
Deep Clustering-Based Immunotherapy Prediction for Gastric Cancer mRNA Vaccine Development


Abstract
1. Introduction
2. Results
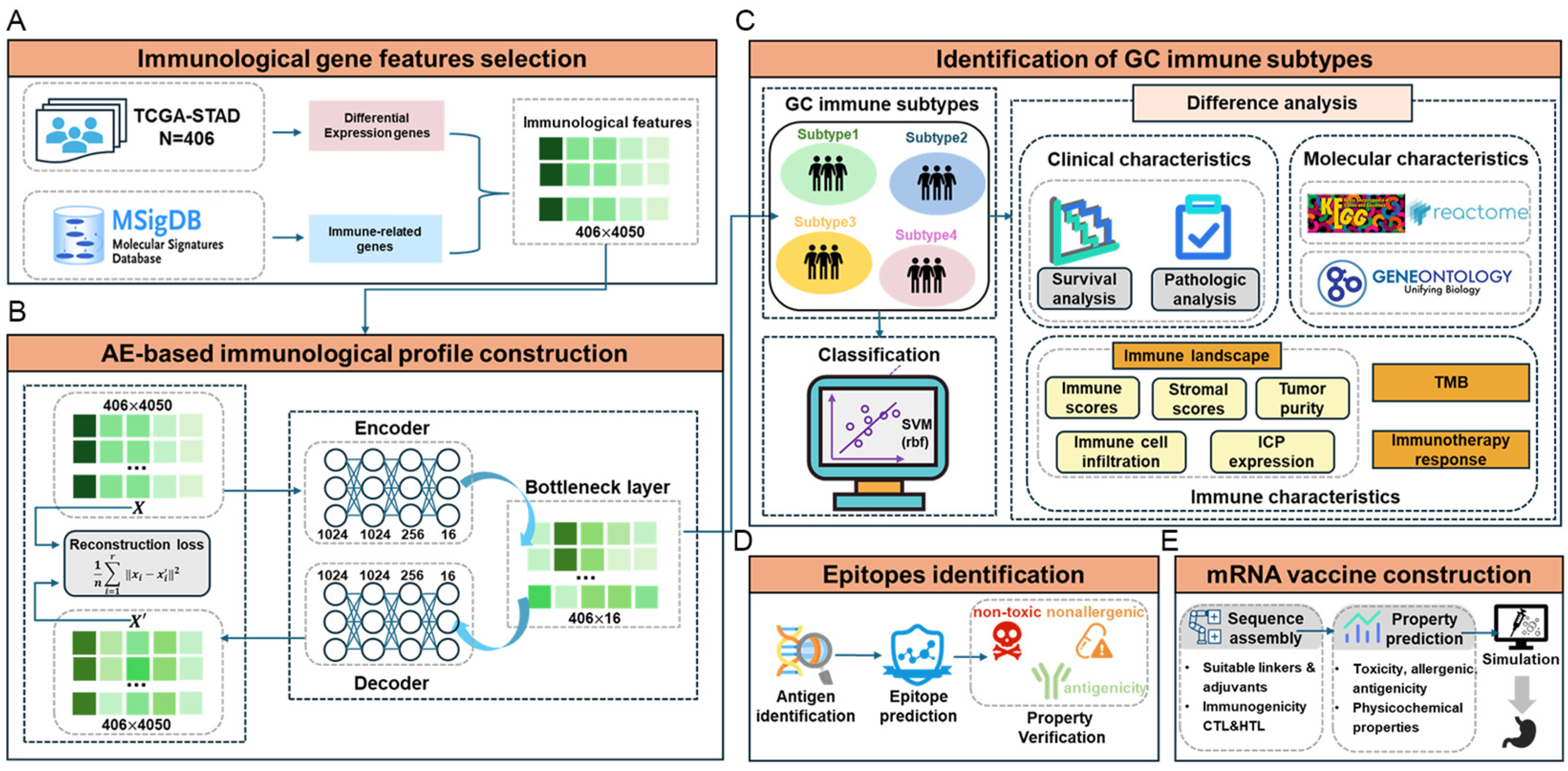
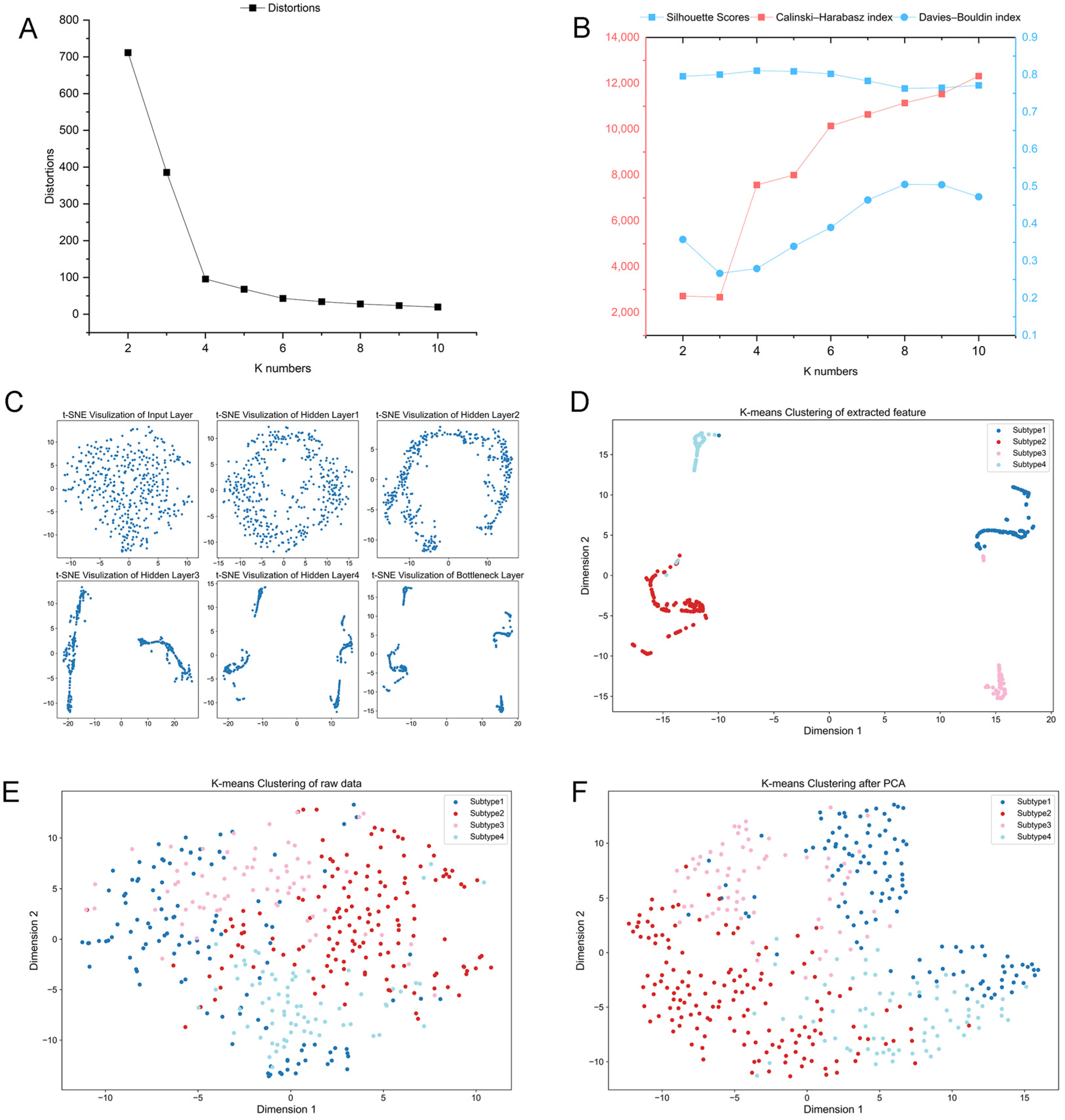
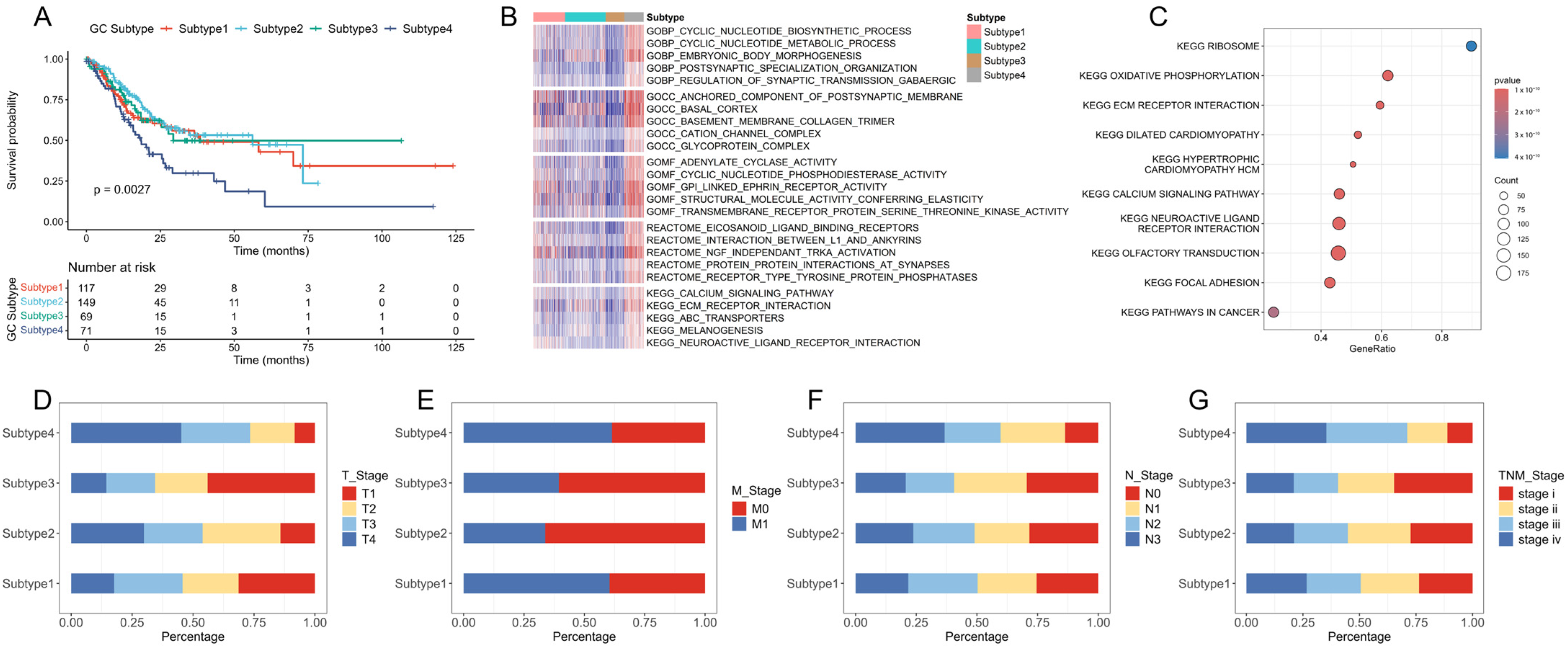
2.1. Identification of New Immune Subtypes of GC and In Silico Prediction
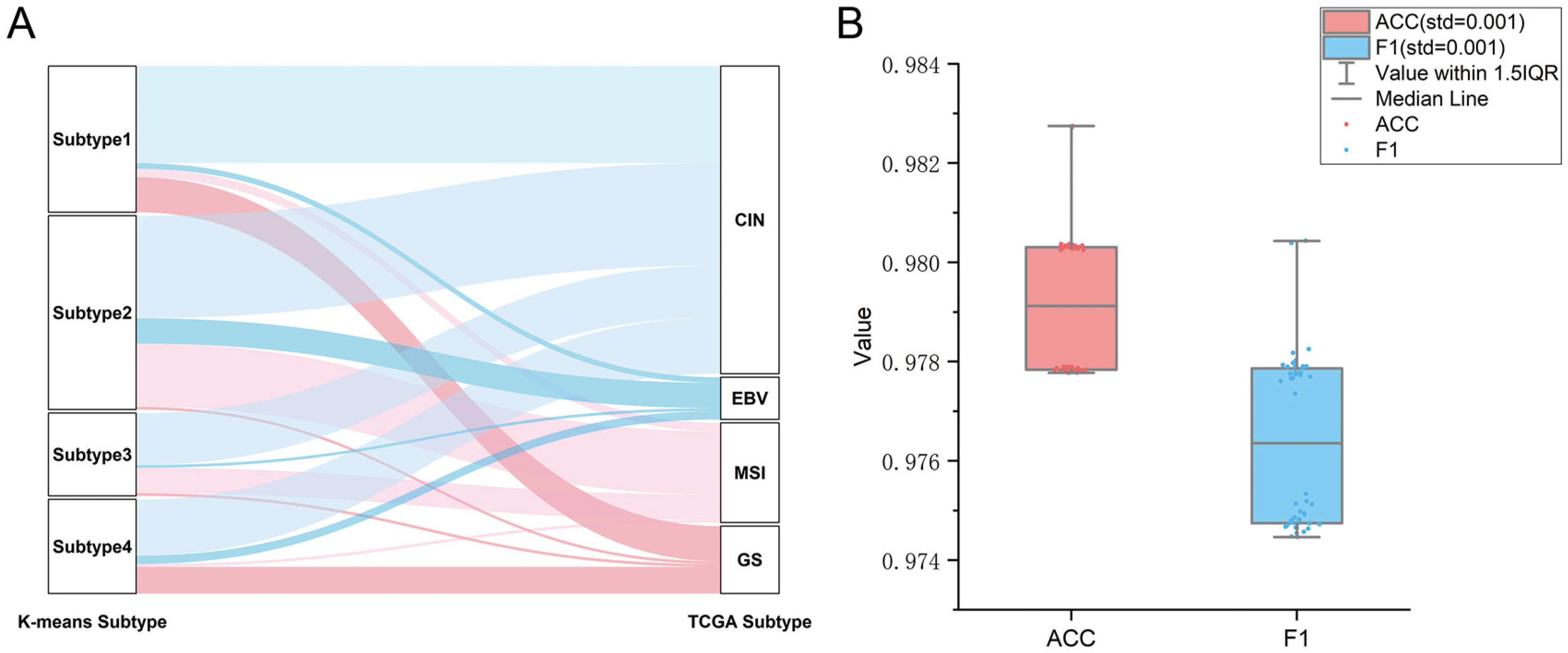
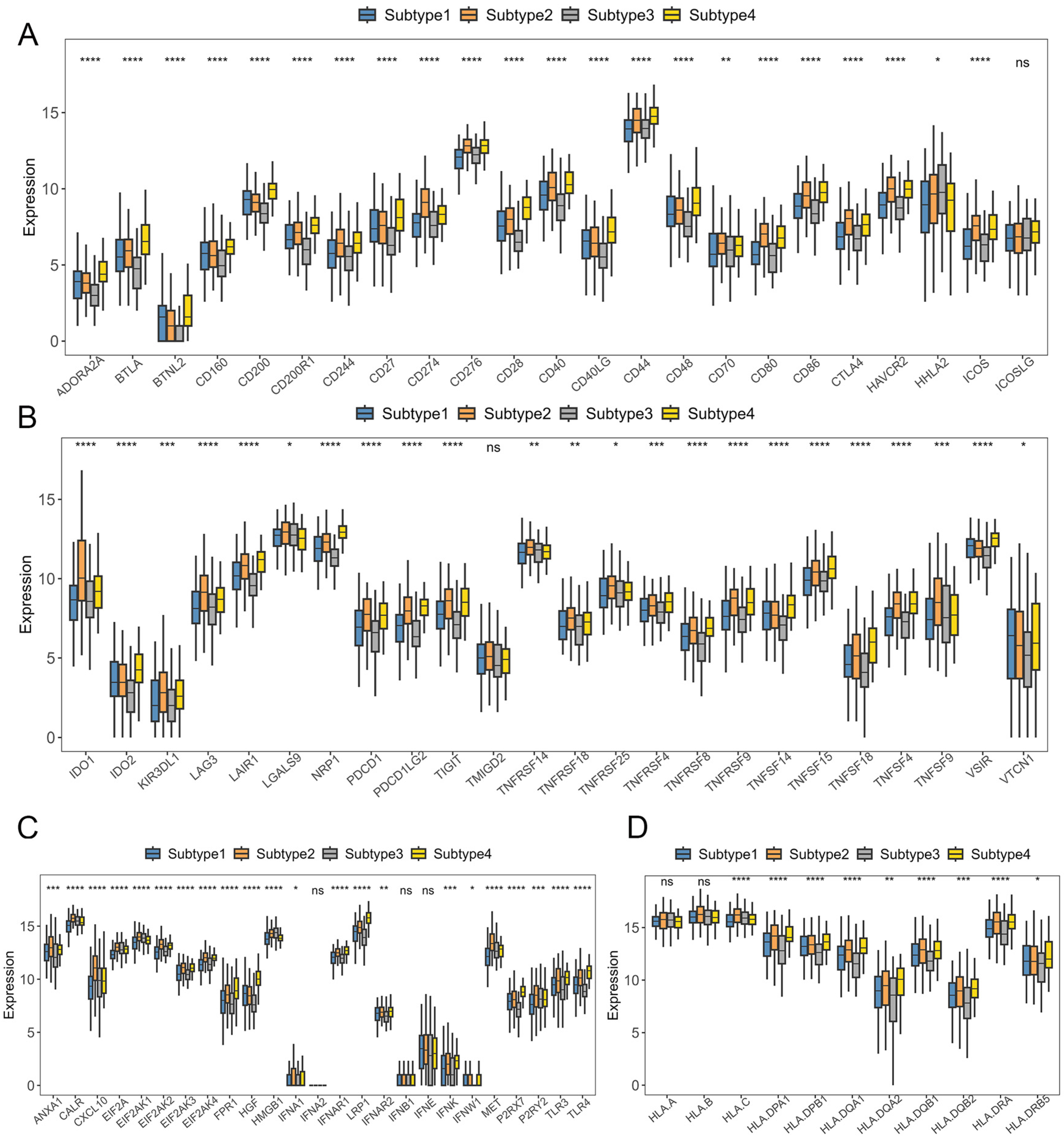
2.2. Verification of GC Immune Subtypes
2.3. Immunotherapy Sensitivity Evaluation
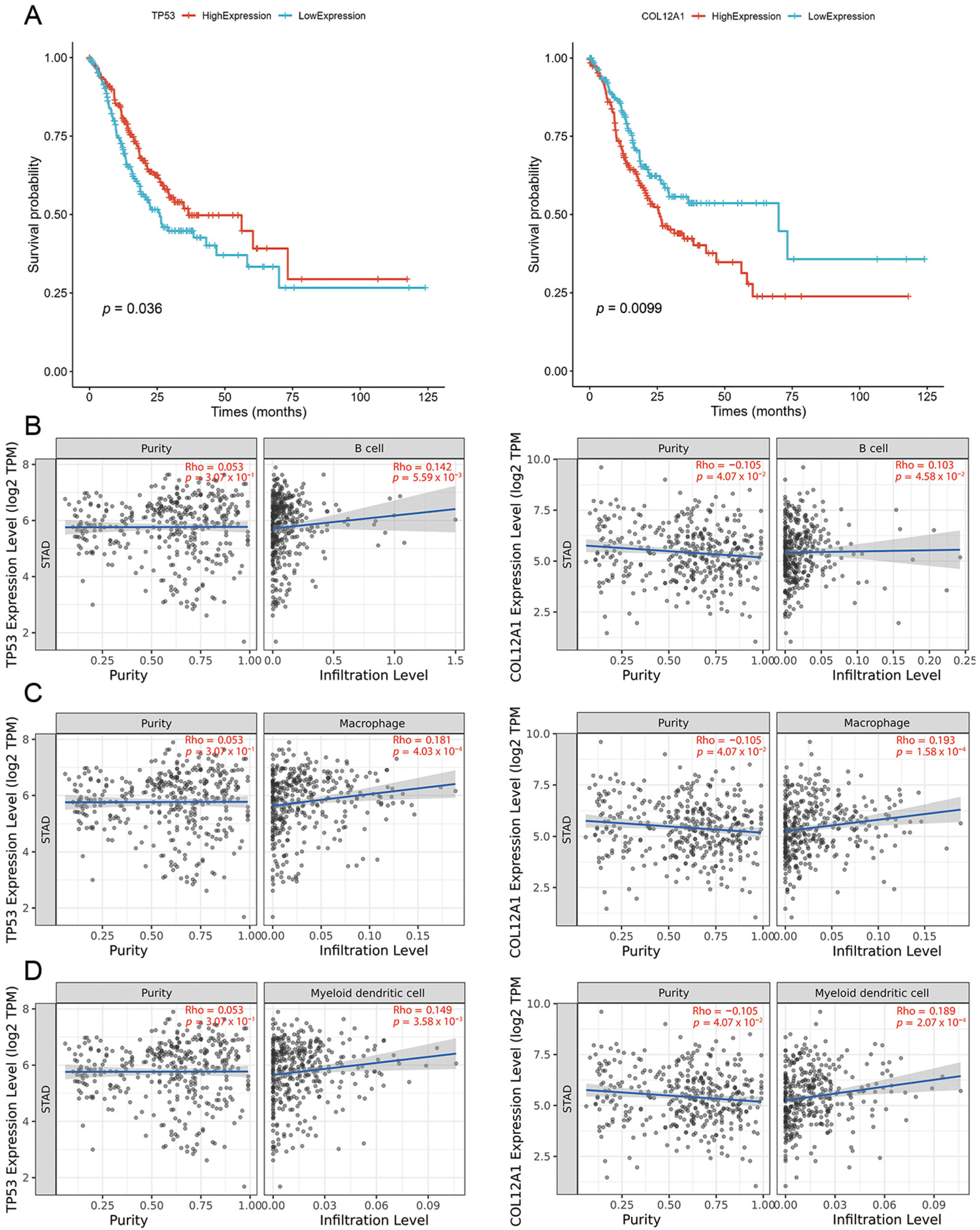
2.4. Tumor Vaccine Selection
