
Utilization of Machine Learning in the Prediction, Diagnosis, Prognosis, and Management of Chronic Myeloid Leukemia
 ,
,  ,
,
Abstract
1. Introduction
General Consideration on Chronic Myeloid Leukemia and Artificial Intelligence
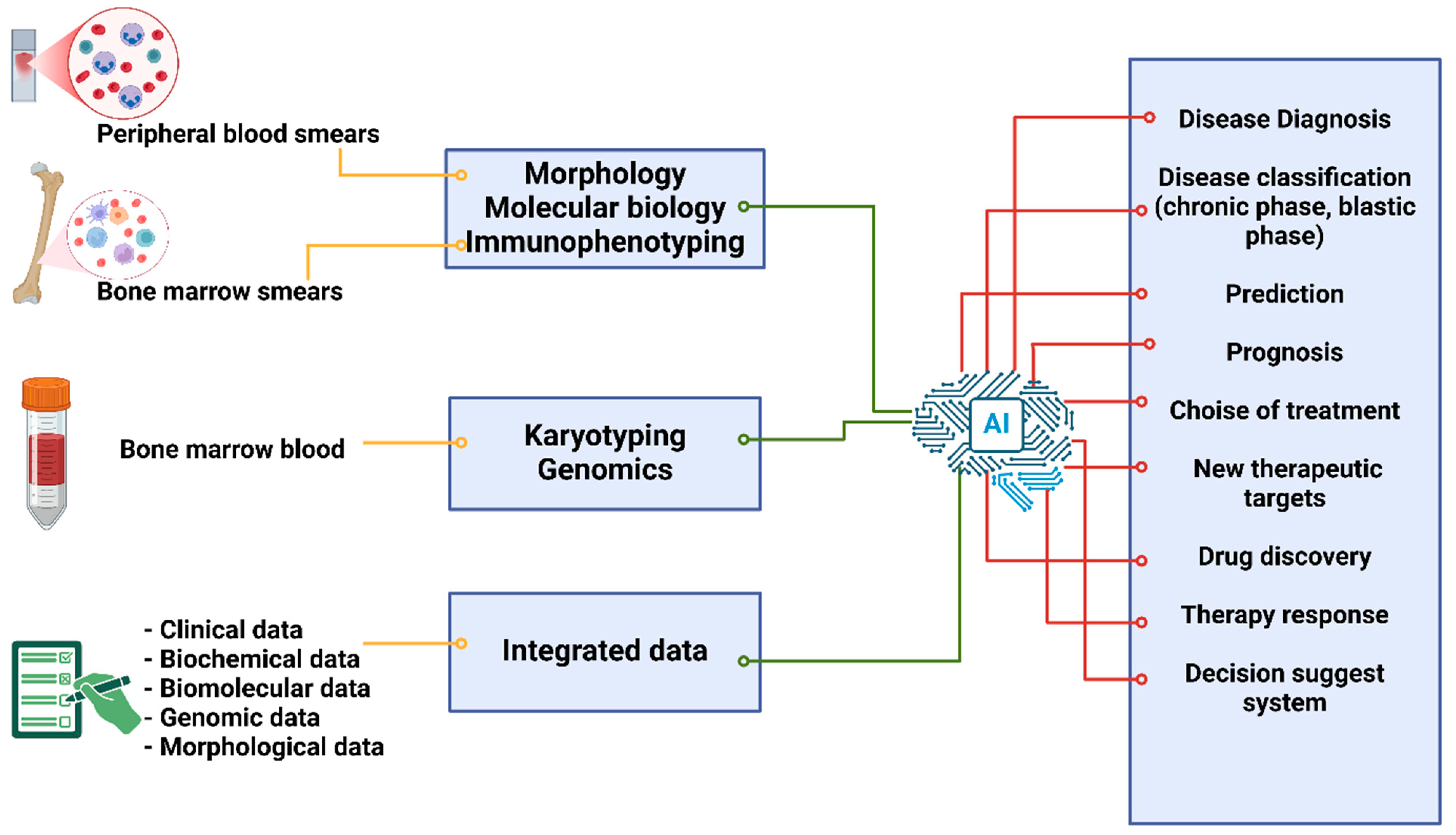
2. Use of AI in CML
2.1. Diagnosis Through a Morphological Analysis of Peripheral and Bone Marrow Smears
2.2. AI’s Application in Immunophenotyping for CML Diagnosis
2.3. Using AI for Karyotyping in the Diagnosis of CML
3. AI for Improving Adult CML Treatment Response and Disease Progression Prediction Using Biochemical, Biomolecular, and Clinical Data
4. AI-Based Assessment of Drug Resistance in CML Patients
AI and Treatment Side Effects in Individuals with CML
5. AI-Powered Drug Discovery for CML Patients
Applying AI to the Study of CML’s Molecular Space
6. Prospects for the Application of AI in CML in the Future
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- American Cancer Society. Cancer Facts & Figures 2023; American Cancer Society: Atlanta, GA, USA, 2023; Available online: https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2023/2023-cancer-facts-and-figures.pdf (accessed on 2 March 2025).
