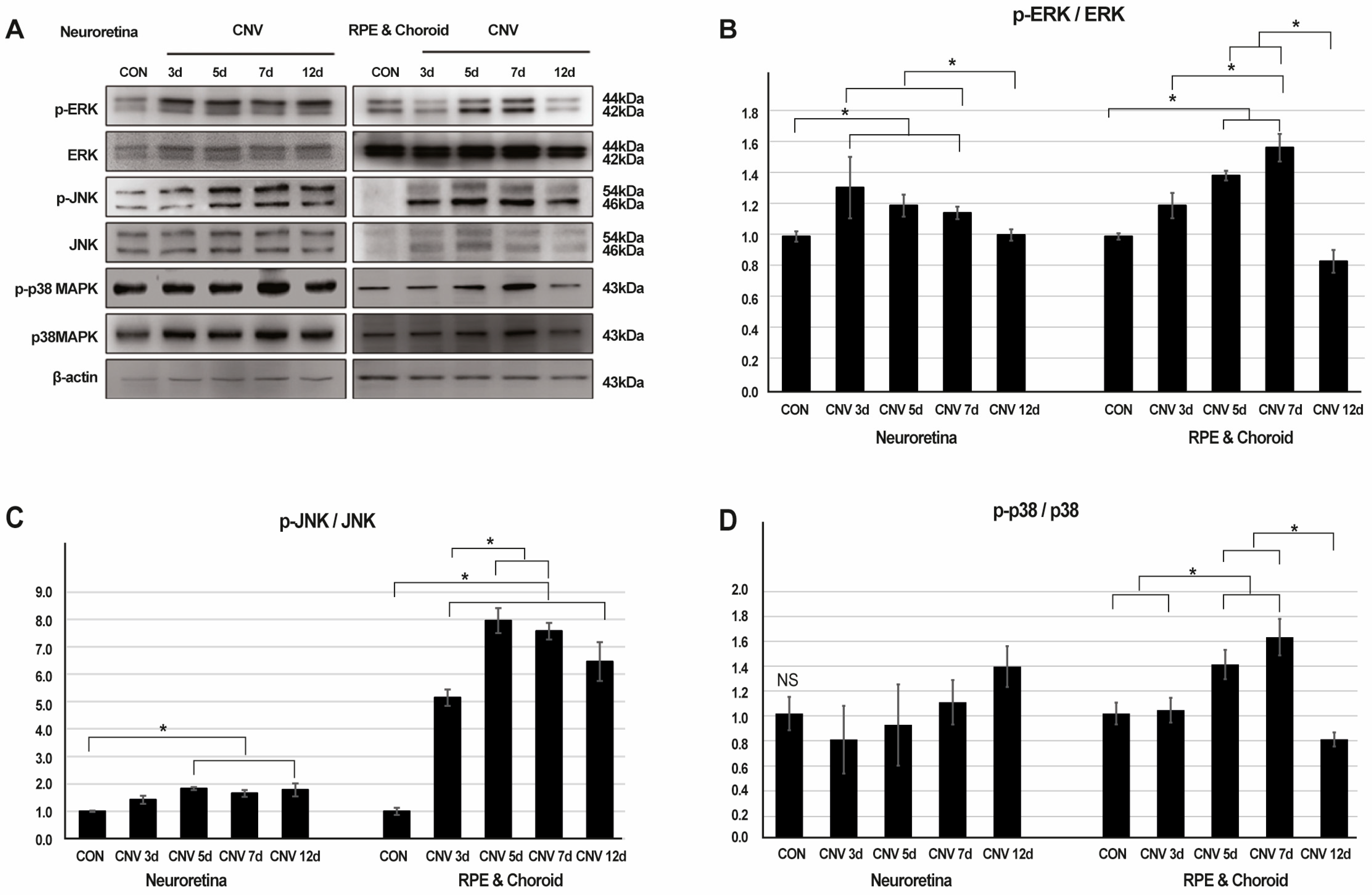
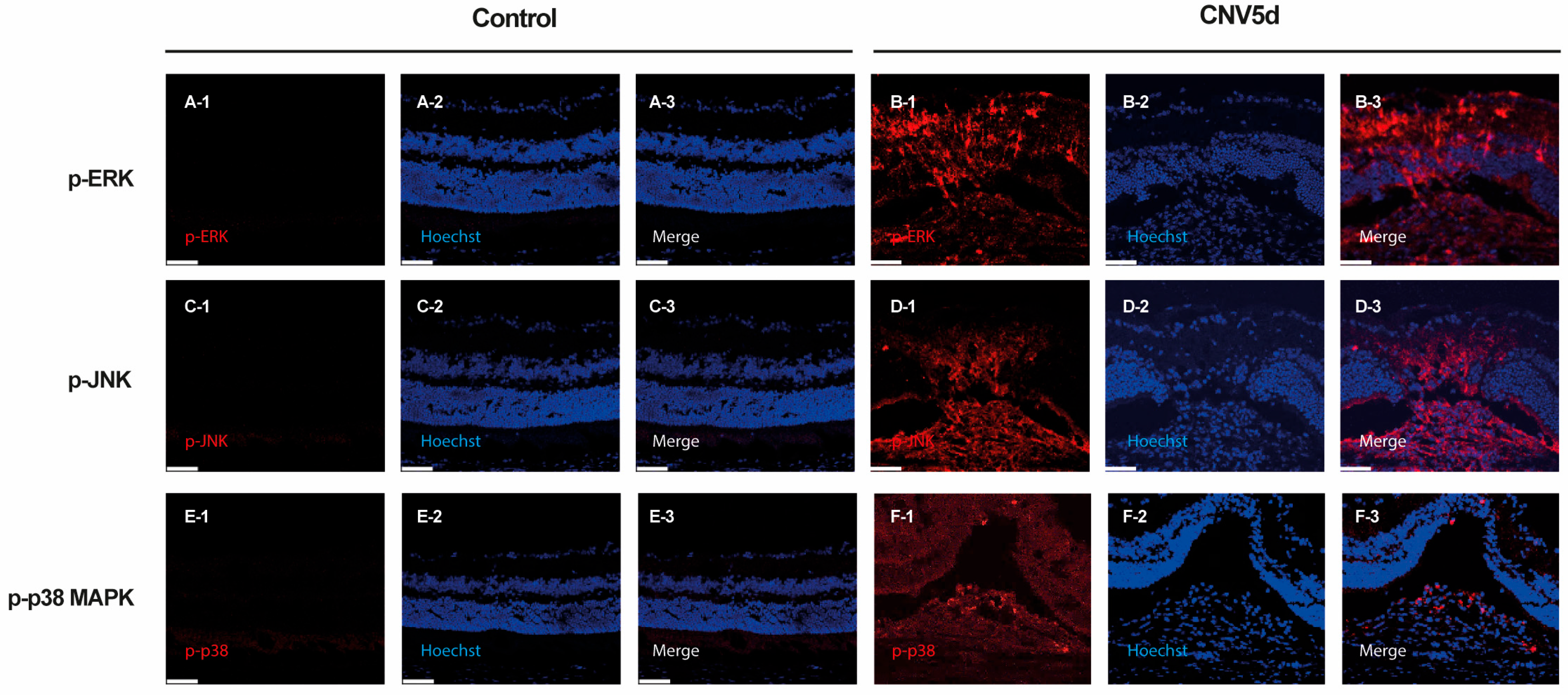
2.1. Activation of ERK, JNK, and p38-MAPK Signaling in Response to CNV Induction
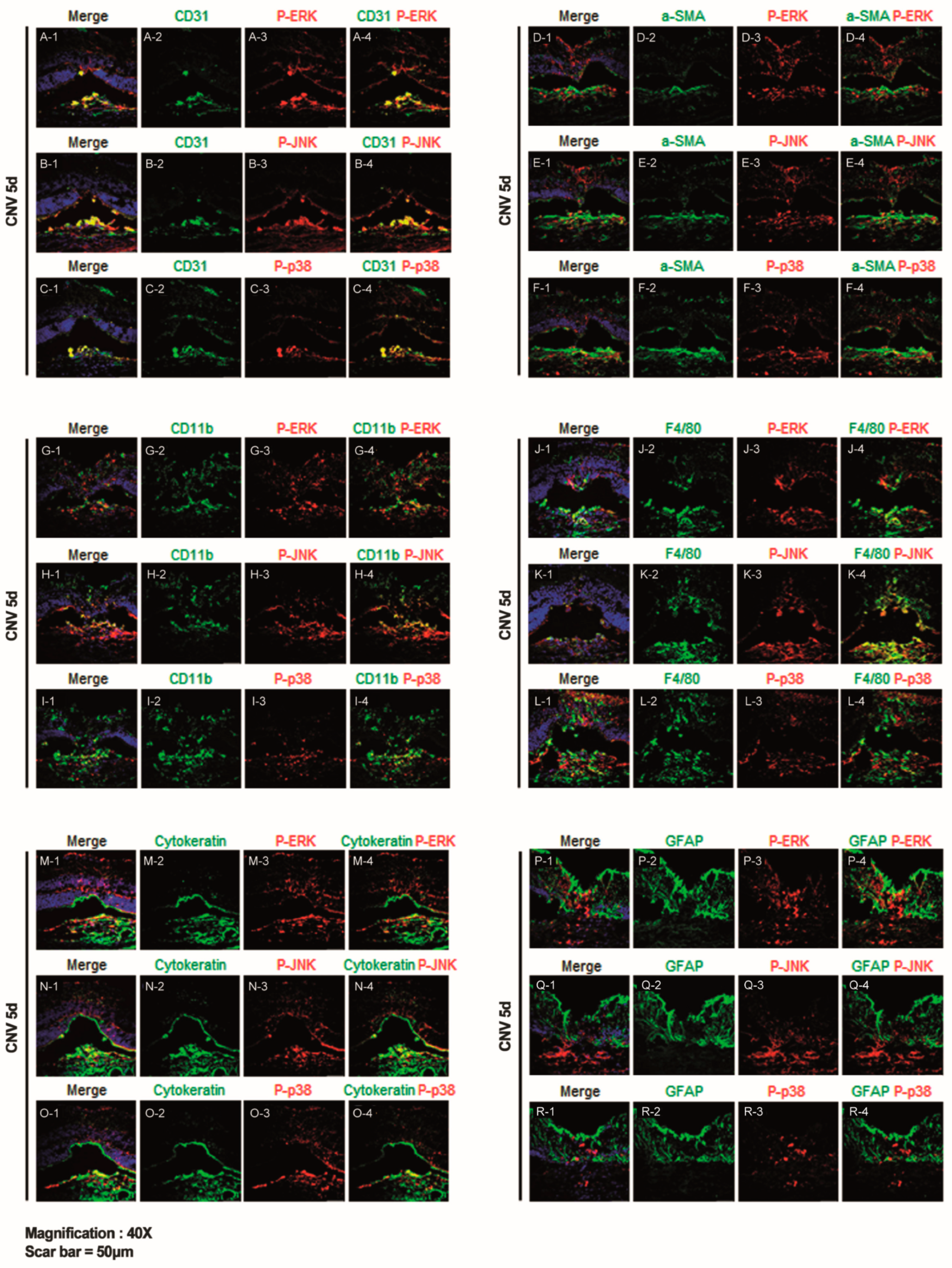
2.2. ERK, JNK, and p38-MAPK Signalling Pathway Differentially Induce Angiogenesis, Fibrosis, Inflammation, and Gliosis After Laser-Induced CNV
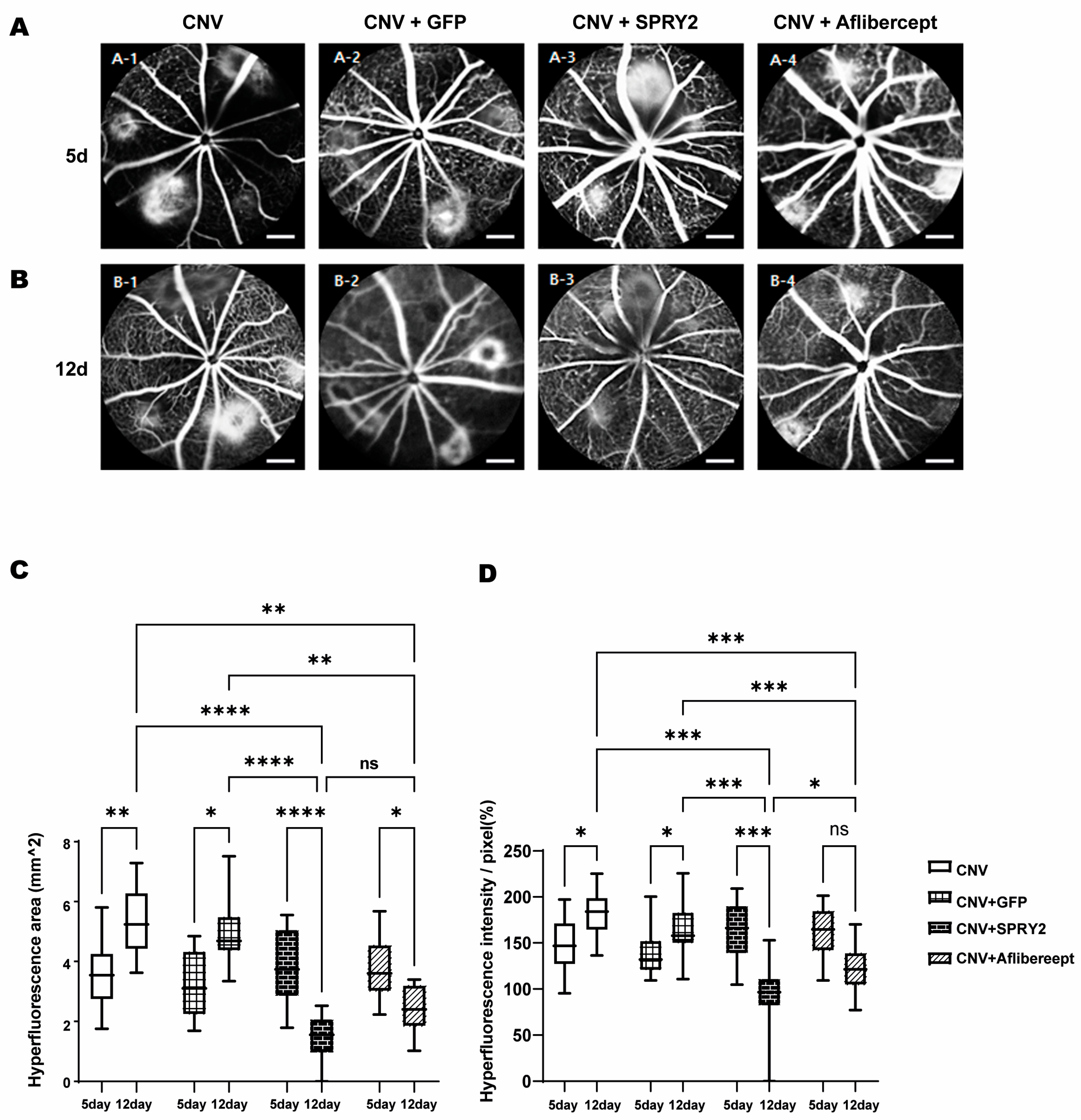
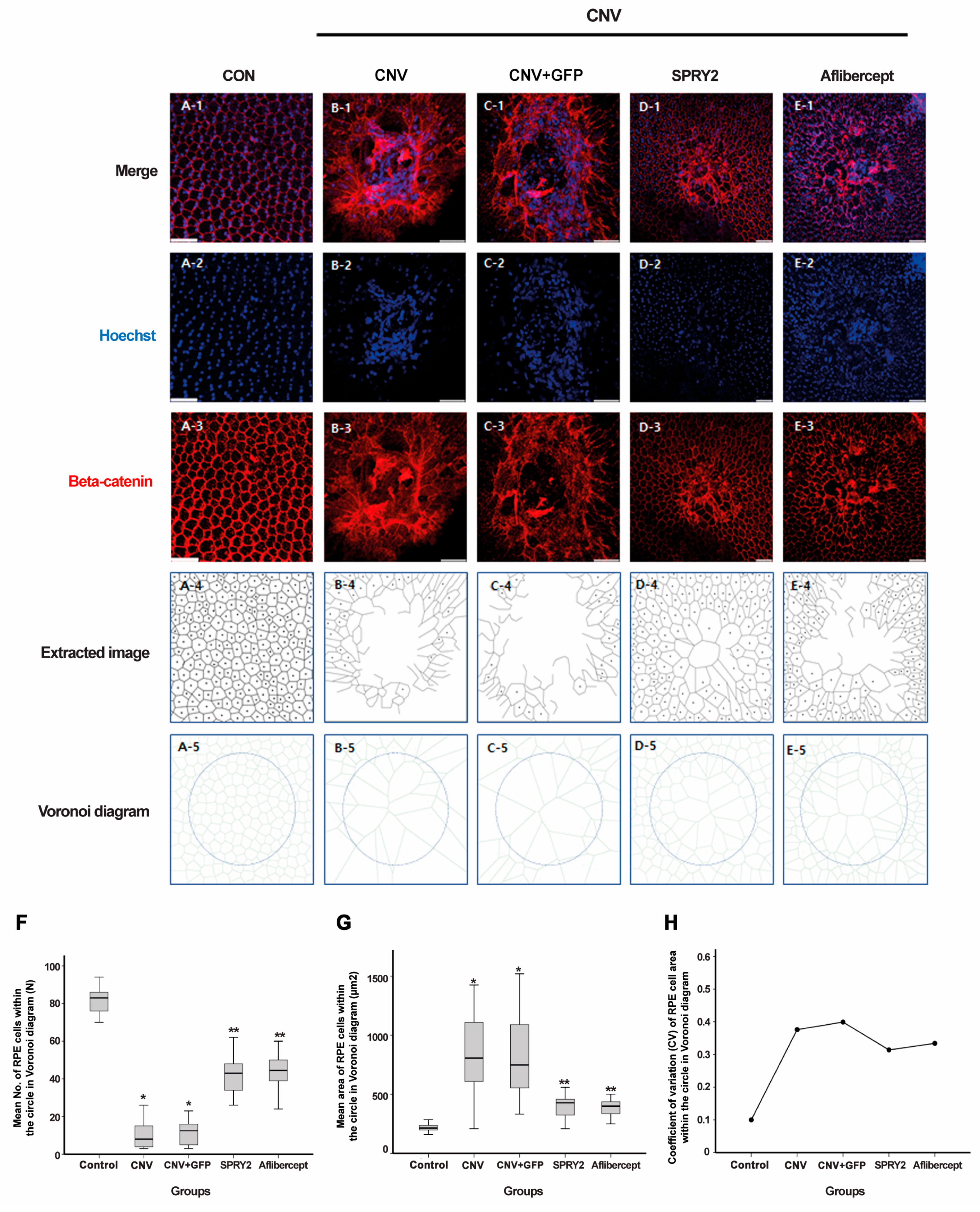
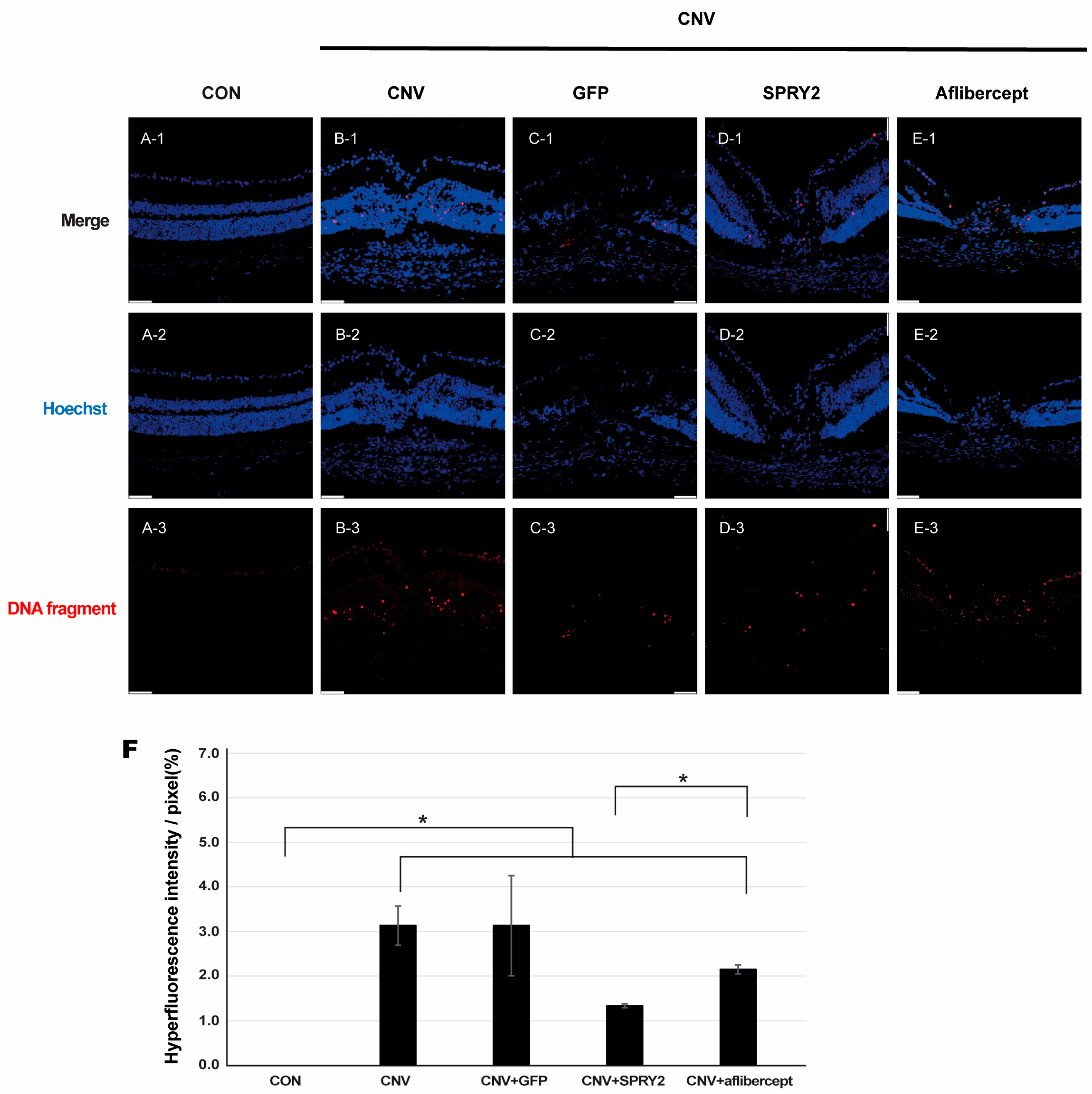
2.3. SPRY2 Reduce CNV Induction by Inhibiting Endothelial Cell Proliferation, Epithelial–Mesenchymal Transition (EMT), and Apoptosis
3. Discussion
4. Materials and Methods
4.1. Animal Care and Use
4.2. Induction of CNV in Mice
4.3. Experimental Design
4.4. FFA
4.5. Intravitreal Adeno-Associated Virus (AAV) and Aflibercept Administration
4.6. Immunofluorescence
4.7. Terminal Deoxynucleotidyl Transferase dUTP Nick End Labeling (TUNEL) Assays
4.8. Western Blot Analysis
4.9. Voronoi Diagram
4.10. Image Analysis and Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lim, L.S.; Mitchell, P.; Seddon, J.M.; Holz, F.G.; Wong, T.Y. Age-related macular degeneration. Lancet 2012, 379, 1728–1738. [Google Scholar] [CrossRef] [PubMed]
- Fine, S.L.; Berger, J.W.; Maguire, M.G.; Ho, A.C. Age-related macular degeneration. N. Engl. J. Med. 2000, 342, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Bakri, S.J.; Thorne, J.E.; Ho, A.C.; Ehlers, J.P.; Schoenberger, S.D.; Yeh, S.; Kim, S.J. Safety and Efficacy of Anti-Vascular Endothelial Growth Factor Therapies for Neovascular Age-Related Macular Degeneration: A Report by the American Academy of Ophthalmology. Ophthalmology 2019, 126, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Ba, J.; Peng, R.S.; Xu, D.; Li, Y.H.; Shi, H.; Wang, Q.; Yu, J. Intravitreal anti-VEGF injections for treating wet age-related macular degeneration: A systematic review and meta-analysis. Drug Des. Dev. Ther. 2015, 9, 5397–5405. [Google Scholar]
- Li, E.; Donati, S.; Lindsley, K.B.; Krzystolik, M.G.; Virgili, G. Treatment regimens for administration of anti-vascular endothelial growth factor agents for neovascular age-related macular degeneration. Cochrane Database Syst. Rev. 2020, 5, CD012208. [Google Scholar]
- Kent, D.L. Age-related macular degeneration: Beyond anti-angiogenesis. Mol. Vis. 2014, 20, 46–55. [Google Scholar]
- Yang, S.; Zhao, J.; Sun, X. Resistance to anti-VEGF therapy in neovascular age-related macular degeneration: A comprehensive review. Drug Des. Dev. Ther. 2016, 10, 1857–1867. [Google Scholar]
- Ford, K.M.; Saint-Geniez, M.; Walshe, T.; Zahr, A.; D’Amore, P.A. Expression and role of VEGF in the adult retinal pigment epithelium. Investig. Ophthalmol. Vis. Sci. 2011, 52, 9478–9487. [Google Scholar] [CrossRef]
- Brinkmann, A.; Winkelmann, K.; Käckenmeister, T.; Roider, J.; Klettner, A. Effect of Long-term Anti-VEGF Treatment on Viability and Function of RPE Cells. Curr. Eye Res. 2022, 47, 127–134. [Google Scholar] [CrossRef]
- Danis, R.P.; Lavine, J.A.; Domalpally, A. Geographic atrophy in patients with advanced dry age-related macular degeneration: Current challenges and future prospects. Clin. Ophthalmol. 2015, 9, 2159–2174. [Google Scholar] [CrossRef]
- Gemenetzi, M.; Lotery, A.J.; Patel, P.J. Risk of geographic atrophy in age-related macular degeneration patients treated with intravitreal anti-VEGF agents. Eye 2017, 31, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Enslow, R.; Bhuvanagiri, S.; Vegunta, S.; Cutler, B.; Neff, M.; Stagg, B. Association of Anti-VEGF Injections with Progression of Geographic Atrophy. Ophthalmol. Eye Dis. 2016, 8, 31–32. [Google Scholar] [CrossRef] [PubMed]
- Moustardas, P.; Aberdam, D.; Lagali, N. MAPK Pathways in Ocular Pathophysiology: Potential Therapeutic Drugs and Challenges. Cells 2023, 12, 617. [Google Scholar] [CrossRef]
- Peti, W.; Page, R. Molecular basis of MAP kinase regulation. Protein Sci. A Publ. Protein Soc. 2013, 22, 1698–1710. [Google Scholar] [CrossRef] [PubMed]
- Kyosseva, S.V. Targeting MAPK Signaling in Age-Related Macular Degeneration. Ophthalmol. Eye Dis. 2016, 8, 23–30. [Google Scholar] [CrossRef]
- Kyriakis, J.M.; Avruch, J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: A 10-year update. Physiol. Rev. 2012, 92, 689–737. [Google Scholar] [CrossRef]
- Cruickshanks, K.J.; Klein, R.; Klein, B.E.; Nondahl, D.M. Sunlight and the 5-year incidence of early age-related maculopathy: The beaver dam eye study. Arch. Ophthalmol. 2001, 119, 246–250. [Google Scholar]