2.5. Neoantigens with CTL Epitopes
2.6. Construction of the mRNA Vaccine
- MAKLSTDELLDAFKEMTLLELSDFVKKFEETFEVTAAAPVAVAAAGAAPAGAAVEAAEEQSEFDVILEAAGDKKIGVIKVVREIVSGLGLKEAKDLVDGAPKPLLEKVAKEAADEAKAKLEAAGATVTVKEAAAKHMTEVVRRYAAYTTIHTNTMYAAYAAIKKIPYKAAPVPGKVHKYAAYLAAIKKIPYAAYRTVRSSISRAAYKQIAYTPSLAAYTAQETTRPMAAYMRMVHLERLCPCPGDLPIGINITRFQTLL
2.7. Simulated Immune Response Against Neoantigen Vaccine
3. Discussion
4. Materials and Methods
4.1. Immunological Gene Feature Selection
4.2. Immunological Profile Construction
4.3. Unsupervised Clustering and Supervised Identification of New Immunological Subtypes
4.4. Immune Difference Analysis
4.5. Neoantigen Vaccine Development
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACC | Accuracy |
| AE | Autoencoder |
| CTL | Cytotoxic T lymphocyte |
| DEGs | Differentially expressed genes |
| GC | Gastric cancer |
| HLA | Human leukocyte antigen |
| HTL | Helper T lymphocyte |
| ICD | Immunogenic cell death |
| ICIs | Immune checkpoint inhibitors |
| ICP | Immune checkpoint |
| MHC | Major histocompatibility complex |
| ML | Machine learning |
| SVM | Support vector machine |
| TIME | Tumor immune microenvironment |
