
Next-Generation Analogues of AC265347 as Positive Allosteric Modulators of the Calcium-Sensing Receptor: Pharmacological Investigation of Structural Modifications at the Stereogenic Centre

 , , and
, , and 
Abstract

1. Introduction
2. Results and Discussion
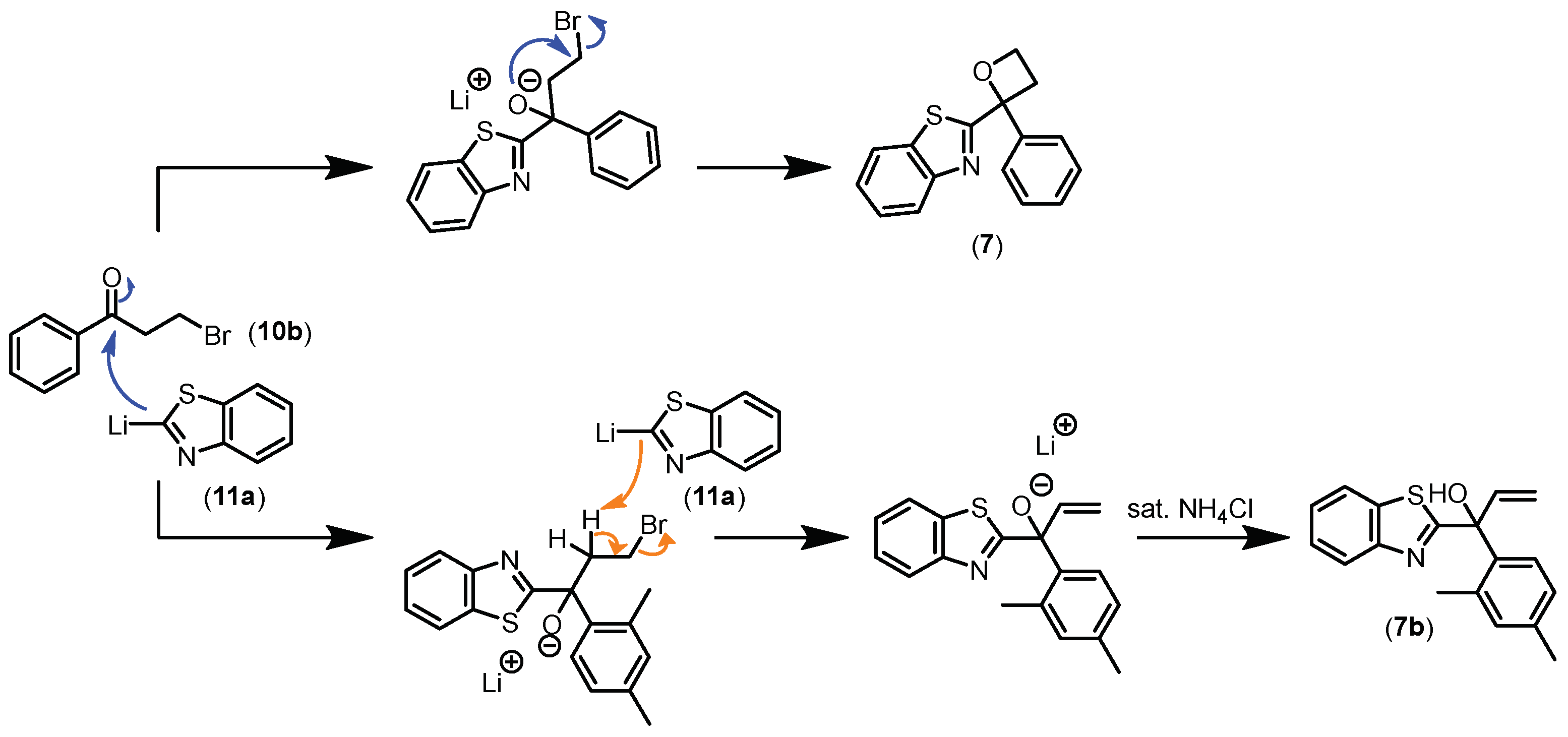
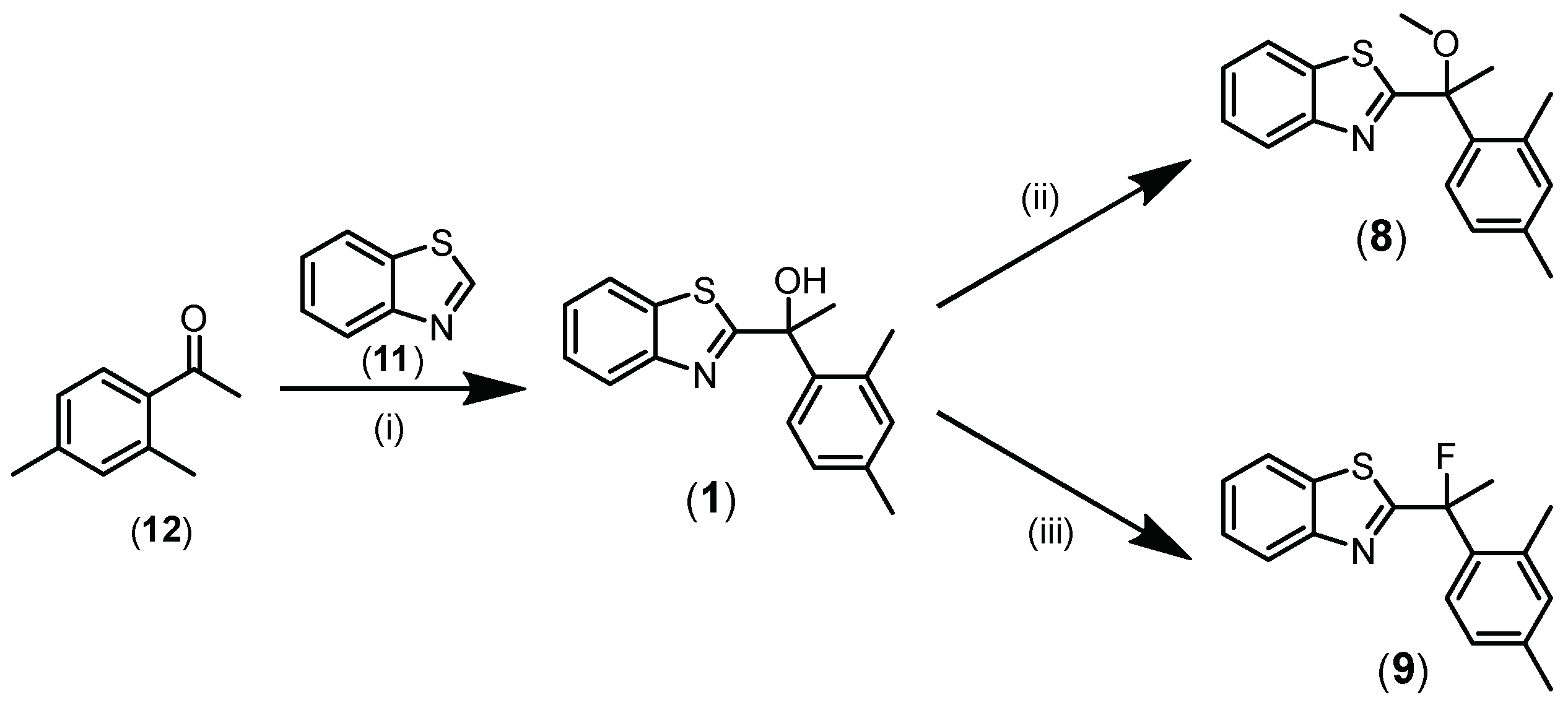
2.1. Chemistry
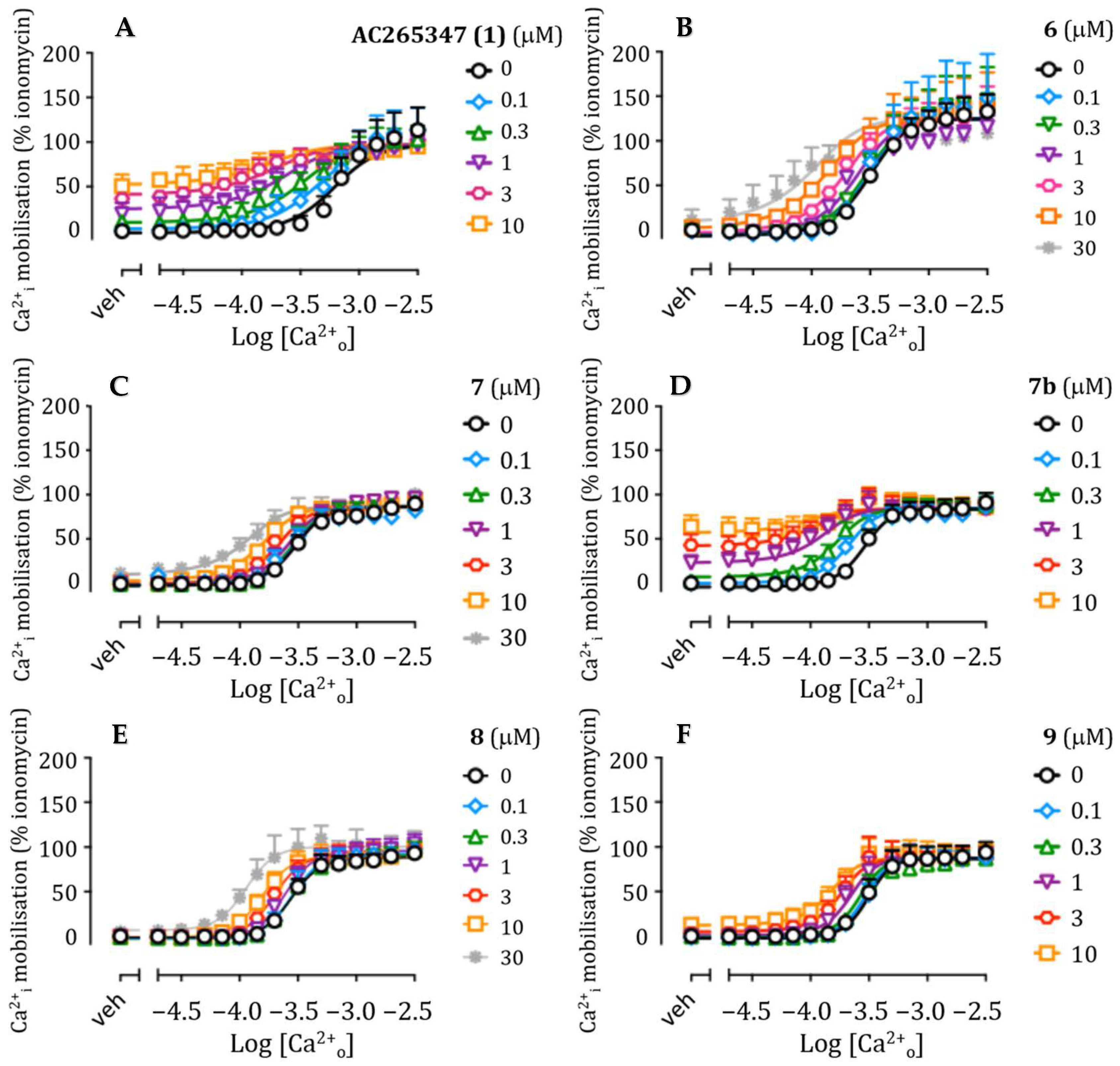
2.2. Pharmacology

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Structure | pKB ± SEM (KB, μM) | Logαβ ± SEM (αβ) | LogτB ± SEM (τB) | n |
|---|---|---|---|---|---|
| AC265347 (1) a |  | 5.47 ± 0.11 (3.4) | 1.63 ± 0.16 (43) | 0.12 ± 0.08 (1.3) | 5 |
| 5 a | 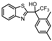 | 5.30 ± 0.23 (5.0) | 0.94 ± 0.27 (8.7) | 0.53 ± 0.18 (3.4) | 5 |
| 6 |  | 4.54 ± 0.49 (29) | 1.45± 0.41 (28) | ND b | 3–7 |
| 7 | 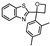 | 3.91 ± 0.66 (123) | 1.95 ± 0.61 (89) | −0.29 ± 0.54 (0.5) | 3–7 |
| 7b | 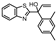 | 5.09 ± 0.27 (8.1) | 2.24 ± 0.37 (174) | 0.58 ± 0.20 (3.8) | 4 |
| 8 c | 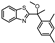 | ND b | ND b | ND b | 3 |
| 9 |  | 4.98 ± 0.41 (10) | 1.59 ± 0.37 (39) | −0.43 ± 0.26 (0.4) | 4 |
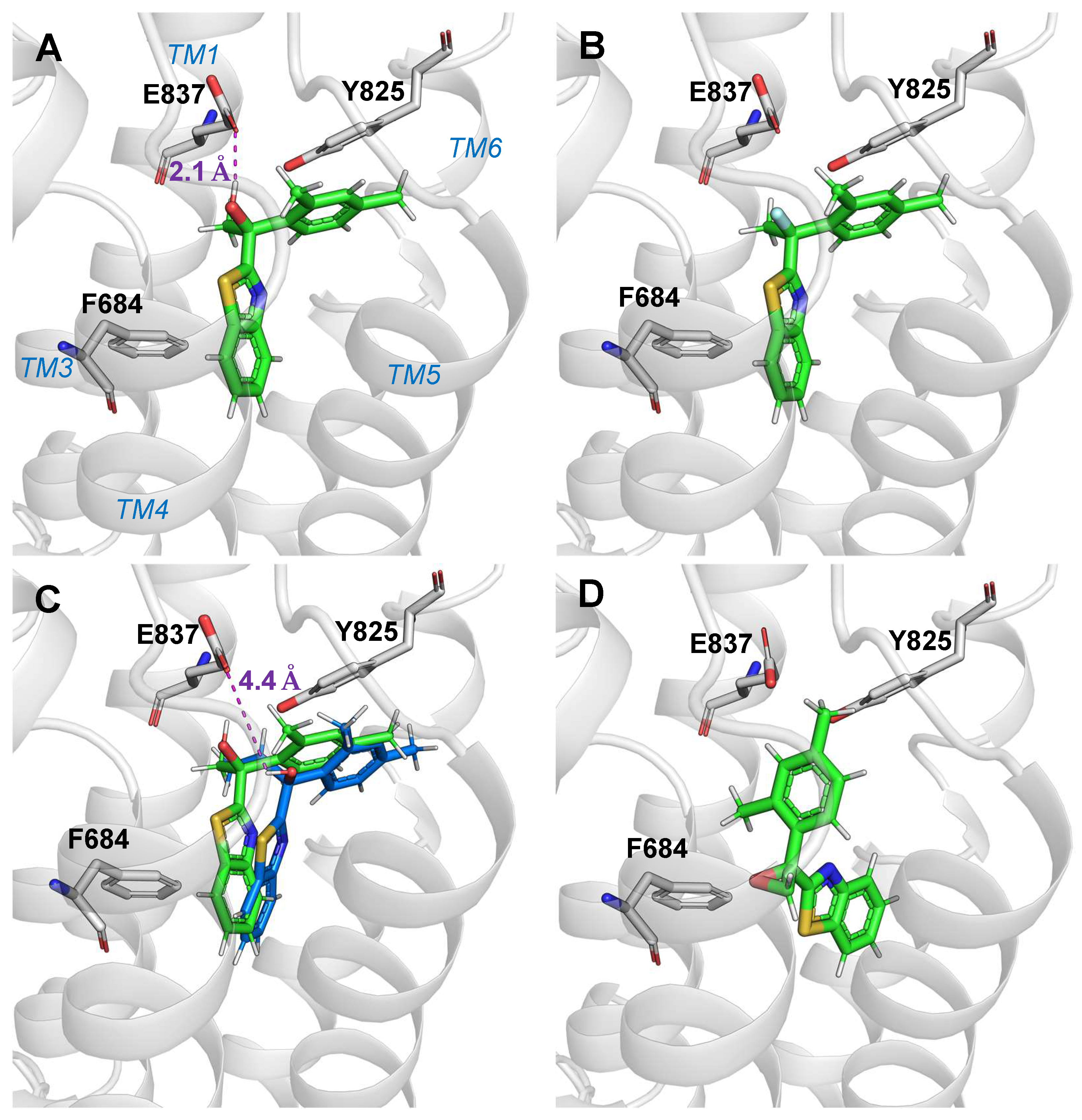
2.3. Molecular Docking Study
3. Materials and Methods
3.1. Materials
3.1.1. General Chemistry
3.1.2. Cell Culture
3.2. Chemical Synthesis of Compounds
- General procedure A [34] (lithiation)
- General procedure B [17] (O-cyclisation)
- Synthesis of 1-(Benzo[d]thiazol-2-yl)-1-(2,4-dimethylphenyl)ethan-1-ol (1) [4]
- Synthesis of 2-(2-(2,4-Dimethylphenyl)oxiran-2-yl)benzo[d]thiazole (6)