- Skorski, T. Genetic mechanisms of chronic myeloid leukemia blastic transformation. Curr. Hematol. Malig. Rep. 2012, 7, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.M.; Keating, M.J.; Talpaz, M.; Walters, R.S.; Smith, T.L.; Cork, A.; McCredie, K.B.; Freireich, E.J. Chronic myelogenous leukemia in blast crisis. Analysis of 242 patients. Am. J. Med. 1987, 83, 445–454. [Google Scholar] [CrossRef] [PubMed]
- DeFilipp, Z.; Khoury, H.J. Management of advanced-phase chronic myeloid leukemia. Curr. Hematol. Malig. Rep. 2015, 10, 173–181. [Google Scholar] [CrossRef]
- Yohanan, B.; George, B. Current Management of Chronic Myeloid Leukemia Myeloid Blast Phase. Clin. Med. Insights Oncol. 2022, 16, 11795549221139357. [Google Scholar] [CrossRef]
- Melo, J.V.; Hughes, T.P.; Apperley, J.F. Chronic myeloid leukemia. Hematol. Am. Soc. Hematol. Educ. Program 2003, 2003, 132–152. [Google Scholar] [CrossRef]
- Silver, R.T.; Woolf, S.H.; Hehlmann, R.; Appelbaum, F.R.; Anderson, J.; Bennett, C.; Goldman, J.M.; Guilhot, F.; Kantarjian, H.M.; Lichtin, A.E.; et al. An evidence-based analysis of the effect of busulfan, hydroxyurea, interferon, and allogeneic bone marrow transplantation in treating the chronic phase of chronic myeloid leukemia: Developed for the American Society of Hematology. Blood 1999, 94, 1517–1536. [Google Scholar] [PubMed]
- Senapati, J.; Sasaki, K.; Issa, G.C.; Lipton, J.H.; Radich, J.P.; Jabbour, E.; Kantarjian, H.M. Management of chronic myeloid leukemia in 2023—Common ground and common sense. Blood Cancer J. 2023, 13, 58. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jabbour, E.; Cortes, J.E.; Kantarjian, H.M. Molecular monitoring in chronic myeloid leukemia. Cancer 2008, 112, 2112–2118. [Google Scholar] [CrossRef]
- Wang, W.; Cortes, J.E.; Tang, G.; Khoury, J.D.; Wang, S.; Bueso-Ramos, C.E.; DiGiuseppe, J.A.; Chen, Z.; Kantarjian, H.M.; Medeiros, L.J.; et al. Risk stratification of chromosomal abnormalities in chronic myelogenous leukemia in the era of tyrosine kinase inhibitor therapy. Blood 2016, 127, 2742–2750. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Abdelmagid, M.G.; Litzow, M.R.; McCullough, K.B.; Gangat, N.; Pardanani, A.; Murthy, H.S.; Foran, J.M.; Ketterling, R.P.; Viswanatha, D.; Begna, K.H.; et al. Chronic phase CML with sole P190 (e1a2) BCR::ABL1: Long-term outcome among ten consecutive cases. Blood Cancer J. 2022, 12, 103. [Google Scholar] [CrossRef]
- Verma, D.; Kantarjian, H.M.; Jones, D.; Luthra, R.; Borthakur, G.; Verstovsek, S.; Rios, M.B.; Cortes, J. Chronic myeloid leukemia (CML) with P190BCR-ABL: Analysis of characteristics, outcomes, and prognostic significance. Blood 2009, 114, 2232–2235. [Google Scholar] [CrossRef] [PubMed]
- Ashida, T.; Kanamaru, A. Differential diagnosis of chronic myeloid leukemia and the related disorders. Nihon Rinsho. Jpn. J. Clin. Med. 2001, 59, 2358–2362. [Google Scholar] [PubMed]
- Walter, W.; Haferlach, C.; Nadarajah, N.; Schmidts, I.; Kühn, C.; Kern, W.; Haferlach, T. How artificial intelligence might disrupt diagnostics in hematology in the near future. Oncogene 2021, 40, 4271–4280. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lin, E.; Fuda, F.; Luu, H.S.; Cox, A.M.; Fang, F.; Feng, J.; Chen, M. Digital pathology and artificial intelligence as the next chapter in diagnostic hematopathology. Semin. Diagn. Pathol. 2023, 40, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Bera, K.; Schalper, K.A.; Rimm, D.L.; Velcheti, V.; Madabhushi, A. Artificial intelligence in digital pathology—New tools for diagnosis and precision oncology. Nat. Rev. Clin. Oncol. 2019, 16, 703–715. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Swolin, B.; Simonsson, P.; Backman, S.; Lofqvist, I.; Bredin, I.; Johnsson, M. Differential Counting of Blood Leukocytes Using Automated Microscopy and a Decision Support System Based on Artificial Neural Networks—Evaluation of DiffMasterTM Octavia. Clin. Lab. Haematol. 2003, 25, 139–147. [Google Scholar] [CrossRef]