- Roduit, R.; Schorderet, D.F. MAP kinase pathways in UV-induced apoptosis of retinal pigment epithelium ARPE19 cells. Apoptosis Int. J. Program. Cell Death 2008, 13, 343–353. [Google Scholar] [CrossRef]
- Chan, C.M.; Huang, J.H.; Lin, H.H.; Chiang, H.S.; Chen, B.H.; Hong, J.Y.; Hung, C.F. Protective effects of (-)-epigallocatechin gallate on UVA-induced damage in ARPE19 cells. Mol. Vis. 2008, 14, 2528–2534. [Google Scholar]
- Galán, A.; Jmaeff, S.; Barcelona, P.F.; Brahimi, F.; Sarunic, M.V.; Saragovi, H.U. In retinitis pigmentosa TrkC.T1-dependent vectorial Erk activity upregulates glial TNF-α, causing selective neuronal death. Cell Death Dis. 2017, 8, 3222. [Google Scholar] [CrossRef]
- Plössl, K.; Weber, B.H.; Friedrich, U. The X-linked juvenile retinoschisis protein retinoschisin is a novel regulator of mitogen-activated protein kinase signalling and apoptosis in the retina. J. Cell. Mol. Med. 2017, 21, 768–780. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.J. Signal transduction by the JNK group of MAP kinases. Cell 2000, 103, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Kamata, H.; Honda, S.; Maeda, S.; Chang, L.; Hirata, H.; Karin, M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell 2005, 120, 649–661. [Google Scholar] [CrossRef] [PubMed]
- Guma, M.; Rius, J.; Duong-Polk, K.X.; Haddad, G.G.; Lindsey, J.D.; Karin, M. Genetic and pharmacological inhibition of JNK ameliorates hypoxia-induced retinopathy through interference with VEGF expression. Proc. Natl. Acad. Sci. USA 2009, 106, 8760–8765. [Google Scholar] [CrossRef]