- Synthesis of 2-(2-(2,4-Dimethylphenyl)oxetan-2-yl)benzo[d]thiazole (7)
- 1-(Benzo[d]thiazol-2-yl)-1-(2,4-dimethylphenyl)prop-2-en-1-ol (7b)
- Synthesis of 2-(1-(2,4-Dimethylphenyl)-1-methoxyethyl)benzo[d]thiazole (8)
- Synthesis of 2-(1-(2,4-Dimethylphenyl)-1-fluoroethyl)benzo[d]thiazole (9)
3.3. Pharmacological Evaluation of Compounds
3.3.1. Functional Assay—Ca2+i Mobilisation
3.3.2. Data Statistical Analysis
3.4. Computational Modelling of Compounds
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ma, J.N.; Owens, M.; Gustafsson, M.; Jensen, J.; Tabatabaei, A.; Schmelzer, K.; Olsson, R.; Burstein, E.S. Characterization of highly efficacious allosteric agonists of the human calcium-sensing receptor. J. Pharmacol. Exp. Ther. 2011, 337, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, M.; Jensen, J.; Bertozzi, S.M.; Currier, E.A.; Ma, J.N.; Burstein, E.S.; Olsson, R. Discovery of a class of calcium sensing receptor positive allosteric modulators; 1-(benzothiazol-2-yl)-1-phenylethanols. Bioorg. Med. Chem. Lett. 2010, 20, 5918–5921. [Google Scholar] [CrossRef]
- Wada, M.; Kawata, T.; Nagano, N. Calcimimetics for Treating Hyperparathyroidism. In Encyclopedia of Bone Biology; Academic Press: Cambridge, MA, USA, 2020; pp. 697–710. [Google Scholar]
- Dinh, L.V.; DeBono, A.; Keller, A.N.; Josephs, T.M.; Gregory, K.J.; Leach, K.; Capuano, B. Development of AC265347-Inspired Calcium-Sensing Receptor Ago-Positive Allosteric Modulators. ChemMedChem 2021, 16, 3451–3462. [Google Scholar] [CrossRef]