| TMB | Tumor mutational burden |
| TME | Tumor microenvironment |
References
- Hiam-Galvez, K.J.; Allen, B.M.; Spitzer, M.H. Systemic immunity in cancer. Nat. Rev. Cancer 2021, 21, 345–359. [Google Scholar] [CrossRef] [PubMed]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA Vaccines—A New Era in Vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef]
- Lorentzen, C.L.; Haanen, J.B.; Met, Ö.; Svane, I.M. Clinical Advances and Ongoing Trials of mRNA Vaccines for Cancer Treatment. Lancet Oncol. 2022, 23, e450–e458. [Google Scholar] [CrossRef]
- Cafri, G.; Gartner, J.J.; Zaks, T.; Hopson, K.; Levin, N.; Paria, B.C.; Parkhurst, M.R.; Yossef, R.; Lowery, F.J.; Jafferji, M.S.; et al. mRNA Vaccine–Induced Neoantigen-Specific T Cell Immunity in Patients with Gastrointestinal Cancer. J. Clin. Investig. 2020, 130, 5976–5988. [Google Scholar] [CrossRef]
- Rojas, L.A.; Sethna, Z.; Soares, K.C.; Olcese, C.; Pang, N.; Patterson, E.; Lihm, J.; Ceglia, N.; Guasp, P.; Chu, A.; et al. Personalized RNA Neoantigen Vaccines Stimulate T Cells in Pancreatic Cancer. Nature 2023, 618, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ye, Z.; Huang, C.; Qiu, M.; Song, D.; Li, Y.; Xu, Q. Lipid Nanoparticle-Mediated Lymph Node–Targeting Delivery of mRNA Cancer Vaccine Elicits Robust CD8 + T Cell Response. Proc. Natl. Acad. Sci. USA 2022, 119, e2207841119. [Google Scholar] [CrossRef]
- Li, X.; Ma, S.; Gao, T.; Mai, Y.; Song, Z.; Yang, J. The Main Battlefield of mRNA Vaccine—Tumor Immune Microenvironment. Int. Immunopharmacol. 2022, 113, 109367. [Google Scholar] [CrossRef]
- Heine, A.; Juranek, S.; Brossart, P. Clinical and Immunological Effects of mRNA Vaccines in Malignant Diseases. Mol. Cancer 2021, 20, 52. [Google Scholar] [CrossRef] [PubMed]
- Lv, B.; Wang, Y.; Ma, D.; Cheng, W.; Liu, J.; Yong, T.; Chen, H.; Wang, C. Immunotherapy: Reshape the Tumor Immune Microenvironment. Front. Immunol. 2022, 13, 844142. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Comprehensive Molecular Characterization of Gastric Adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef]
- Liu, Z.; Jiang, Z.; Wu, N.; Zhou, G.; Wang, X. Classification of Gastric Cancers Based on Immunogenomic Profiling. Transl. Oncol. 2021, 14, 100888. [Google Scholar] [CrossRef]
- Li, S.; Gao, J.; