
Recent Evidences of Epigenetic Alterations in Chronic Obstructive Pulmonary Disease (COPD): A Systematic Review

 , ,
, ,  ,
,  and
and
Abstract

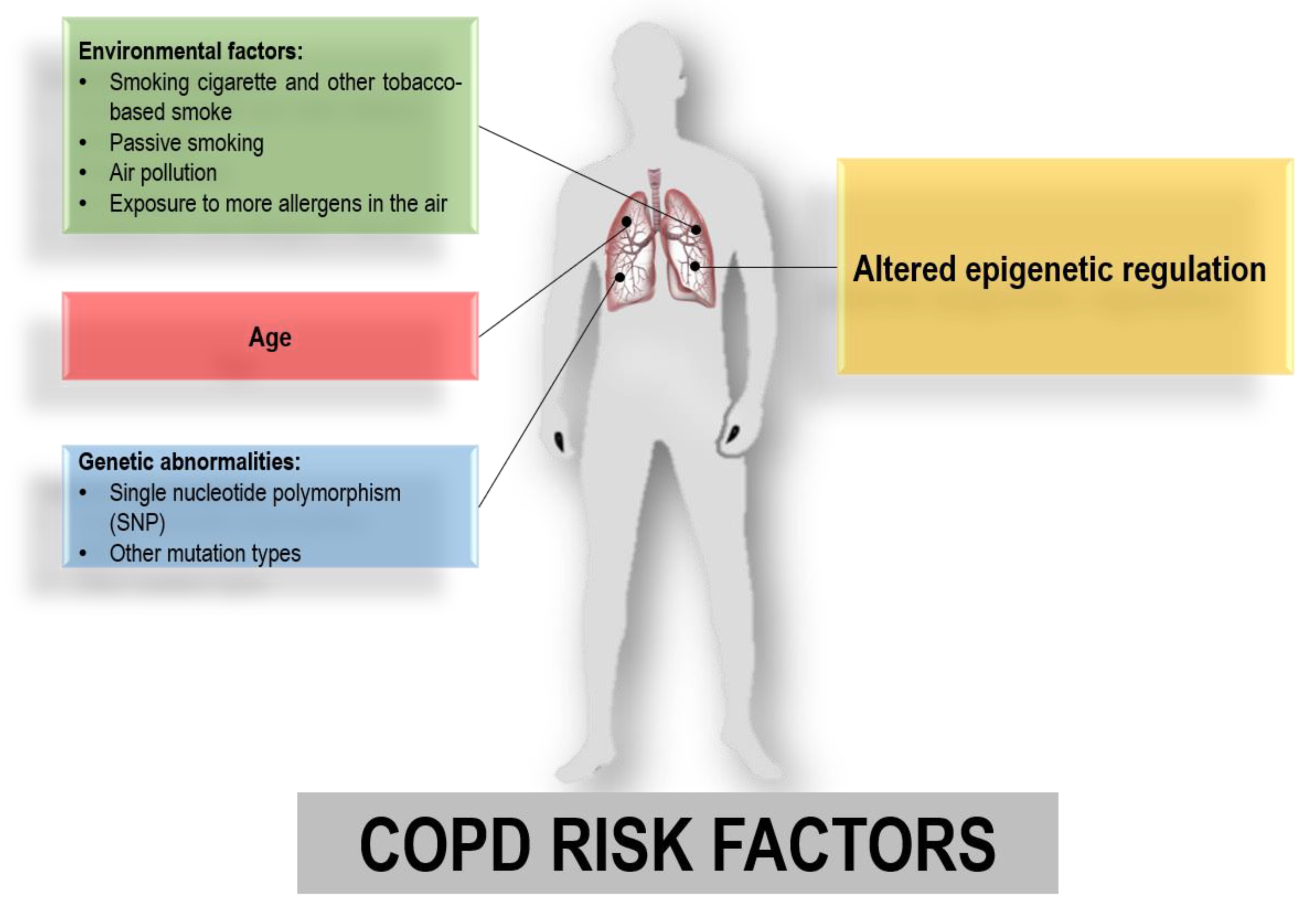
1. Introduction
2. Materials and Methods
2.1. Study Design and Search Strategy
2.1.1. Systematic Search Phases
2.1.2. Title and Abstract Selection
2.1.3. Full-Text Selection According to PICOS Criteria
2.1.4. Synthesis Method
2.1.5. Study Risk of Bias Assessment
3. Results
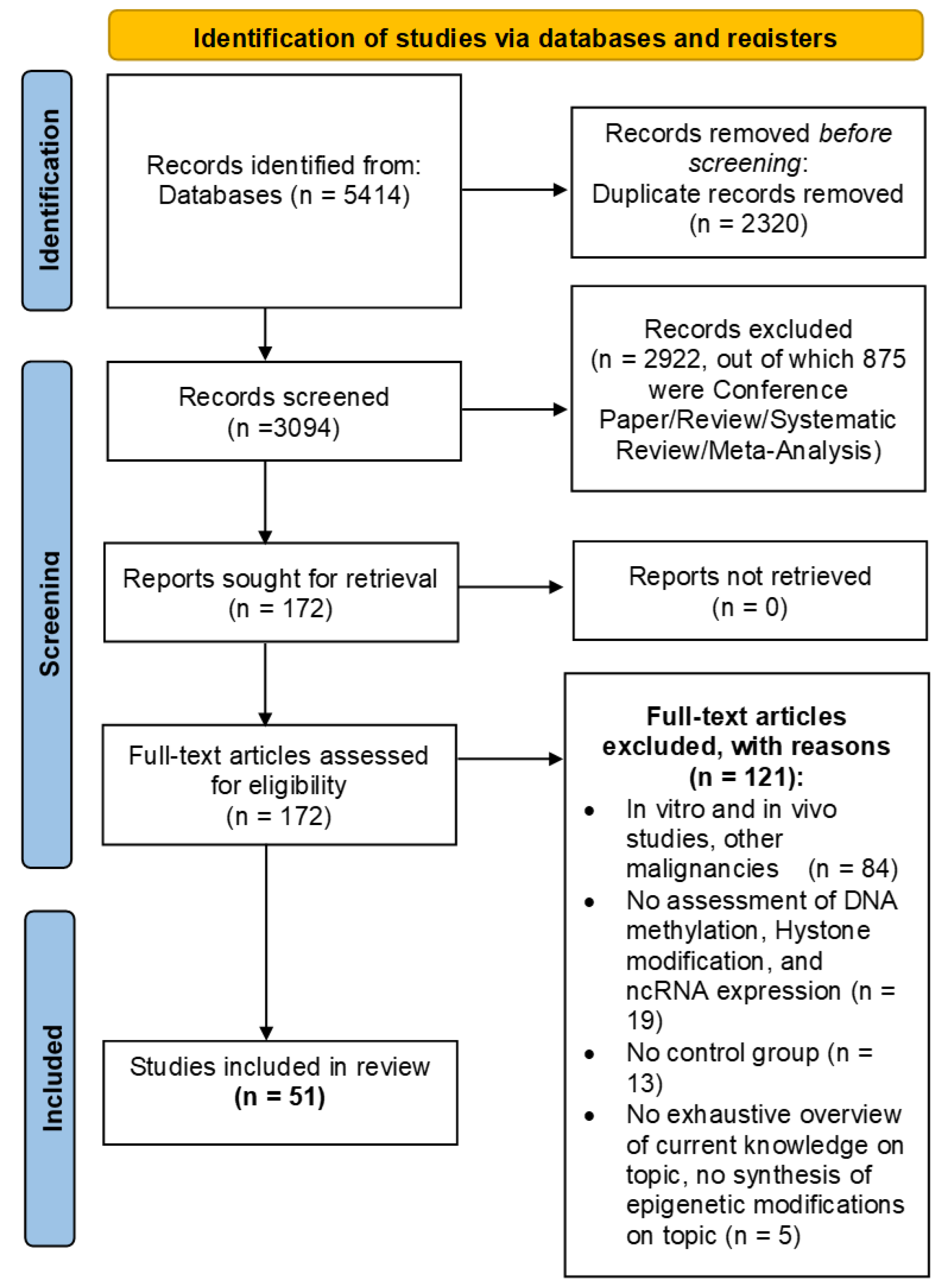
3.1. Flow Diagram
3.2. Study Selection and Characteristics
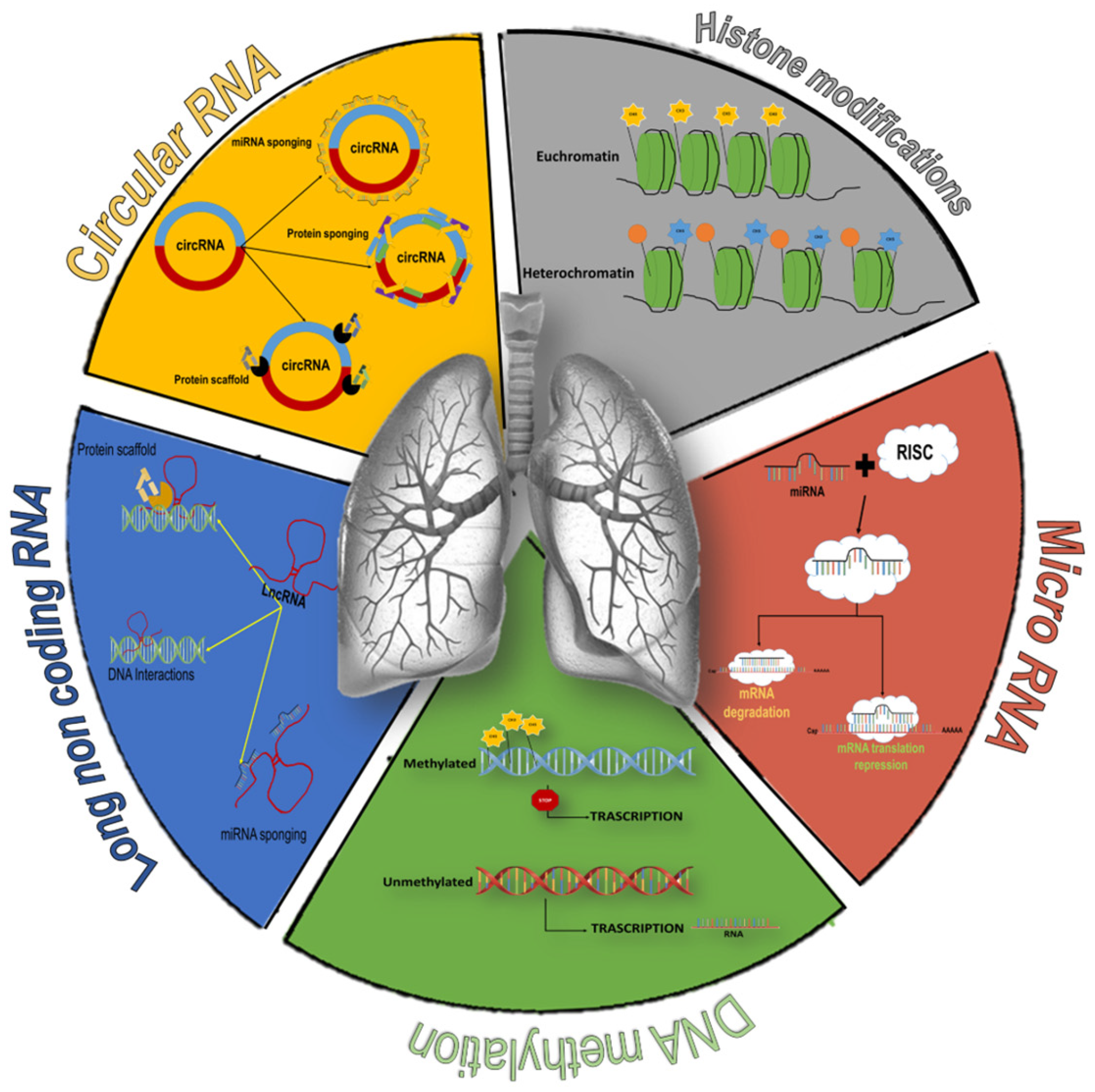
3.3. Synthesized Findings
3.4. DNA Methylation
3.5. Histone Modifications
3.6. Non-Coding RNA
3.6.1. Circular RNA
3.6.2. Long Non-Coding RNA
3.6.3. MicroRNA
3.6.4. Competing Endogenous RNA
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Disclaimer
References
- Barnes, P.J. Inflammatory Mechanisms in Patients with Chronic Obstructive Pulmonary Disease. J. Allergy Clin. Immunol. 2016, 138, 16–27. [Google Scholar] [CrossRef] [PubMed]
- WHO. 2023. Available online: https://www.who.int/News-Room/Fact-Sheets/Detail/Chronic-Obstructive-Pulmonary-Disease-(Copd) (accessed on 6 November 2024).