- Leach, K.; Sexton, P.M.; Christopoulos, A. Allosteric GPCR modulators: Taking advantage of permissive receptor pharmacology. Trends Pharmacol. Sci. 2007, 28, 382–389. [Google Scholar] [CrossRef]
- Shen, S.; Zhao, C.; Wu, C.; Sun, S.; Li, Z.; Yan, W.; Shao, Z. Allosteric modulation of G protein-coupled receptor signaling. Front. Endocrinol. 2023, 14, 1137604. [Google Scholar] [CrossRef] [PubMed]
- Conn, P.J.; Christopoulos, A.; Lindsley, C.W. Allosteric modulators of GPCRs: A novel approach for the treatment of CNS disorders. Nat. Rev. Drug Discov. 2009, 8, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Kirk, K.L. Selective Fluorination in Drug Design and Development: An Overview of Biochemical Rationales. Curr. Top Med. Chem. 2006, 6, 1447–1456. [Google Scholar] [CrossRef] [PubMed]
- Glyn, R.J.; Pattison, G. Effects of Replacing Oxygenated Functionality with Fluorine on Lipophilicity. J. Med. Chem. 2021, 64, 10246–10259. [Google Scholar] [CrossRef]
- Meanwell, N.A. Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design. J. Med. Chem. 2018, 61, 5822–5880. [Google Scholar] [CrossRef]
- Welch, J.T. Advances in the Preparation of Biologically Active Organofluorine Compounds. Tetrahedron 1987, 43, 3123–3197. [Google Scholar] [CrossRef]
- Wang, J.; Sanchez-Rosello, M.; Acena, J.L.; del Pozo, C.; Sorochinsky, A.E.; Fustero, S.; Soloshonok, V.A.; Liu, H. Fluorine in pharmaceutical industry: Fluorine-containing drugs introduced to the market in the last decade (2001–2011). Chem. Rev. 2014, 114, 2432–2506. [Google Scholar] [CrossRef] [PubMed]
- Hagman, W.K. The Many Roles for Fluorine in Medicinal Chemistry. J. Med. Chem. 2008, 51, 4359–4369. [Google Scholar] [CrossRef]