- Du, H.; Sun, X.; Guma, M.; Luo, J.; Ouyang, H.; Zhang, X.; Zeng, J.; Quach, J.; Nguyen, D.H.; Shaw, P.X.; et al. JNK inhibition reduces apoptosis and neovascularization in a murine model of age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2013, 110, 2377–2382. [Google Scholar] [CrossRef]
- Kyosseva, S.V.; Chen, L.; Seal, S.; McGinnis, J.F. Nanoceria inhibit expression of genes associated with inflammation and angiogenesis in the retina of Vldlr null mice. Exp. Eye Res. 2013, 116, 63–74. [Google Scholar] [CrossRef]
- Liu, X.; Yang, X.; Zhu, R.; Dai, M.; Zhu, M.; Shen, Y.; Fang, H.; Sang, A.; Chen, H. Involvement of Fra-1 in Retinal Ganglion Cell Apoptosis in Rat Light-Induced Retina Damage Model. Cell. Mol. Neurobiol. 2017, 37, 83–92. [Google Scholar] [CrossRef]
- Ding, X.Y.; Gu, R.P.; Tang, W.Y.; Shu, Q.M.; Xu, G.Z.; Zhang, M. Effect of Phosphorylated-Extracellular Regulated Kinase 1/2 Inhibitor on Retina from Light-induced Photoreceptor Degeneration. Chin. Med. J. 2018, 131, 2836–2843. [Google Scholar]
- Tezel, G.; Chauhan, B.C.; LeBlanc, R.P.; Wax, M.B. Immunohistochemical assessment of the glial mitogen-activated protein kinase activation in glaucoma. Investig. Ophthalmol. Vis. Sci. 2003, 44, 3025–3033. [Google Scholar] [CrossRef]
- Gao, F.; Li, F.; Miao, Y.; Xu, L.J.; Zhao, Y.; Li, Q.; Zhang, S.H.; Wu, J.; Sun, X.H.; Wang, Z. Involvement of the MEK-ERK/p38-CREB/c-fos signaling pathway in Kir channel inhibition-induced rat retinal Müller cell gliosis. Sci. Rep. 2017, 7, 1480. [Google Scholar] [CrossRef]