- Agustí, A.; Celli, B.R.; Criner, G.J.; Halpin, D.; Anzueto, A.; Barnes, P.; Bourbeau, J.; Han, M.L.K.; Martinez, F.J.; de Oca, M.M.; et al. Global Initiative for Chronic Obstructive Lung Disease 2023 Report: GOLD Executive Summary. Eur. Respir. J. 2023, 61, 2300239. [Google Scholar] [CrossRef] [PubMed]
- Safiri, S.; Carson-Chahhoud, K.; Noori, M.; Nejadghaderi, S.A.; Sullman, M.J.M.; Ahmadian Heris, J.; Ansarin, K.; Mansournia, M.A.; Collins, G.S.; Kolahi, A.A.; et al. Burden of Chronic Obstructive Pulmonary Disease and Its Attributable Risk Factors in 204 Countries and Territories, 1990–2019: Results from the Global Burden of Disease Study 2019. BMJ 2022, 378, e069679. [Google Scholar] [CrossRef]
- Lu, W.; Aarsand, R.; Schotte, K.; Han, J.; Lebedeva, E.; Tsoy, E.; Maglakelidze, N.; Soriano, J.B.; Bill, W.; Halpin, D.M.G.; et al. Tobacco and COPD: Presenting the World Health Organization (WHO) Tobacco Knowledge Summary. Respir. Res. 2024, 25, 338. [Google Scholar] [CrossRef]
- Lee, Y.J.; Choi, S.; Kwon, S.Y.; Lee, Y.; Lee, J.K.; Heo, E.Y.; Chung, H.S.; Kim, D.K. A Genome-Wide Association Study in Early Copd: Identification of One Major Susceptibility Loci. Int. J. Chronic Obstr. Pulm. Dis. 2020, 15, 2967–2975. [Google Scholar] [CrossRef]
- Yang, J.; Bakshi, A.; Zhu, Z.; Hemani, G.; Vinkhuyzen, A.A.E.; Lee, S.H.; Robinson, M.R.; Perry, J.R.B.; Nolte, I.M.; Van Vliet-Ostaptchouk, J.V.; et al. Genetic Variance Estimation with Imputed Variants Finds Negligible Missing Heritability for Human Height and Body Mass Index. Nat. Genet. 2015, 47, 1114–1120. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.H.; Hobbs, B.D.; Silverman, E.K. Genetics of Chronic Obstructive Pulmonary Disease: Understanding the Pathobiology and Heterogeneity of a Complex Disorder. Lancet Respir. Med. 2022, 10, 485–496. [Google Scholar] [CrossRef]
- Feng, X.; Dong, H.; Li, B.; Yu, L.; Zhu, J.; Lou, C.; Zhang, J. Integrative Analysis of the Expression Profiles of Whole Coding and Non-Coding RNA Transcriptomes and Construction of the Competing Endogenous RNA Networks for Chronic Obstructive Pulmonary Disease. Front. Genet. 2023, 14, 1050783. [Google Scholar] [CrossRef]
- Hernandez Cordero, A.I.; Yang, C.X.; Li, X.; Yang, J.; Shaipanich, T.; MacIsaac, J.L.; Lin, D.T.S.; Kobor, M.S.; Horvath, S.; Man, S.F.P.; et al. The Blood DNA Methylation Clock GrimAge Is a Robust Surrogate for Airway Epithelia Aging. Biomedicines 2022, 10, 3094. [Google Scholar] [CrossRef]
- Li, B.; Zhang, J.; Dong, H.; Feng, X.; Yu, L.; Zhu, J.; Zhang, J. Systematic Analysis of Various RNA Transcripts and Construction of Biological Regulatory Networks at the Post-Transcriptional Level for Chronic Obstructive Pulmonary Disease. J. Transl. Med. 2023, 21, 790. [Google Scholar] [CrossRef]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 Statement: An Updated Guideline for Reporting Systematic Reviews. Syst. Rev. 2021, 10, 89. [Google Scholar] [CrossRef] [PubMed]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G.; Antes, G.; Atkins, D.; Barbour, V.; Barrowman, N.; Berlin, J.A.; Clark, J.; et al. Preferred Reporting Items for Systematic Reviews and Meta-Analyses: The PRISMA Statement. PLoS Med. 2009, 6, e1000097. [Google Scholar] [CrossRef] [PubMed]
- Kachroo, P.; Morrow, J.D.; Vyhlidal, C.A.; Gaedigk, R.; Silverman, E.K.; Weiss, S.T.; Tantisira, K.G.; DeMeo, D.L. DNA methylation perturbations may link altered development and aging in the lung. Aging 2021, 13, 1742–1764. [Google Scholar] [CrossRef]
- Kachroo, P.; Morrow, J.D.; Kho, A.T.; Vyhlidal, C.A.; Silverman, E.K.; Weiss, S.T.; Tantisira, K.G.; DeMeo, D.L. Co-Methylation Analysis in Lung Tissue Identifies Pathways for Fetal Origins of COPD. Eur. Respir. J. 2020, 56, 1902347. [Google Scholar] [CrossRef]
- Schwartz, U.; Llamazares Prada, M.; Pohl, S.T.; Richter, M.; Tamas, R.; Schuler, M.; Keller, C.; Mijosek, V.; Muley, T.; Schneider, M.A.; et al. High-resolution Transcriptomic and Epigenetic Profiling Identifies Novel Regulators of COPD. EMBO J. 2023, 42, e111272. [Google Scholar] [CrossRef]
- Strom, J.E.; Merid, S.K.; Pourazar, J.; Blomberg, A.; Lindberg, A.; Ringh, M.V.; Hagemann-Jensen, M.; Ekstrom, T.J.; Behndig, A.F.; Melen, E. Chronic Obstructive Pulmonary Disease Is Associated with Epigenome-Wide Differential Methylation in BAL Lung Cells. Am. J. Respir. Cell Mol. Biol. 2022, 66, 638–647. [Google Scholar] [CrossRef] [PubMed]
- Morrow, J.D.; Make, B.; Regan, E.; Han, M.L.; Hersh, C.P.; Tal-Singer, R.; Quackenbush, J.; Choi, A.M.K.; Silverman, E.K.; DeMeo, D.L. DNA Methylation Is Predictive of Mortality in Current and Former Smokers. Am. J. Respir. Crit. Care Med. 2020, 201, 1099–1109. [Google Scholar] [CrossRef]
- Zhang, Z.; Fu, C.; Liu, J.; Sai, X.; Qin, C.; Di, T.; Yang, Y.; Wu, Y.; Bian, T. Hypermethylation of the Nrf2 Promoter Induces Ferroptosis by Inhibiting the Nrf2-GPX4 Axis in COPD. Int. J. Chronic Obstr. Pulm. Dis. 2021, 16, 3347–3362. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Nwozor, K.O.; van den Berge, M.; Slebos, D.J.; Faiz, A.; Jonker, M.R.; Boezen, H.M.; Heijink, I.H.; de Vries, M. From Differential DNA Methylation in COPD to Mitochondria: Regulation of AHRR Expression Affects Airway Epithelial Response to Cigarette Smoke. Cells 2022, 11, 3423. [Google Scholar] [CrossRef]
- Günes Günsel, G.; Conlon, T.M.; Jeridi, A.; Kim, R.; Ertüz, Z.; Lang, N.J.; Ansari, M.; Novikova, M.; Jiang, D.; Strunz, M.; et al. The Arginine Methyltransferase PRMT7 Promotes Extravasation of Monocytes Resulting in Tissue Injury in COPD. Nat. Commun. 2022, 13, 1303. [Google Scholar] [CrossRef]
- Duan, R.; Niu, H.; Yu, T.; Cui, H.; Yang, T.; Hao, K.; Wang, C. Identification and Bioinformatic Analysis of Circular RNA Expression in Peripheral Blood Mononuclear Cells from Patients with Chronic Obstructive Pulmonary Disease. Int. J. Chronic Obstr. Pulm. Dis. 2020, 15, 1391–1401. [Google Scholar] [CrossRef] [PubMed]