- Johnson, B.M.; Shu, Y.Z.; Zhuo, X.; Meanwell, N.A. Metabolic and Pharmaceutical Aspects of Fluorinated Compounds. J. Med. Chem. 2020, 63, 6315–6386. [Google Scholar] [CrossRef] [PubMed]
- Kaur, B.; Singh, P. Epoxides: Developability as active pharmaceutical ingredients and biochemical probes. Bioorg. Chem. 2022, 125, 105862. [Google Scholar] [CrossRef]
- Bull, J.A.; Croft, R.A.; Davis, O.A.; Doran, R.; Morgan, K.F. Oxetanes: Recent Advances in Synthesis, Reactivity, and Medicinal Chemistry. Chem. Rev. 2016, 116, 12150–12233. [Google Scholar] [CrossRef]
- Chikashita, H.; Ishibaba, M.; Ori, K.; Itoh, K. General Reactivity of 2-Lithiobenzothiazole to Various Electrophiles and the Use as a Formyl Anion Equivalent in the Synthesis of α-Hydroxy Carbonyl Compounds. Bull. Chem. Soc. Jpn. 1988, 61, 3637–3648. [Google Scholar] [CrossRef]
- Morrill, C.; Grubbs, R.H. Highly Selective 1,3-Isomerization of Allylic Alcohols via Rhenium Oxo Catalysis. J. Am. Chem. Soc. 2005, 127, 2842–2843. [Google Scholar] [CrossRef]
- Bird, T.G.C.; Bruneau, P.; Crawley, G.C.; Edwards, M.P.; Foster, S.J.; Girodeau, J.-M.; Kingston, J.F.; McMillan, R.M. (Methoxyalkyl)Thiazoles: A new series of potent, selective, and orally active 5-lipoxygenase inhibitors displaying high enantioselectivity. J. Med. Chem. 1991, 34, 2176–2186. [Google Scholar] [CrossRef]
- L’Heureux, A.; Beaulieu, F.; Bennett, C.; Bill, D.R.; Clayton, S.; Laflamme, F.; Mirmehrabi, M.; Tadayon, S.; Tovell, D.; Couturier, M. Aminodifluorosulfinium salts: Selective fluorination reagents with enhanced thermal stability and ease of handling. J. Org. Chem. 2010, 75, 3401–3411. [Google Scholar] [CrossRef]