- Zeng, S.; Zhang, T.; Chen, Y.; Chu-Tan, J.; Jin, K.; Lee, S.R.; Yam, M.X.; Madigan, M.C.; Fernando, N.; Cioanca, A.; et al. Inhibiting the activation of MAPK (ERK1/2) in stressed Müller cells prevents photoreceptor degeneration. Theranostics 2022, 12, 6705–6722. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Hu, J. The role of JNK in prostate cancer progression and therapeutic strategies. Biomed. Pharmacother. Biomed. Pharmacother. 2020, 121, 109679. [Google Scholar] [CrossRef] [PubMed]
- Uchida, C.; Gee, E.; Ispanovic, E.; Haas, T.L. JNK as a positive regulator of angiogenic potential in endothelial cells. Cell Biol. Int. 2008, 32, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.M.; Huang, C.H.; Li, H.J.; Hsiao, C.Y.; Su, C.C.; Lee, P.L.; Hung, C.F. Protective effects of resveratrol against UVA-induced damage in ARPE19 cells. Int. J. Mol. Sci. 2015, 16, 5789–5802. [Google Scholar] [CrossRef]
- Glotin, A.L.; Calipel, A.; Brossas, J.Y.; Faussat, A.M.; Tréton, J.; Mascarelli, F. Sustained versus transient ERK1/2 signaling underlies the anti- and proapoptotic effects of oxidative stress in human RPE cells. Investig. Ophthalmol. Vis. Sci. 2006, 47, 4614–4623. [Google Scholar] [CrossRef]
- SanGiovanni, J.P.; Lee, P.H. AMD-associated genes encoding stress-activated MAPK pathway constituents are identified by interval-based enrichment analysis. PLoS ONE 2013, 8, e71239. [Google Scholar] [CrossRef]