- Xie, T.; Yang, Z.; Xian, S.; Lin, Q.; Huang, L.; Ding, Y. Hsa_circ_0008833 Promotes COPD Progression via Inducing Pyroptosis in Bronchial Epithelial Cells. Exp. Lung Res. 2024, 50, 1–14. [Google Scholar] [CrossRef]
- Liu, P.; Wang, Y.; Zhang, N.; Zhao, X.; Li, R.; Wang, Y.; Chen, C.; Wang, D.; Zhang, X.; Chen, L.; et al. Comprehensive Identification of RNA Transcripts and Construction of RNA Network in Chronic Obstructive Pulmonary Disease. Respir. Res. 2022, 23, 154. [Google Scholar] [CrossRef]
- Zhang, C.; Gu, S.; Kang, X. CircRNA Circ_0006892 Regulates MiR-24/PHLPP2 Axis to Mitigate Cigarette Smoke Extract-Induced Bronchial Epithelial Cell Injury. Biotechnol. Appl. Biochem. 2022, 69, 735–748. [Google Scholar] [CrossRef]
- Wang, Z.; Zuo, Y.; Gao, Z. CircANKRD11 Knockdown Protects HPMECs from Cigarette Smoke Extract-Induced Injury by Regulating MiR-145-5p/BRD4 Axis. Int. J. Chronic Obstr. Pulm. Dis. 2021, 16, 887–899. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Ding, Y.; Zhou, Z.; Yang, W. Identification and Bioinformatic Analysis of CircRNAs in the Plasma of Patients with Very Severe Chronic Obstructive Pulmonary Disease. BMC Pulm. Med. 2023, 23, 211. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.R.; Liu, Y.Y.; Qian, R.Q.; Zhang, W.Y.; Huang, J.A.; Zhang, X.Q.; Zeng, D.X. Circular RNA Expression of Peripheral Blood Mononuclear Cells Associated with Risk of Acute Exacerbation in Smoking Chronic Obstructive Pulmonary Disease. Int. J. Chronic Obstr. Pulm. Dis. 2024, 19, 789–797. [Google Scholar] [CrossRef]
- Wu, S.M.; Feng, P.H.; Chuang, H.C.; Ho, S.C.; Fan Chung, K.; Chen, K.Y.; Wu, G.S.; Chen, T.T.; Tseng, C.H.; Liu, W.T.; et al. Impaired Lnc-IL7R Modulatory Mechanism of Toll-like Receptors Is Associated with an Exacerbator Phenotype of Chronic Obstructive Pulmonary Disease. FASEB J. 2020, 34, 13317–13332. [Google Scholar] [CrossRef]
- Zhou, A.Y.; Zhao, Y.Y.; Zhou, Z.J.; Duan, J.X.; Zhu, Y.Z.; Cai, S.; Chen, P. Microarray Analysis of Long Non-Coding RNAs in Lung Tissues of Patients with Copd and Hoxa-As2 Promotes Hpmecs Proliferation via Notch1. Int. J. Chronic Obstr. Pulm. Dis. 2020, 15, 2449–2460. [Google Scholar] [CrossRef]
- Wang, Y.; Lyu, X.; Wu, X.; Yu, L.; Hu, K. Long Non-Coding RNA PVT1, a Novel Biomarker for Chronic Obstructive Pulmonary Disease Progression Surveillance and Acute Exacerbation Prediction Potentially through Interaction with MicroRNA-146a. J. Clin. Lab. Anal. 2020, 34, e23346. [Google Scholar] [CrossRef]
- Liu, S.; Liu, M.; Dong, L. The Clinical Value of LncRNA MALAT1 and Its Targets MiR-125b, MiR-133, MiR-146a, and MiR-203 for Predicting Disease Progression in Chronic Obstructive Pulmonary Disease Patients. J. Clin. Lab. Anal. 2020, 34, e23410. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Zhang, H.; Zeng, H.; Meng, Y.; Gao, H.; Zhang, M.; Zhao, L. LncRNA CASC2 Is Involved in the Development of Chronic Obstructive Pulmonary Disease via Targeting MiR-18a-5p/IGF1 Axis. Ther. Adv. Respir. Dis. 2021, 15, 17534666211028072. [Google Scholar] [CrossRef]
- Zhao, S.; Lin, C.; Yang, T.; Qian, X.; Lu, J.; Cheng, J. Expression of Long Non-Coding RNA LUCAT1 in Patients with Chronic Obstructive Pulmonary Disease and Its Potential Functions in Regulating Cigarette Smoke Extract-Induced 16HBE Cell Proliferation and Apoptosis. J. Clin. Lab. Anal. 2021, 35, e23823. [Google Scholar] [CrossRef]
- Dai, Z.; Liu, X.; Zeng, H.; Chen, Y. Long Noncoding RNA HOTAIR Facilitates Pulmonary Vascular Endothelial Cell Apoptosis via DNMT1 Mediated Hypermethylation of Bcl-2 Promoter in COPD. Respir. Res. 2022, 23, 356. [Google Scholar] [CrossRef] [PubMed]
- Zong, D.; Liu, X.; Li, J.; Long, Y.; Ouyang, R.; Chen, Y. LncRNA-CCAT1/MiR-152-3p Is Involved in CSE-Induced Inflammation in HBE Cells via Regulating ERK Signaling Pathway. Int. Immunopharmacol. 2022, 109, 108818. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhao, H.; Raman, I.; Yan, M.; Chen, Q.; Li, Q.Z. Peripheral Blood Mononuclear Cell Gene Expression in Chronic Obstructive Pulmonary Disease: MiRNA and MRNA Regulation. J. Inflamm. Res. 2022, 15, 2167–2180. [Google Scholar] [CrossRef]
- Hu, J.; Wang, W.; Lu, Q.; Du, L.; Qin, T. Differential Expression of MiRNAs in Bronchoalveolar Lavage Fluid and Plasma from Patients with Chronic Obstructive Pulmonary Disease. Medicine 2022, 101, e30969. [Google Scholar] [CrossRef]
- Zhang, J.; Xu, Z.; Kong, L.; Gao, H.; Zhang, Y.; Zheng, Y.; Wan, Y. Mirna-486-5p Promotes Copd Progression by Targeting Hat1 to Regulate the Tlr4-Triggered Inflammatory Response of Alveolar Macrophages. Int. J. Chronic Obstr. Pulm. Dis. 2020, 15, 2991–3001. [Google Scholar] [CrossRef]
- Yang, H.; Zhang, L.; Wang, Q. MicroRNA-221-3p Alleviates Cell Apoptosis and Inflammatory Response by Targeting Cyclin Dependent Kinase Inhibitor 1B in Chronic Obstructive Pulmonary Disease. Bioengineered 2021, 12, 5705–5715. [Google Scholar] [CrossRef]
- Chang, C.; Huang, K.; Xu, X.; Duan, R.; Yu, T.; Chu, X.; Chen, C.; Li, B.; Yang, T. MiR-23a-5p Alleviates Chronic Obstructive Pulmonary Disease through Targeted Regulation of RAGE-ROS Pathway. Respir. Res. 2024, 25, 93. [Google Scholar] [CrossRef]
- Kim, R.Y.; Sunkara, K.P.; Bracke, K.R.; Jarnicki, A.G.; Donovan, C.; Hsu, A.C.; Ieni, A.; Beckett, E.L.; Galvão, I.; Wijnant, S.; et al. A MicroRNA-21-Mediated SATB1/S100A9/NF-κB Axis Promotes Chronic Obstructive Pulmonary Disease Pathogenesis. Sci. Transl. Med. 2021, 13, eaav7223. [Google Scholar] [CrossRef]
- De Smet, E.G.; Van Eeckhoutte, H.P.; Avila Cobos, F.; Blomme, E.; Verhamme, F.M.; Provoost, S.; Verleden, S.E.; Venken, K.; Maes, T.; Joos, G.F.; et al. The Role of MiR-155 in Cigarette Smoke-Induced Pulmonary Inflammation and COPD. Mucosal Immunol. 2020, 13, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Han, Y.; Almuntashiri, S.; Dutta, S.; Wang, X.; Owen, C.A.; Zhang, D. Dysregulation of MiR-103a Mediates Cigarette Smoking–Induced Lipid-Laden Macrophage Formation. Am. J. Respir. Cell Mol. Biol. 2022, 67, 695–707. [Google Scholar] [CrossRef]