- Franconetti, A. Dialkylaminodifluorosulfinium Salts: XtalFluor-E and XtalFluor-M. Synlett 2013, 24, 891–892. [Google Scholar] [CrossRef]
- Bondi, A. van der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Hevey, R. Bioisosteres of Carbohydrate Functional Groups in Glycomimetic Design. Biomimetics 2019, 4, 53. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Rychlewski, J.; Rychlewska, U. Effects of Substitution of OH Group by F Atom for Conformational Preferences of Fluorine-Substituted Analogues of (R,R)-Tartaric Acid, Its Dimethyl Diester, Diamide, and N,N,N′,N′-Tetramethyl Diamide. Ab Initio Conformational Analysis. J. Am. Chem. Soc. 1999, 121, 1912–1921. [Google Scholar] [CrossRef]
- Josephs, T.M.; Zhang, F.; Dinh, L.V.; Keller, A.N.; Conigrave, A.D.; Capuano, B.; Gregory, K.J.; Leach, K. Personalised medicines for familial hypercalcemia and hyperparathyroidism. J. Mol. Endocrinol. 2022, 69, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Walter, S.; Baruch, A.; Dong, J.; Tomlinson, J.E.; Alexander, S.T.; Janes, J.; Hunter, T.; Yin, Q.; Maclean, D.; Bell, G.; et al. Pharmacology of AMG 416 (Velcalcetide), a novel peptide agonist of the calcium-sensing receptor, for the treatment of secondary hyperparathyroidism in hemodialysis patients. J. Pharmacol. Exp. Ther. 2013, 346, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Gregory, K.J.; Giraldo, J.; Diao, J.; Christopoulos, A.; Leach, K. Evaluation of Operational Models of Agonism and Allosterism at Receptors with Multiple Orthosteric Binding Sites. Mol. Pharmacol. 2020, 97, 35–45. [Google Scholar] [CrossRef]