- Haferlach, T.; Pohlkamp, C.; Heo, I.; Drescher, R.; Hänselmann, S.; Lörch, T.; Kern, W.; Haferlach, C.; Nadarajah, N. Automated peripheral blood cell differentiation using artificial intelligence-a study with more than 10,000 routine samples in a specialized leukemia laboratory. Blood 2021, 138, 103. [Google Scholar] [CrossRef]
- Ahmed, N.; Yigit, A.; Isik, Z.; Alpkocak, A. Identification of Leukemia Subtypes from Microscopic Images Using Convolutional Neural Network. Diagnostics 2019, 9, 104. [Google Scholar] [CrossRef]
- Huang, F.; Guang, P.; Li, F.; Liu, X.; Zhang, W.; Huang, W. AML, ALL, and CML Classification and Diagnosis Based on Bone Marrow Cell Morphology Combined with Convolutional Neural Network: A STARD Compliant Diagnosis Research. Medicine 2020, 99, e23154. [Google Scholar] [CrossRef]
- Murković, M.; Babarović, E.; Marijić, B.; Grohovac, D.; Hadžisejdić, I. Association of pre-treatment bone marrow morphology and achievement of BCR-ABL1 transcript milestones in CML. Pathol. Res. Pract. 2023, 246, 154517. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Huang, X.; Yan, Q.; Lin, Y.; Liu, E.; Mi, Y.; Liang, S.; Wang, H.; Xu, J.; Ru, K. The Diagnosis of Chronic Myeloid Leukemia with Deep Adversarial Learning. Am. J. Pathol. 2022, 192, 1083–1091. [Google Scholar] [CrossRef] [PubMed]
- Dese, K.; Raj, H.; Ayana, G.; Yemane, T.; Adissu, W.; Krishnamoorthy, J.; Kwa, T. Accurate Machine-Learning-Based Classification of Leukemia from Blood Smear Images. Clin. Lymphoma Myeloma Leuk. 2021, 21, e903–e914. [Google Scholar] [CrossRef] [PubMed]
- Wood, B.L.; Arroz, M.; Barnett, D.; DiGiuseppe, J.; Greig, B.; Kussick, S.J.; Oldaker, T.; Shenkin, M.; Stone, E.; Wallace, P. 2006 Bethesda International Consensus recommendations on the immunophenotypic analysis of hematolymphoid neoplasia by flow cytometry: Optimal reagents and reporting for the flow cytometric diagnosis of hematopoietic neoplasia. Cytom. B Clin. Cytom. 2007, 72 (Suppl. S1), S14–S22. [Google Scholar] [CrossRef]
- Raspadori, D.; Pacelli, P.; Sicuranza, A.; Abruzzese, E.; Iurlo, A.; Cattaneo, D.; Gozzini, A.; Galimberti, S.; Barate, C.; Pregno, P.; et al. Flow Cytometry Assessment of CD26(+) Leukemic Stem Cells in Peripheral Blood: A Simple and Rapid New Diagnostic Tool for Chronic Myeloid Leukemia. Cytom. B Clin. Cytom. 2019, 96, 294–299. [Google Scholar] [CrossRef]
- Cerrato, T.R. Use of artificial intelligence to improve access to initial leukemia diagnosis in low- and middle-income countries. J. Clin. Oncol. 2020, 38, e14117. [Google Scholar] [CrossRef]
- Ni, W.; Tong, X.; Qian, W.; Jin, J.; Zhao, H. Discrimination of malignant neutrophils of chronic myelogenous leukemia from normal neutrophils by support vector machine. Comput. Biol. Med. 2013, 43, 1192–1195. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zheng, B.; Li, S.; Mulvihill, J.J.; Chen, X.; Liu, H. Automated identification of abnormal metaphase chromosome cells for the detection of chronic myeloid leukemia using microscopic images. J. Biomed. Opt. 2010, 15, 046026. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ram, M.; Afrash, M.R.; Moulaei, K.; Parvin, M.; Esmaeeli, E.; Karbasi, Z.; Heydari, S.; Sabahi, A. Application of artificial intelligence in chronic myeloid leukemia (CML) disease prediction and management: A scoping review. BMC Cancer 2024, 24, 1026. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Shanbehzadeh, M.; Afrash, M.R.; Mirani, N.; Kazemi-Arpanahi, H. Comparing Machine Learning Algorithms to Predict 5-Year Survival in Patients with Chronic Myeloid Leukemia. BMC Med. Inform. Decis. Mak. 2022, 22, 236. [Google Scholar] [CrossRef]