- Yang, Z.; Li, P.; Yuan, Q.; Wang, X.; Ma, H.H.; Zhuan, B. Inhibition of MiR-4640-5p Alleviates Pulmonary Hypertension in Chronic Obstructive Pulmonary Disease Patients by Regulating Nitric Oxide Synthase 1. Respir. Res. 2023, 24, 92. [Google Scholar] [CrossRef]
- Tasena, H.; Timens, W.; Van Den Berge, M.; Van Broekhuizen, J.; Kennedy, B.K.; Hylkema, M.N.; Brandsma, C.A.; Heijink, I.H. MicroRNAs Associated with Chronic Mucus Hypersecretion in COPD Are Involved in Fibroblast–Epithelium Crosstalk. Cells 2022, 11, 526. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Li, F.J.; Dsouza, K.; Stephens, C.T.; Zheng, H.; Kumar, A.; Dransfield, M.T.; Antony, V.B. Low Dose Cadmium Exposure Regulates MiR-381–ANO1 Interaction in Airway Epithelial Cells. Sci. Rep. 2024, 14, 246. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Jiang, Y.L.; Fei, J.; Cao, P.; Zhang, C.; Xie, G.F.; Wang, L.X.; Cao, W.; Fu, L.; Zhao, H. Circulatory Cadmium Positively Correlates with Epithelial-Mesenchymal Transition in Patients with Chronic Obstructive Pulmonary Disease. Ecotoxicol. Environ. Saf. 2021, 215, 112164. [Google Scholar] [CrossRef]
- Shi, X.F.; He, X.; Sun, Z.R.; Wang, J.X.; Gu, Y.H.; Xie, Y.B.; Duo, J. Different Expression of Circulating MicroRNA Profile and Plasma SP-D in Tibetan COPD Patients. Sci. Rep. 2022, 12, 3388. [Google Scholar] [CrossRef]
- Shen, Y.; Lu, H.; Song, G. MiR-221-3p and MiR-92a-3p Enhances Smoking-Induced Inflammation in COPD. J. Clin. Lab. Anal. 2021, 35, e23857. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, X.; Zheng, Y.; Du, F.; He, G. MiR-548ar-3p Increases Cigarette Smoke Extract-Induced Chronic Obstructive Pulmonary Disease (COPD) Injury through Solute Carrier Family 17 Member 9 (SLC17A9). Arch. Biol. Sci. 2022, 74, 97–105. [Google Scholar] [CrossRef]
- Nadi, E.; Geramirad, G.; Kahramfar, Z.; Rasouli-Saravani, A.; Solgi, G. Peripheral Blood Expressions of MicroRNA-146a and MicroRNA-218 in Chronic Obstructive Pulmonary Disease with/without Cigarette Smoke Exposure. Iran. J. Allergy Asthma Immunol. 2022, 21, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Tang, S.; Zhou, Z.; Wei, H.; Yang, W. Plasma MiR-150-5p as a Biomarker for Chronic Obstructive Pulmonary Disease. Int. J. Chronic Obstr. Pulm. Dis. 2023, 18, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Shi, Q.; Xiong, L.; Shi, S.; Li, Y.; Wang, Y.; Zhang, M. Clinical Relevance of MiR-423-5p Levels in Chronic Obstructive Pulmonary Disease Patients. Clinics 2022, 77, 100102. [Google Scholar] [CrossRef]
- Tao, S.; Liao, C.; Wang, Y.; Xu, D.; Li, Z.; Li, F. Differential MiRNA Profiling Reveals MiR-4433a-5p as a Key Regulator of Chronic Obstructive Pulmonary Disease Progression via PIK3R2- Mediated Phenotypic Modulation. Comb. Chem. High Throughput Screen. 2024, 27, 2323–2334. [Google Scholar] [CrossRef] [PubMed]
- Cazorla-Rivero, S.; Mura-Escorche, G.; Gonzalvo-Hernández, F.; Mayato, D.; Córdoba-Lanús, E.; Casanova, C. Circulating Mir-1246 in the Progression of Chronic Obstructive Pulmonary Disease (Copd) in Patients from the Bode Cohort. Int. J. Chronic Obstr. Pulm. Dis. 2020, 15, 2727–2737. [Google Scholar] [CrossRef]
- Wang, C.; Feng, D.; Dong, S.; He, R.; Fan, B. Dysregulated Circulating MicroRNA-126 in Chronic Obstructive Pulmonary Disease: Linkage with Acute Exacerbation Risk, Severity Degree, and Inflammatory Cytokines. J. Clin. Lab. Anal. 2022, 36, e24204. [Google Scholar] [CrossRef]
- Huang, H.; Wu, F.; Yang, J.; Li, H.; Cai, M.; Shan, K.; Shi, S. Increased Plasma Level of MiR-210 as a Potential Diagnostic Marker for Chronic Obstructive Pulmonary Disease Induced Pulmonary Hypertension. Clin. Lab. 2020, 66, 971–977. [Google Scholar] [CrossRef]
- Burke, H.; Cellura, D.; Freeman, A.; Hicks, A.; Ostridge, K.; Watson, A.; Williams, N.P.; Spalluto, C.M.; Staples, K.J.; Wilkinson, T.M.A. Pulmonary EV MiRNA Profiles Identify Disease and Distinct Inflammatory Endotypes in COPD. Front. Med. 2022, 9, 1039702. [Google Scholar] [CrossRef]
- Wang, F.; Yang, B.; Qiao, J.; Bai, L.; Li, Z.; Sun, W.; Liu, Q.; Yang, S.; Cui, L. Serum Exosomal MicroRNA-1258 May as a Novel Biomarker for the Diagnosis of Acute Exacerbations of Chronic Obstructive Pulmonary Disease. Sci. Rep. 2023, 13, 18332. [Google Scholar] [CrossRef]
- Shen, H.F.; Liu, Y.; Qu, P.P.; Tang, Y.; Li, B.B.; Cheng, G.L. Mir-361-5p/Abca1 and Mir-196-5p/Arhgef12 Axis Involved in γ-Sitosterol Inducing Dual Anti-Proliferative Effects on Bronchial Epithelial Cells of Chronic Obstructive Pulmonary Disease. Int. J. Chronic Obstr. Pulm. Dis. 2021, 16, 2741–2753. [Google Scholar] [CrossRef]
- Downes, M.J.; Brennan, M.L.; Williams, H.C.; Dean, R.S. Development of a Critical Appraisal Tool to Assess the Quality of Cross-Sectional Studies (AXIS). BMJ Open 2016, 6, e011458. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef]
- Kodal, J.B.; Kobylecki, C.J.; Vedel-Krogh, S.; Nordestgaard, B.G.; Bojesen, S.E. AHRR Hypomethylation, Lung Function, Lung Function Decline and Respiratory Symptoms. Eur. Respir. J. 2018, 51, 1701512. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Rahman, I. Role of Histone Deacetylase 2 in Epigenetics and Cellular Senescence: Implications in Lung Inflammaging and COPD. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, 557–566. [Google Scholar] [CrossRef]
- Santer, L.; Bär, C.; Thum, T. Circular RNAs: A Novel Class of Functional RNA Molecules with a Therapeutic Perspective. Mol. Ther. 2019, 27, 1350–1363. [Google Scholar] [CrossRef]
- Mattick, J.S.; Amaral, P.P.; Carninci, P.; Carpenter, S.; Chang, H.Y.; Chen, L.L.; Chen, R.; Dean, C.; Dinger, M.E.; Fitzgerald, K.A.; et al. Long Non-Coding RNAs: Definitions, Functions, Challenges and Recommendations. Nat. Rev. Mol. Cell Biol. 2023, 24, 430–447. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Shen, Z.; Guo, J.; Sun, S. Screening of Long Non-Coding RNA and TUG1 Inhibits Proliferation with TGF-β Induction in Patients with COPD. Int. J. Chronic Obstr. Pulm. Dis. 2016, 11, 2951–2964. [Google Scholar] [CrossRef]
- Devadoss, D.; Long, C.; Langley, R.J.; Manevski, M.; Nair, M.; Campos, M.A.; Borchert, G.; Rahman, I.; Chand, H.S. Long Noncoding Transcriptome in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Cell Mol. Biol. 2019, 61, 678–688. [Google Scholar] [CrossRef]
- Feng, F.; Qi, Y.; Dong, C.; Yang, C. Pvt1 Regulates Inflammation and Cardiac Functiovia the Mapk/Nf-Κb Pathway in a Sepsis Model. Exp. Ther. Med. 2018, 16, 4471–4478. [Google Scholar] [CrossRef]
- Chen, Y.; Zeng, Z.; Shen, X.; Wu, Z.; Dong, Y.; Cheng, J.C.H. MicroRNA-146a-5p Negatively Regulates pro-Inflammatory Cytokine Secretion and Cell Activation in Lipopolysaccharide Stimulated Human Hepatic Stellate Cells through Inhibition of Toll-like Receptor 4 Signaling Pathways. Int. J. Mol. Sci. 2016, 17, 1076. [Google Scholar] [CrossRef]
- Billi, M.; De Marinis, E.; Gentile, M.; Nervi, C.; Grignani, F. Nuclear MiRNAs: Gene Regulation Activities. Int. J. Mol. Sci. 2024, 25, 6066. [Google Scholar] [CrossRef] [PubMed]
- Arif, R.; Pandey, A.; Zhao, Y.; Arsenault-Mehta, K.; Khoujah, D.; Mehta, S. Treatment of Pulmonary Hypertension Associated with COPD: A Systematic Review. ERJ Open Res. 2022, 8, 00348-2021. [Google Scholar] [CrossRef] [PubMed]
- Tasena, H.; Faiz, A.; Timens, W.; Noordhoek, J.; Hylkema, M.N.; Gosens, R.; Hiemstra, P.S.; Spira, A.; Postma, D.S.; Tew, G.W.; et al. MicroRNA–MRNA Regulatory Networks Underlying Chronic Mucus Hypersecretion in COPD. Eur. Respir. J. 2018, 52, 1701556. [Google Scholar] [CrossRef] [PubMed]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A CeRNA Hypothesis: The Rosetta Stone of a Hidden RNA Language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Key Words |
|---|
| Chronic obstructive pulmonary disease, COPD |
| Biomarker*, therapeutic target* |
| Epigenome-Wide Association, sequencing, epigenetic*, DNA methylation*, long noncoding RNA*, circRNA*, miRNA*, histone* deacetylation*, histone* protein*, HDAC |
| Molecular mechanism*, extracellular vesicle* |
| Database | Steps | Query | Research in | Items Found |
|---|---|---|---|---|
| PubMed | #1 | (((((((((((((Biomarker*[Title/Abstract]) OR (“therapeutic target*”[Title/Abstract])) OR (“Epigenome-Wide Association*”[Title/Abstract])) OR (sequencing[Title/Abstract])) OR (epigenetic*[Title/Abstract])) OR (“DNA methylation*”[Title/Abstract])) OR (“long noncoding RNA*”[Title/Abstract])) OR (circRNA*[Title/Abstract])) OR (miRNA*[Title/Abstract])) OR (“histone* deacetylation*”[Title/Abstract])) OR (“histone* protein*”[Title/Abstract])) OR (HDAC[Title/Abstract])) OR (“Molecular mechanism*”[Title/Abstract])) OR (“extracellular vesicle*”[Title/Abstract]) | Title/Abstract | 1,460,188 |
| #2 | “COPD”[Title/Abstract] OR “Chronic obstructive pulmonary disease”[Title/Abstract] | Title/Abstract | 83,977 | |
| #3 | Combine #1 AND #2 | 5834 | ||
| #4 | Limit to (English) | 5663 | ||
| #5 | Limit after 2020 | 2509 | ||
| Scopus | #1 | TITLE-ABS-KEY (“Biomarker*” OR “therapeutic target*” OR “Epigenome-Wide Association*” OR “sequencing” OR “epigenetic*” OR “ DNA methylation*” OR “long noncoding RNA*” OR “circRNA*” OR “miRNA*” OR “histone* deacetylation*” OR “histone* protein*” OR “HDAC” OR “molecular mechanism*” OR “extracellular vesicle*”) | Title/Abstract/Keywords | 1,989,075 |
| #2 | TITLE-ABS-KEY (“COPD” OR “Chronic Obstructive Pulmonary Disease”) | Title/Abstract/Keywords | 102,296 | |
| #3 | Combine #1 AND #2 | 7433 | ||
| #4 | Limit to (English) and (Italian) | 7078 | ||
| #5 | Limit after 2020 | 2906 |
| Parameters | Inclusion Criteria | Exclusion Criteria |
|---|---|---|
| Participants | Studies in humans Studies including COPD patients | In vitro and in vivo studies Participants with other malignancies |
| Interventions | Assessment of DNA methylation, Histone modification, and ncRNA expression | Others |
| Comparisons | Control Group | Others |
| Outcomes | (1) to provide unbiased and exhaustive overview of the current knowledge on the epigenetic modification associated COPD; (2) to summarize the epigenetic modifications translated into clinical therapeutic interventions and biomarkers for COPD. | Others |
| Study Design | Original studies in English | Review, Scoping Review, Narrative Review, Systematic Review, Meta-Analysis, Editorial, Book, Case Report, Conference Review, and Conference Paper |
| Study | Country | Number of Participants | Type of Sample | Gene Affected | Epigenetic Alteration | Activity in COPD | Role of Epigenetic Mechanisms |
|---|---|---|---|---|---|---|---|
| Kachroo P, et al., 2021 [14] | Boston (USA) | N = 78 fetal N = 160 adult COPD | Lung tissue | Transcription factors, oxido-reductase, VEGFA-VEGFR2 | Hyper-/hypo-methylation | Air flow limitation, inflammation activation, lung remodeling | Fetal origin of COPD |
| Kachroo P, et al., 2020 [15] | Boston (USA) | N = 78 fetal N = 160 adult COPD | Lung tissue | Co-methylation: Wnt, Pi3K/AKT, MAPK, Hippo | DNA methylation imbalance | Low lung function | Fetal origin of COPD |
| Schwartz U, et al., 2023 [16] | Heidelberg and Munich (Germany) Huston (USA) | N = 3 control N = 3 COPD I N = 5 COPD II-IV | Parenchymal fibroblasts (lung tissue) | 3 cluster of genes involved in cell proliferation, DNA repair and extracellular matrix organization | Hyper-/hypo-methylation | Low lung function | Kinetics of DNA methylation in COPD |
| Strom JE, et al., 2022 [17] | Northern Sweden | N = 15 control N = 18 COPD | Macrophage from Broncho alveolar lavage (BAL) | DMPs co-localized with COPD-associated SNPs | DNA methylation imbalance | --- | Pathophysiology of COPD |
| Cordero AIH, et al., 2022 [10] | Vancouver (Canada) | N = 27 control N = 15 COPD | Small airway epithelial brushings and buffy coat blood | DNAmGrimAge | DNA methylation imbalance | Biomarker for assessing accelerated aging in the airways of individuals with COPD | Biomarker |
| Morrow JD, et al., 2020 [18] | Boston (USA) | N = 336 control N = 331 COPD | Blood samples | Pi3KCD cg03971555 cg12033075 | Hyper methylation | Predictive biomarker | Biomarker |