- Keller, A.N.; Kufareva, I.; Josephs, T.M.; Diao, J.; Mai, V.T.; Conigrave, A.D.; Christopoulos, A.; Gregory, K.J.; Leach, K. Identification of Global and Ligand-Specific Calcium Sensing Receptor Activation Mechanisms. Mol. Pharmacol. 2018, 93, 619–630. [Google Scholar] [CrossRef]
- Leach, K.; Gregory, K.J.; Kufareva, I.; Khajehali, E.; Cook, A.E.; Abagyan, R.; Conigrave, A.D.; Sexton, P.M.; Christopoulos, A. Towards a structural understanding of allosteric drugs at the human calcium-sensing receptor. Cell Res. 2016, 26, 574–592. [Google Scholar] [CrossRef]
- Gao, Y.; Robertson, M.J.; Rahman, S.N.; Seven, A.B.; Zhang, C.; Meyerowitz, J.G.; Panova, O.; Hannan, F.M.; Thakker, R.V.; Brauner-Osborne, H.; et al. Asymmetric activation of the calcium-sensing receptor homodimer. Nature 2021, 595, 455–459. [Google Scholar] [CrossRef]
- Fulmer, G.R.; Miller, A.J.M.; Sherden, N.H.; Gottlieb, H.E.; Nudelman, A.; Stoltz, B.M.; Bercaw, J.E.; Goldberg, K.I. NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar] [CrossRef]
- Babij, N.R.; McCusker, E.O.; Whiteker, G.T.; Canturk, B.; Choy, N.; Creemer, L.C.; Amicis, C.V.D.; Hewlett, N.M.; Johnson, P.L.; Knobelsdorf, J.A.; et al. NMR Chemical Shifts of Trace Impurities: Industrially Preferred Solvents Used in Process and Green Chemistry. Org. Process. Res. Dev. 2016, 20, 661–667. [Google Scholar] [CrossRef]
- Gottlieb, H.G.; Kotlyar, V.; Nudelman, A. NMR Chemical Shifts of Common Laboratory Solvents as Trace Impurities. J. Org. Chem. 1997, 62, 7512–7515. [Google Scholar] [CrossRef] [PubMed]