- Dey, P.; Lamba, A.; Kumari, S.; Marwaha, N. Application of an Artificial Neural Network in the Prognosis of Chronic Myeloid Leukemia. Anal. Quant. Cytol. Histol. 2011, 33, 335–339. [Google Scholar]
- Banjar, H.; Ranasinghe, D.; Brown, F.; Adelson, D.; Kroger, T.; Leclercq, T.; White, D.; Hughes, T.; Chaudhri, N. Modelling Predictors of Molecular Response to Frontline Imatinib for Patients with Chronic Myeloid Leukaemia. PLoS ONE 2017, 12, e0168947. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Jabbour, E.J.; Ravandi, F.; Konopleva, M.; Borthakur, G.; Wierda, W.G.; Daver, N.; Takahashi, K.; Naqvi, K.; DiNardo, C.; et al. The LEukemia Artificial Intelligence Program (LEAP) in Chronic Myeloid Leukemia in Chronic Phase: A Model to Improve Patient Outcomes. Am. J. Hematol. 2021, 96, 241. [Google Scholar] [CrossRef] [PubMed]
- Borisov, N.; Tkachev, V.; Suntsova, M.; Kovalchuk, O.; Zhavoronkov, A.; Muchnik, I.; Buzdin, A. A Method of Gene Expression Data Transfer from Cell Lines to Cancer Patients for Machine-Learning Prediction of Drug Efficiency. Cell Cycle 2018, 17, 486. [Google Scholar] [CrossRef]
- Yen, R.; Grasedieck, S.; Wu, A.; Lin, H.; Su, J.; Rothe, K.; Nakamoto, H.; Forrest, D.L.; Eaves, C.J.; Jiang, X. Identification of Key MicroRNAs as Predictive Biomarkers of Nilotinib Response in Chronic Myeloid Leukemia: A Sub-Analysis of the ENESTxtnd Clinical Trial. Leukemia 2022, 36, 2443–2452. [Google Scholar] [CrossRef]
- Alves, R.; Gonçalves, A.C.; Rutella, S.; Almeida, A.M.; De Las Rivas, J.; Trougakos, I.P.; Sarmento Ribeiro, A.B. Resistance to Tyrosine Kinase Inhibitors in Chronic Myeloid Leukemia-From Molecular Mechanisms to Clinical Relevance. Cancers 2021, 13, 4820. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhou, Y.; Portelli, S.; Pat, M.; Rodrigues, C.H.M.; Nguyen, T.B.; Pires, D.E.V.; Ascher, D.B. Structure-Guided Machine Learning Prediction of Drug Resistance Mutations in Abelson 1 Kinase. Comput. Struct. Biotechnol. J. 2021, 19, 5381. [Google Scholar] [CrossRef]
- Su, J.; Fu, C.; Zhang, S.; Chen, X.; Wang, R.; Shi, H.; Li, J.; Wang, X. Screening and Activity Evaluation of Novel BCR-ABL/T315I Tyrosine Kinase Inhibitors. Curr. Med. Chem. 2024, 31, 2872–2894. [Google Scholar] [CrossRef]
- Mehra, N.; Varmeziar, A.; Chen, X.; Kronick, O.; Fisher, R.; Kota, V.; Mitchell, C.S. Cross-Domain Text Mining to Predict Adverse Events from Tyrosine Kinase Inhibitors for Chronic Myeloid Leukemia. Cancers 2022, 14, 4686. [Google Scholar] [CrossRef]
- Cortes, J.; Lang, F. Third-line therapy for chronic myeloid leukemia: Current status and future directions. J. Hematol. Oncol. 2021, 14, 44. [Google Scholar] [CrossRef]
- Combes, F.P.; Li, Y.F.; Hoch, M.; Lorenzo, S.; Ho, Y.; Sy, S.K.B. Exposure–Efficacy Analysis of Asciminib in Philadelphia Chromosome–Positive Chronic Myeloid Leukemia in Chronic Phase. Clin. Pharmacol. Ther. 2022, 112, 1040–1050. [Google Scholar] [CrossRef]
- Naveed, M.; Ain, N.U.; Aziz, T.; Javed, K.; Shabbir, M.A.; Alharbi, M.; Alsahammari, A.; Alasmari, A.F. Artificial Intelligence Assisted Pharmacophore Design for Philadelphia Chromosome-Positive Leukemia with Gamma-Tocotrienol: A Toxicity Comparison Approach with Asciminib. Biomedicines 2023, 11, 1041. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nicolotti, O.; Giangreco, I.; Introcaso, A.; Leonetti, F.; Stefanachi, A.; Carotti, A. Strategies of multi-objective optimization in drug discovery and development. Expert Opin. Drug Discov. 2011, 6, 871–884. [Google Scholar] [CrossRef]
- Cavalluzzi, M.M.; Imbrici, P.; Gualdani, R.; Stefanachi, A.; Mangiatordi, G.F.; Lentini, G.; Nicolotti, O. Human ether-à-go-go-related potassium channel: Exploring SAR to improve drug design. Drug Discov. Today 2020, 25, 344–366. [Google Scholar] [CrossRef] [PubMed]