| Zhang Z, et al., 2021 [19] | Wuxi (China) | N = 18/17 control N = 8/16 COPD | Lung tissue/bronchoscopies (bronco epithelial cells) | Nfr2 | Hyper methylation | Increased oxidative stress and cell death | Therapeutic target |
| Chen Q, et al., 2022 [20] | Groningen (The Netherlands) | N = 966/8 control N = 595/14 COPD | whole blood/airway epithelial cells | AHRR cg05575921 cg21161138 | Hypo methylation | Airway epithelial cell proliferation, dysregulate mitochondrial function, and reduce apoptotic processes | Therapeutic target |
| Study | Country | Number of Participants | Type of Sample | Gene Affected | Epigenetic Alteration | Activity in COPD | Role of Epigenetic Mechanisms |
| Günes GG, et al., 2022 [21] | Ghent (Belgium) | N = 40 control N = 111 COPD | Monocytes/lung tissue | PRMT7 | Histone methylation | Chronic inflammation | Pathophysiology of COPD/Therapeutic target |
| Study | Country | Number of Participants | Type of Sample | Gene Affected | Epigenetic Alteration | Activity in COPD | Role of Epigenetic Mechanisms |
|---|---|---|---|---|---|---|---|
| Duan R, et al., 2020 [22] | Beijing (China) | N = 21 control N = 21 COPD | Peripheral blood mononuclear cells | Gene involved in natural killer T cell activation and T-helper cell differentiation | Differential expression in COPD compared to control | Immune balance alteration | Pathophysiology of COPD/Therapeutic target |
| Xie T, et al., 2024 [23] | Hainan (China) | N = 5 control N = 10 (acute and stable) COPD | Peripheral blood mononuclear cells | Caspase 1, IL-18, IL-1β | Upregulation hsa-circ_0008833-57aa | Pyroptosis | Pathophysiology of COPD |
| Liu P, et al., 2022 [24] | Anhui (China) | N = 3 control N = 3 COPD | Blood samples | miR-1273h-3p; miR-411-5p; miR-122-5p; miR-615-5p; miR-519d-3p; miR-485-3p; miR-3646; miR-4714-5p; miR-203b-5p; miR-193a-5p; miR-1261; miR-4690-5p; miR-939-5p; miR-9-5p; miR-2113; miR-7977 | Upregulation circFCHO2; circMBOAT2, circPTPN22; circTBC1D22A; circACADM; circCKAP5 | ---- | Pathophysiology of COPD |
| Zhang C, et al., 2022 [25] | Jiangsu (China) | N = 17 COPD non-smoker N = 23 COPD smoker | Lung tissue | miR-24/PHPPL2 axis | Downregulation Circ_0006892 | Inflammatory injury | Pathophysiology of COPD |
| Wang Z, et al., 2021 [26] | Hebei (China) | N = 27 control N = 21COPD | Lung tissue | miR-145-5p/BRD4 axis | Upregulation Circ_ANKRDII | Inflammation, apoptosis and oxidative stress | Pathophysiology of COPD |
| Tang S, et al., 2023 [27] | Hefei (China) | N = 30 control N = 30 COPD | Plasma samples | ---- | Differential expression circ_0008882; circ_00089763; circ_00062683; circ_00077607 | Immune balance alteration | Biomarkers |
| Shen X, et al., 2024 [28] | Jiangsu (China) | N = 29 control N = 41 COPD | Peripheral blood mononuclear cells | ---- | Differential expression circ_0049875 and circ_0042590 | Acute exacerbation of COPD | Biomarkers |
| Study | Country | Number of Participants | Type of Sample | Gene Affected | Epigenetic Alteration | Activity in COPD | Role of Epigenetic Mechanisms |
|---|---|---|---|---|---|---|---|
| Wu S, et al., 2020 [29] | Taipei (Taiwan) | N = 35 control N = 64 COPD | Peripheral blood mononuclear cells | IL-8, VCAM1, E-SEL | Downregulation lncRNA-IL7R | Inflammatory processes | Pathophysiology of COPD |
| Zhou AY, et al., 2020 [30] | Xiangya (China) | N = 3 control N = 7COPD | Lung tissue | Notch1 | Downregulation lncRNA- HOXA-AS2 | Cell viability and Inflammatory processes | Pathophysiology of COPD |
| Wang Y, et al., 2020 [31] | Wuhan (China) | N = 80 control N = 80 stable COPD N = 80 AECOPD | Peripheral blood mononuclear cells | Mir-146a/TNFα, IL-6, IL-8, IL-1β, IL-17 | Upregulation LncRNA-PVT1 | Inflammatory processes | Prognostic biomarker |
| Liu S, et al., 2020 [32] | Wuhan (China) | N = 120control N = 120 stable COPD N = 120 AECOPD | Blood samples | miR-125b, miR-133, miR-146a, miR-203/TNFα, IL-6, IL-8, IL-1β, IL-17, IL-23 | Upregulation LncRNA-MALAT1 | Inflammatory processes | Prognostic biomarker |
| Liu P, et al., 2021 [33] | Shanghai (China) | N = 90 control N = 50 COPD | Blood samples | Mir-18a-5p/TNFα, IL-6, IL-8, IL-1β | Downregulation lncRNA-CASC2 | Inflammatory processes | Diagnostic biomarker |
| Zhao S, et al., 2021 [34] | Jiangsu (China) | N = 150 control N = 70 COPD | Blood samples | Mir-181-5p/Wnt/b-catenin axis | Upregulation LncRNA-LUCAT1 | Apoptotic/Inflammatory processes | Biomarker/therapeutic target |
| Dai Z, et al., 2022 [35] | Xiangya (China) | N = 8 control N = 5COPD | Lung tissue | Bcl-2 | Upregulation lncRNA-HOTAIR | Apoptotic processes | Therapeutic target |
| Zong D, et al., 2022 [36] | Xiangya (China) | N = 10 control N = 10 COPD | Lung tissue | Mir-152-3p/ERK | Upregulation lncRNA-CCT1 | Inflammatory processes | Therapeutic target |
| Study | Country | Number of Participants | Type of Sample | Gene Affected | Epigenetic Alteration | Activity in COPD | Role of Epigenetic Mechanisms |
|---|---|---|---|---|---|---|---|
| Wang L, et al., 2022 [37] | Xiangya (China) | N = 12 control N = 12 COPD | Peripheral blood mononuclear cells | IL-8 signaling; ------ iCOS-iCOSL signaling | Aberrant expression: miR-4453; miR-4736; miR-3118; miR-6967-5p; miR-132-3p; miR-96-5p; miR-4497 ------ miR-16-5p; miR-1964-5p; miR-29b-3p; miR-2355-3p; miR-18a-5p; miR-1234-3p; miR-148-3p; miR-21-5p; miR-1184; miR-140-5p; miR-19b-3p; miR-223-3p; miR-1246; miR-130a-3p | Inflammatory processes | Pathophysiology of COPD |
| Hu J, et al., 2022 [38] | Wuhan (China) | N = 3/9 control N = 3/9 COPD | BALF/ blood samples | MAPK, RAS, FOXO | miR-129-5p; miR-3529-3p; miR-365b-3p; miR-6503-5p; miR-26-3p; miR-34b-5p; miR-4748; miR-491-5p; miR-158-3p | Oxidative/inflammatory process | Pathophysiology of COPD |
| Zhang J, et al., 2020 [39] | Xuzhou (China) | N = 14/75 control N = 36/53 COPD | Alveolar macrophages/Peripheral blood mononuclear cells | HAT1/TNFα-IL-6-IL-8 | Upregulation miR-486-5p | Inflammatory processes | Pathophysiology of COPD |