- Anthony, N.J.; Andresen, B.M.; Northrup, A.B.; Childers, K.K.; Donofrio, A.; Miller, T.A.; Liu, Y.; Machacek, M.R.; Woo, H.C.; Spencer, K.B.; et al. Thiazole-substituted Aminoheteroaryls as spleen Tyrosine Kinase Inhibitors. WO/2014/176210 A1, 18 December 2014. [Google Scholar]
- Leach, K.; Loiacono, R.E.; Felder, C.C.; McKinzie, D.L.; Mogg, A.; Shaw, D.B.; Sexton, P.M.; Christopoulos, A. Molecular mechanisms of action and in vivo validation of an M4 muscarinic acetylcholine receptor allosteric modulator with potential antipsychotic properties. Neuropsychopharmacology 2010, 35, 855–869. [Google Scholar] [CrossRef] [PubMed]
- Christopoulos, A. Assessing the distribution of parameters in models of ligand-receptor interaction: To log or not to log. Trends Pharmacol. Sci. 1998, 19, 351–357. [Google Scholar] [CrossRef]
- Totrov, M.; Abagyan, R. Derivation of sensitive discrimination potential for virtual ligand screening. In Proceedings of the RECOMB ’99: Proceedings of the third annual International Conference on Computational Molecular Biology, Lyon, France, 11–14 April 1999; pp. 312–320. [Google Scholar]
- Abagyan, R.; Totrov, M. Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J. Mol. Biol. 1994, 235, 983–1002. [Google Scholar] [CrossRef]
- Neves, M.A.; Totrov, M.; Abagyan, R. Docking and scoring with ICM: The benchmarking results and strategies for improvement. J. Comput. Aided Mol. Des. 2012, 26, 675–686. [Google Scholar] [CrossRef]