- Impagnatiello, M.A.; Weitzer, S.; Gannon, G.; Compagni, A.; Cotten, M.; Christofori, G. Mammalian sprouty-1 and -2 are membrane-anchored phosphoprotein inhibitors of growth factor signaling in endothelial cells. J. Cell Biol. 2001, 152, 1087–1098. [Google Scholar] [CrossRef]
- Yusoff, P.; Lao, D.H.; Ong, S.H.; Wong, E.S.; Lim, J.; Lo, T.L.; Leong, H.F.; Fong, C.W.; Guy, G.R. Sprouty2 inhibits the Ras/MAP kinase pathway by inhibiting the activation of Raf. J. Biol. Chem. 2002, 277, 3195–3201. [Google Scholar] [CrossRef]
- Lee, C.T.; Chu, C.A.; Wang, Y.M.; Wang, Y.W.; Chen, Y.L.; Ho, C.L.; Yeh, Y.M.; Lin, P.C.; Lin, B.W.; Chen, P.C.; et al. Dual role of sprouty2 as an inhibitor of RAS/ERK-driven proliferation and a promoter of cancer invasion in KRAS wild-type colorectal cancer. Mol. Carcinog. 2023, 62, 951–962. [Google Scholar] [CrossRef]
- Milillo, A.; La Carpia, F.; Costanzi, S.; D’Urbano, V.; Martini, M.; Lanuti, P.; Vischini, G.; Larocca, L.M.; Marchisio, M.; Miscia, S.; et al. A SPRY2 mutation leading to MAPK/ERK pathway inhibition is associated with an autosomal dominant form of IgA nephropathy. European journal of human genetics. Eur. J. Hum. Genet. 2015, 23, 1673–1678. [Google Scholar] [CrossRef]
- Sun, J.; Yoon, J.; Lee, M.; Hwang, Y.S.; Daar, I.O. Sprouty2 regulates positioning of retinal progenitors through suppressing the Ras/Raf/MAPK pathway. Sci. Rep. 2020, 10, 13752. [Google Scholar] [CrossRef]