| Yang H, et al., 2021 [40] | Xuzhou (China) | N = 27 control N = 21COPD | Lung tissue | CDKN1B | Downregulation miR-221-3p | Apoptotic and Inflammatory processes | Pathophysiology of COPD |
| Chang C, et al., 2024 [41] | Beijing (China) | N = 19 control N = 13 COPD | Lung tissue | RAGE | Downregulation miR-23a-5p | Oxidative/inflammatory process | Pathophysiology of COPD |
| Kim R, et al., 2021 [42] | Newcastle, New South Wales (Australia) | N = 5 control N = 10 COPD | Lung tissue | SATB1/S100A9/NF-kB | Upregulation miR-21 | Inflammatory processes | Pathophysiology of COPD |
| De Smet E, et al., 2020 [43] | Ghent (Belgium) | N= 44 control N= 48 COPD | Lung tissue | MMP12, ADAM19 | Upregulation miR-155 | Inflammatory processes | Pathophysiology of COPD |
| Zhu Y, et al., 2022 [44] | Brigham (USA) | N= 17control N = 8COPD | Alveolar macrophages | LDLR | Downregulation miR-103a | Oxidative/inflammatory process | Pathophysiology of COPD |
| Yang Z, et al., 2023 [45] | Suzhou (China) | N = 14 control N = 14 COPD | Lung tissue | NOS1 | Upregulation miR-4640-5p | Pulmonary hypertension | Pathophysiology of COPD |
| Tasena H, et al., 2022 [46] | Groningen (The Netherlands) | N = 6 control N = 3 COPD | Lung tissue | MICS5AC, COL4A1, COL5A1 | Upregulation miR-708-5p, let-7a-5p, miR-31-5p, miR-146a-5p | Epithelial differentiation, chronic mucus hypersecretion (CMH) | Pathophysiology of COPD |
| Singh P, et al., 2024 [47] | Birmingham (USA) | N = 9 control N = 13 COPD | Lung tissue | ANO1 | Downregulation miR-381 | Mucus production and secretion | Pathophysiology/therapeutic target |
| Zheng L, et al., 2021 [48] | Hefei (China) | N = 400 control N = 400 COPD N = 50 control + COPD | Blood samples/ Lung tissue | E-cadherin, α-SMA, Vimentin, N-cadherin | Downregulation miR-30 | Epithelial-mesenchymal transition | Pathophysiology of COPD |
| Shi X, et al., 2022 [49] | Qinghai (China) | N = 40 control N = 40 COPD | Blood samples | ------ | Upregulation miR-486-5p; miR-106b-5p | Hypoxia/Pulmonary Hypertension | Biomarkers |
| Shen Y, et al., 2021 [50] | Nanjing (China) | N = 77 control N = 155 COPD | Blood samples | TNFα, IL-6, IL-8, IL-1β | Upregulation miR-221-3p; miR-92a-3p | Inflammatory processes | Biomarkers |
| Zhang L, et al., 2022 [51] | Guizhou (China) | N = 6 control N = 6 COPD | Blood samples | SLC17A9 | Downregulation miR-548ar-3p | ------ | Biomarkers |
| Nadi E, et al., 2022 [52] | Hamadan (Iran) | N = 60 control N = 60 COPD | Blood samples | ------ | Downregulation miR-146a; miR-216 | Oxidative/inflammatory process | Biomarkers |
| Ding Y, et al., 2023 [53] | Hefei (China) | N = 26 control N = 59 COPD | Blood samples | ------ | Downregulation miR-150-5p | Inflammatory processes | Biomarkers |
| Zhang X, et al., 2022 [54] | ChongQing (China) | N = 33 control N = 36 COPD | Blood samples | ------ | Downregulation miR-423-5p | ------ | Biomarkers |
| Tao S, et al., 2024 [55] | Xiangya (China) | N = 23 control N = 240 COPD | Blood samples | ------ ------ PIK3R2 | Downregulation miR-1290; miR-1246 ------ Upregulation miR-4433a-5p | ------ ------ Apoptotic and Inflammatory processes | Biomarkers |
| Cazola-Rivero S, et al., 2020 [56] | Tenerife (Spain) | N = 13 control N = 24 COPD | Blood samples | MAPK, chemokines, Wnt | Downregulation miR-1246 | Emphysema development | Biomarkers |
| Wang C, et al., 2022 [57] | Jiaozuo (China) | N = 70 control N = 140 COPD | Blood samples | TNFα, IL-1β, IL-6 | Upregulation miR-126 | Inflammatory processes | Biomarkers |
| Huang H, et al., 2020 [58] | Kunshan (China) | N = 80 control N = 160 COPD | Blood samples | ------ | Upregulation miR-210 | Pulmonary hypertension | Biomarkers |
| Burke H, et al., 2022 [59] | Southampton (United Kingdom) | N = 20 control N = 24 COPD | BALF/Extra vesicles | ------ | Downregulation miR-338-3p; miR-204-5p ------ Upregulation miR-223-3p; miR-182-5p; miR-2110 | Inflammatory patterns | Biomarkers |
| Wang F, et al., 2023 [60] | Peking (China) | N = 42 control N = 111COPD | Blood samples/Exosome | ------ | Upregulation miR-1258 | Inflammatory processes | Biomarkers |
| Shen HF, et al., 2021 [61] | Binzhou (China) | N = 20 control N = 20 COPD | Blood samples | ARHGEF12, BCAT1 | Upregulation miR-196-5p Downregulation miR-361-5p | Epithelial hyperplasia | Therapeutic target |
| Study | Country | Number of Participants | Type of Sample | Gene Affected | Epigenetic Alteration | Activity in COPD | Role of Epigenetic Mechanisms |
|---|---|---|---|---|---|---|---|
| Li B, et al., 2023 [11] | Ningxia (China) | N = 6 control N = 14 COPD | Lung tissue | 18 hub genes | ceRNA aberrant expression | Immune cells infiltration/differentiation; cell proliferation | Pathophysiology of COPD |
| Feng X, et al., 2023 [9] | Ningxia (China) | N = 6 control N = 7COPD | Lung tissue | TNFα/NF-kb; IL-6/JAK/STAT3 | ceRNA aberrant expression | Inflammatory processes | Pathophysiology of COPD |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ragusa, R.; Bufano, P.; Tognetti, A.; Laurino, M.; Caselli, C. Recent Evidences of Epigenetic Alterations in Chronic Obstructive Pulmonary Disease (COPD): A Systematic Review. Int. J. Mol. Sci. 2025, 26, 2571. https://doi.org/10.3390/ijms26062571
Ragusa R, Bufano P, Tognetti A, Laurino M, Caselli C. Recent Evidences of Epigenetic Alterations in Chronic Obstructive Pulmonary Disease (COPD): A Systematic Review. International Journal of Molecular Sciences. 2025; 26(6):2571. https://doi.org/10.3390/ijms26062571
Chicago/Turabian StyleRagusa, Rosetta, Pasquale Bufano, Alessandro Tognetti, Marco Laurino, and Chiara Caselli. 2025. "Recent Evidences of Epigenetic Alterations in Chronic Obstructive Pulmonary Disease (COPD): A Systematic Review" International Journal of Molecular Sciences 26, no. 6: 2571. https://doi.org/10.3390/ijms26062571
APA StyleRagusa, R., Bufano, P., Tognetti, A., Laurino, M., & Caselli, C. (2025). Recent Evidences of Epigenetic Alterations in Chronic Obstructive Pulmonary Disease (COPD): A Systematic Review. International Journal of Molecular Sciences, 26(6), 2571. https://doi.org/10.3390/ijms26062571