- Schapira, M.; Totrov, M.; Abagyan, R. Prediction of the binding energy for small molecules, peptides and proteins. J. Mol. Recognit. 1999, 12, 177–190. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dinh, L.V.; Dangerfield, J.; DeBono, A.; Keller, A.N.; Josephs, T.M.; Gregory, K.J.; Leach, K.; Capuano, B. Next-Generation Analogues of AC265347 as Positive Allosteric Modulators of the Calcium-Sensing Receptor: Pharmacological Investigation of Structural Modifications at the Stereogenic Centre. Int. J. Mol. Sci. 2025, 26, 2580. https://doi.org/10.3390/ijms26062580
Dinh LV, Dangerfield J, DeBono A, Keller AN, Josephs TM, Gregory KJ, Leach K, Capuano B. Next-Generation Analogues of AC265347 as Positive Allosteric Modulators of the Calcium-Sensing Receptor: Pharmacological Investigation of Structural Modifications at the Stereogenic Centre. International Journal of Molecular Sciences. 2025; 26(6):2580. https://doi.org/10.3390/ijms26062580
Chicago/Turabian StyleDinh, Le Vi, Jesse Dangerfield, Aaron DeBono, Andrew N. Keller, Tracy M. Josephs, Karen J. Gregory, Katie Leach, and Ben Capuano. 2025. "Next-Generation Analogues of AC265347 as Positive Allosteric Modulators of the Calcium-Sensing Receptor: Pharmacological Investigation of Structural Modifications at the Stereogenic Centre" International Journal of Molecular Sciences 26, no. 6: 2580. https://doi.org/10.3390/ijms26062580
APA StyleDinh, L. V., Dangerfield, J., DeBono, A., Keller, A. N., Josephs, T. M., Gregory, K. J., Leach, K., & Capuano, B. (2025). Next-Generation Analogues of AC265347 as Positive Allosteric Modulators of the Calcium-Sensing Receptor: Pharmacological Investigation of Structural Modifications at the Stereogenic Centre. International Journal of Molecular Sciences, 26(6), 2580. https://doi.org/10.3390/ijms26062580