- Cavalluzzi, M.M.; Mangiatordi, G.F.; Nicolotti, O.; Lentini, G. Ligand efficiency metrics in drug discovery: The pros and cons from a practical perspective. Expert Opin. Drug Discov. 2017, 12, 1087–1104. [Google Scholar] [CrossRef]
- Nicolotti, O.; Gillet, V.J.; Fleming, P.J.; Green, D.V.S. Multiobjective optimization in quantitative structure-activity relationships: Deriving accurate and interpretable QSARs. J. Med. Chem. 2002, 45, 5069–5080. [Google Scholar] [CrossRef]
- Alberga, D.; Trisciuzzi, D.; Montaruli, M.; Leonetti, F.; Mangiatordi, G.F.; Nicolotti, O. A New Approach for Drug Target and Bioactivity Prediction: The Multifingerprint Similarity Search Algorithm (MuSSeL). J. Chem. Inf. Model. 2019, 59, 586–596. [Google Scholar] [CrossRef] [PubMed]
- Ruff, K.M.; Pappu, R.V. AlphaFold and implications for intrinsically disordered proteins. J. Mol. Biol. 2021, 433, 167208. [Google Scholar] [CrossRef]
- Jiménez, J.; Doerr, S.; Martínez-Rosell, G.; Rose, A.S.; De Fabritiis, G. DeepSite: Protein-binding site predictor using 3Dconvolutional neural networks. Bioinformatics 2017, 33, 3036–3042. [Google Scholar] [CrossRef]
- Ben Hadda, T.; Rastija, V.; AlMalki, F.; Titi, A.; Touzani, R.; Mabkhot, Y.N.; Khalid, S.; Zarrouk, A.; Siddiqui, B.S. Petra/Osiris/Molinspiration and Molecular Docking Analyses of 3-Hydroxy-Indolin-2-one Derivatives as Potential Antiviral Agents. Curr. Comput. Aided-Drug Des. 2021, 17, 123–133. [Google Scholar] [CrossRef]
- Song, K.; Liu, X.; Huang, W.; Lu, S.; Shen, Q.; Zhang, L.; Zhang, J. Improved Method for the Identification and Validation of Allosteric Sites. J. Chem. Inf. Model. 2017, 57, 2358–2363. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, S.; Hu, Q.; Gao, S.; Ma, X.; Zhang, W.; Shen, Y.; Chen, F.; Lai, L.; Pei, J. CavityPlus: A web server for protein cavity detection with pharmacophore modelling, allosteric site identification and covalent ligand binding ability prediction. Nucleic Acids Res. 2018, 46, W374–W379. [Google Scholar] [CrossRef] [PubMed]
- Yueh, C.; Rettenmaier, J.; Xia, B.; Hall, D.R.; Alekseenko, A.; Porter, K.A.; Barkovich, K.; Keseru, G.; Whitty, A.; Wells, J.A.; et al. Kinase Atlas: Druggability Analysis of Potential Allosteric Sites in Kinases. J. Med. Chem. 2019, 62, 6512–6524. [Google Scholar] [CrossRef] [PubMed]
- Kozakov, D.; Grove, L.E.; Hall, D.R.; Bohnuud, T.; Mottarella, S.E.; Luo, L.; Xia, B.; Beglov, D.; Vajda, S. The FTMap family of web servers for determining and characterizing ligand-binding hot spots of proteins. Nat. Protoc. 2015, 10, 733–755. [Google Scholar] [CrossRef]
- Miljković, F.; Rodríguez-Pérez, R.; Bajorath, J. Machine Learning Models for Accurate Prediction of Kinase Inhibitors with Different Binding Modes. J. Med. Chem. 2019, 63, 8738–8748. [Google Scholar] [CrossRef] [PubMed]
- Alberga, D.; Gambacorta, N.; Trisciuzzi, D.; Ciriaco, F.; Amoroso, N.; Nicolotti, O. De novo drug design of targeted chemical libraries based on artificial intelligence and pair based multiobjective optimization. J. Chem. Inf. Model. 2020, 60, 4582–4593. [Google Scholar]
- Melge, A.R.; Parate, S.; Pavithran, K.; Koyakutty, M.; Mohan, C.G. Discovery of Anticancer Hybrid Molecules by Supervised Machine Learning Models and in Vitro Validation in Drug Resistant Chronic Myeloid Leukemia Cells. J. Chem. Inf. Model. 2022, 62, 1126–1146. [Google Scholar] [CrossRef]
- Desaphy, J.; Bret, G.; Rognan, D.; Kellenberger, E. sc-PDB: A 3D-database of ligandable binding sites—10 years on. Nucleic Acids Res. 2015, 43, D399–D404. [Google Scholar] [CrossRef]
- Schrödinger Release 2020-4: Desmond Molecular Dynamics System, D.E; Maestro-Desmond Interoperability Tools; Shaw Research/Schrödinger: New York, NY, USA, 2020.