- Yang, J.Y.; Madrakhimov, S.B.; Ahn, D.H.; Chang, H.S.; Jung, S.J.; Nah, S.K.; Park, H.Y.; Park, T.K. mTORC1 and mTORC2 are differentially engaged in the development of laser-induced CNV. Cell communication and signaling. Cell Commun. Signal. 2019, 17, 64. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jang, S.Y.; Yang, J.Y.; Park, J.H.; Kim, Y.; An, S.; Jung, W.H.; Park, J.-W.; Han, J.W.; Kim, J.H.; Park, H.S.; et al. The Role of the Mitogen-Activated Protein Kinase Pathway in the Development of Laser-Induced Choroidal Neovascularization. Int. J. Mol. Sci. 2025, 26, 2585. https://doi.org/10.3390/ijms26062585
Jang SY, Yang JY, Park JH, Kim Y, An S, Jung WH, Park J-W, Han JW, Kim JH, Park HS, et al. The Role of the Mitogen-Activated Protein Kinase Pathway in the Development of Laser-Induced Choroidal Neovascularization. International Journal of Molecular Sciences. 2025; 26(6):2585. https://doi.org/10.3390/ijms26062585
Chicago/Turabian StyleJang, Sun Young, Jin Young Yang, Jin Hwan Park, Yeji Kim, Sumin An, Wook Hyun Jung, Jong-Whi Park, Jung Woo Han, Jin Ha Kim, Hyo Song Park, and et al. 2025. "The Role of the Mitogen-Activated Protein Kinase Pathway in the Development of Laser-Induced Choroidal Neovascularization" International Journal of Molecular Sciences 26, no. 6: 2585. https://doi.org/10.3390/ijms26062585
APA StyleJang, S. Y., Yang, J. Y., Park, J. H., Kim, Y., An, S., Jung, W. H., Park, J.-W., Han, J. W., Kim, J. H., Park, H. S., Lyu, J., & Park, T. K. (2025). The Role of the Mitogen-Activated Protein Kinase Pathway in the Development of Laser-Induced Choroidal Neovascularization. International Journal of Molecular Sciences, 26(6), 2585. https://doi.org/10.3390/ijms26062585