- Malik, V.; Radhakrishnan, N.; Kaul, S.C.; Wadhwa, R.; Sundar, D. Computational Identification of BCR-ABL Oncogenic Signaling as a Candidate Target of Withaferin A and Withanone. Biomolecules 2022, 12, 212. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Agrafiotis, D.K.; Lobanov, V.S.; Salemme, F.R. Combinatorial informatics in the post-genomics era. Nature Rev. Drug Discov. 2002, 1, 337–346. [Google Scholar] [CrossRef]
- Ellis, R.J.; Minton, A.P. Join the crowd. Nature 2003, 425, 27–28. [Google Scholar] [CrossRef]
- Hall, D.; Minton, A.P. Macromolecular crowding: Qualitative and semiquantitative successes, quantitative challenges. Biochim. Biophys. Acta. 2003, 1649, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Dobson, C.M. Chemical space and biology. Nature 2004, 16, 824–828. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, H.A.; Baskin, I.I.; Marcou, G.; Horvath, D.; Varnek, A. Stargate GTM: Bridging Descriptor and Activity Spaces. J. Chem. Inf. Model. 2015, 55, 2403–2410. [Google Scholar] [CrossRef] [PubMed]
- Maggiora, G.M.; Bajorath, J. Chemical space networks: A powerful new paradigm for the description of chemical space. J. Comput. Aided Mol. Des. 2014, 28, 795–802. [Google Scholar] [CrossRef] [PubMed]
- Vogt, M.; Stumpfe, D.; Maggiora, G.M.; Bajorath, J. Lessons learned from the design of chemical space networks and opportunities for new applications. J. Comput. Aided Mol. Des. 2016, 30, 191–208. [Google Scholar] [CrossRef] [PubMed]
- Lejmi, M.; Geslin, D.; Bureau, R.; Cuissart, B.; Ben Slima, I.; Meddouri, N.; Borgi, A.; Lamotte, J.L.; Lepailleur, A. Navigating pharmacophore space to identify activity discontinuities: A case study with BCR-ABL. Mol. Inform. 2024, 43, e202400050. [Google Scholar] [CrossRef] [PubMed]
- Métivier, J.P.; Cuissart, B.; Bureau, R.; Lepailleur, A. The Pharmacophore Network: A Computational Method for Exploring Structure-Activity Relationships from a Large Chemical Data Set. J. Med. Chem. 2018, 61, 3551–3564. [Google Scholar] [CrossRef] [PubMed]
- Kerns, J.G.; Gikas, P.D.; Buckley, K.; Shepperd, A.; Birch, H.L.; McCarthy, I.; Miles, J.; Briggs, T.W.; Keen, R.; Parker, A.W.; et al. Evidence from Raman spectroscopy of a putative link between inherent bone matrix chemistry and degenerative joint disease. Arthritis Rheumatol. 2014, 66, 1237–1246. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Carey, P.R. Raman spectroscopy, the sleeping giant in structural biology, awakes. J. Biol. Chem. 1999, 274, 26625–26628. [Google Scholar] [CrossRef]
- Bonsignore, M.; Trusso, S.; De Pasquale, C.; Ferlazzo, G.; Allegra, A.; Innao, V.; Musolino, C.; Franco, D.; De Plano, M.L.; Guglielmino, S.P.P.; et al. A multivariate analysis of Multiple Myeloma subtype plasma cells. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2021, 258, 119813. [Google Scholar] [CrossRef] [PubMed]
- Franco, D.; Trusso, S.; Fazio, E.; Allegra, A.; Musolino, C.; Speciale, A.; Cimino, F.; Saija, A.; Neri, F.; Nicolò, M.S.; et al. Raman spectroscopy differentiates between sensitive and resistant multiple myeloma cell lines. Spectrochim. Acta A Mol Biomol. Spectrosc. 2017, 187, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Fazio, E.; Trusso, S.; Franco, D.; Nicolò, M.S.; Allegra, A.; Neri, F.; Musolino, C.; Guglielmino, S.P. A micro-Raman spectroscopic investigation of leukemic U-937 cells in aged cultures. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2016, 159, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Schlücker, S. Surface-Enhanced Raman Spectroscopy: Concepts and Chemical Applications. Angew. Chem. Int. Ed. 2014, 53, 4756–4795. [Google Scholar] [CrossRef] [PubMed]
- Lentini, G.; Fazio, E.; Calabrese, F.; De Plano, L.M.; Puliafico, M.; Franco, D.; Nicolò, M.S.; Carnazza, S.; Trusso, S.; Allegra, A.; et al. Phage-AgNPs complex as SERS probe for U937 cell identification. Biosens. Bioelectron. 2015, 74, 398–405. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Zhao, J.; Huang, Y.; Sheng, H.; Wang, Z. Combining array-assisted SERS microfluidic chips and machine learning algorithms for clinical leukemia phenotyping. Talanta 2025, 283, 127148. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.J.; Reynolds, D.; Dickinson, I.; Seaborne, A.; Dollin, C.; Wilkinson, K. Jena: Implementing the Semantic Web Recommendations. In Proceedings of the 13th International World Wide Web Conference on Alternate Track, Papers and Posters, New York, NY, USA, 19–21 May 2004; pp. 74–83. [Google Scholar]
- Kozlenkov, A.; Schroeder, M. PROVA: Rule-Based Java-Scripting for a Bioinformatics Semantic Web. In Proceedings of the Data Integration in the Life Sciences: First International Workshop, DILS 2004, Leipzig, Germany, 25–26 March 2004; Volume 2994, pp. 17–30. [Google Scholar]
- Yang, G.; Kifer, M.; Zhao, C.F. Lora-2: A Rule-Based Knowledge Representation and Inference Infrastructure for the Semantic Web. In OTM Confederated International Conferences: On the Move to Meaningful Internet Systems; Springer: Berlin/Heidelberg, Germany, 2003. [Google Scholar]
- Allegra, A.; Tonacci, A.; Sciaccotta, R.; Genovese, S.; Musolino, C.; Pioggia, G.; Gangemi, S. Machine Learning and Deep Learning Applications in Multiple Myeloma Diagnosis, Prognosis, and Treatment Selection. Cancers 2022, 14, 606. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Danieli, M.G.; Brunetto, S.; Gammeri, L.; Palmeri, D.; Claudi, I.; Shoenfeld, Y.; Gangemi, S. Machine learning application in autoimmune diseases: State of art and future prospectives. Autoimmun. Rev. 2024, 23, 103496. [Google Scholar] [CrossRef] [PubMed]
- Simmons, C.P.L.; McMillan, D.C.; McWilliams, K.; Sande, T.A.; Fearon, K.C.; Tuck, S.; Fallon, M.T.; Laird, B.J. Prognostic Tools in Patients with Advanced Cancer: A Systematic Review. J. Pain Symptom Manag. 2017, 53, 962–970.e10. [Google Scholar] [CrossRef] [PubMed]
- Koteluk, O.; Wartecki, A.; Mazurek, S.; Kołodziejczak, I.; Mackiewicz, A. How do machines learn? Artificial intelligence as a new era in medicine. J. Pers. Med. 2021, 11, 32. [Google Scholar] [CrossRef]
- Li, S.; Yi, H.; Leng, Q.; Wu, Y.; Mao, Y. New perspectives on cancer clinical research in the era of big data and machine learning. Surg. Oncol. 2024, 52, 102009. [Google Scholar] [CrossRef] [PubMed]
- Karako, K.; Tang, W. Applications of and issues with machine learning in medicine: Bridging the gap with explainable AI. Biosci. Trends 2025, 18, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, S.; Vallati, M.; Gatta, R. Artificial Intelligence-Based Management of Adult Chronic Myeloid Leukemia: Where Are We and Where Are We Going? Cancers 2024, 16, 848. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Stagno, F.; Breccia, M.; Di Raimondo, F. On the road to treatment-free remission in chronic myeloid leukemia: What about ’the others’? Expert Rev. Anticancer Ther. 2020, 20, 1075–1081. [Google Scholar] [CrossRef] [PubMed]
- Kockerols, C.; Valk, P.J.M.; Hogenbirk, P.; Geelen, I.; Blijlevens, N.M.A.; Janssen, J.J.W.M.; Hoogendoorn, M.; Kersting, S.; Klein, S.K.; Daenen, L.G.M.; et al. Treatment-Free Remission Outcomes in a BCR::ABL1 Digital PCR Selected Clinical Cohort of CML Patients. Eur. J. Haematol. 2025. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Authors—Year | AI | Algorithm | Ref. |
|---|---|---|---|
| Ahmed et al.—2019 | CNN model was trained with 25 epochs and 32 batch size since this setup was more suitable with the sample amount of dataset we used. Various numbers of epochs were experimented with to obtain the best performance results. They tried to increase the number of epochs to 100; however, it took more running time without significant progress in accuracy. | Convolution layers, pooling layers, flattening, and multilayer perceptrons made up the majority of the CNN design. CNNs used fully connected neural networks to classify the input photos after automatically extracting features from them. The convolution and pooling layers were used to extract features. Following the application of filters to these layers, the image’s features were acquired, and the classification phase began. | [19] |
| Huang et al.—2020 | Deep Learning (CNN) They employed transfer learning technology. With this method knowledge from other tasks is transferred to the current task, requiring less data for learning and adaptation to the target task. | GoogLeNet ResNet DenseNet | [20] |
| Zhang et al.—2022 | Deep Learning The UNet22 design offered a precise location for picture semantic segmentation in medical image segmentation. High segmentation and classification accuracy were achieved by the deformable convolution layer, which implemented the free-form deformation of the feature learning process. UNet introduced skip connections between the encoder feature maps and the decoder feature maps at the same scale in contrast to the standard encoder–decoder segmentation models. | The multiclass bone marrow cell segmentation performance of CMLcGAN was satisfactory. Following segmentation, five statistical features were chosen, and a conventional threefold cross-validation with 100 repetitions was carried out. | [22] |
| Dese et al.—2021 | k-means clustering Marker-controlled watershed segmentation Morphological operations | SVM classifies the provided inputs using optimum hyperplanes. Together with the class descriptors, the hyper plane—also known as support vectors—is constructed from the training instances. Like a line dividing a 2D plane into two sections, these hyper planes separate the positive samples from the negative samples. | [23] |
| Morphometric Analysis | |||
|---|---|---|---|
| Material | AI | Ref. | |
| Peripheral blood | CNN | [18] | |
| Blood | CNN | [19] | |
| Bone marrow blood | CNN | [20] | |
| Bone marrow biopsy | cGAN | [22,23] | |
| Immunophenotyping | |||
| Periferal and bone marrow blood | ML | [26] | |
| Bone marrow blood | SVM | [27] | |
| Karyotyping | |||
| Bone marrow blood | Specific algorithm | [28] | |
| Integrated data | |||
| Aim | |||
| Prognosis | ML | [30] | |
| Prediction | ANN | [31] | |
| Prediction | ML | [32] | |
| Response to treatment | LEAP | [33] | |
| Response to treatment | SVM | [34] | |
| Response to treatment | RF | [35] |
| Toxicity and Side Effects | |||
|---|---|---|---|
| Target | Drug | AI | Ref. |
| Gamma-Tocotrienol | Asciminib | Deep learning | [42] |
| Drug resistance | |||
| Aim | |||
| Resistance profiles against 8 drugs | Axitinib, Bosutinib, Dasatinib, Erlotinib, Gefitinib, Imatinib, Nilotinib, and Ponatinib | Machine learning | [37] |
| Overcome T315I resistance | Imatinib mesylate, nilotinib, dasatinib | Machine learning | [38] |
| Generation of new drugs | |||
| Evaluation of bioactivity scores | Support Vector Machine | [51] | |
| Potential binding sites | Support Vector Machine Cavity Plus | [52] | |
| Allosteric drug development | FTMap | [53] | |
| Identification of binding hot spot | FTMap | [54] | |
| Evaluation of allosteric and non-allosteric inhibitors | Random forest, Support Vector Machine, Deep neural network | [55] | |
| Production of TKIs inhibiting TKI-resistant cells | Machine learning | [57] | |
| Evaluation of Withaferin A as TKi | Algorithm of Glide, Visual Molecular Dynamics | [60] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stagno, F.; Russo, S.; Murdaca, G.; Mirabile, G.; Alvaro, M.E.; Nasso, M.E.; Zemzem, M.; Gangemi, S.; Allegra, A. Utilization of Machine Learning in the Prediction, Diagnosis, Prognosis, and Management of Chronic Myeloid Leukemia. Int. J. Mol. Sci. 2025, 26, 2535. https://doi.org/10.3390/ijms26062535
Stagno F, Russo S, Murdaca G, Mirabile G, Alvaro ME, Nasso ME, Zemzem M, Gangemi S, Allegra A. Utilization of Machine Learning in the Prediction, Diagnosis, Prognosis, and Management of Chronic Myeloid Leukemia. International Journal of Molecular Sciences. 2025; 26(6):2535. https://doi.org/10.3390/ijms26062535
Chicago/Turabian StyleStagno, Fabio, Sabina Russo, Giuseppe Murdaca, Giuseppe Mirabile, Maria Eugenia Alvaro, Maria Elisa Nasso, Mohamed Zemzem, Sebastiano Gangemi, and Alessandro Allegra. 2025. "Utilization of Machine Learning in the Prediction, Diagnosis, Prognosis, and Management of Chronic Myeloid Leukemia" International Journal of Molecular Sciences 26, no. 6: 2535. https://doi.org/10.3390/ijms26062535
APA StyleStagno, F., Russo, S., Murdaca, G., Mirabile, G., Alvaro, M. E., Nasso, M. E., Zemzem, M., Gangemi, S., & Allegra, A. (2025). Utilization of Machine Learning in the Prediction, Diagnosis, Prognosis, and Management of Chronic Myeloid Leukemia. International Journal of Molecular Sciences, 26(6), 2535. https://doi.org/10.3390/ijms26062535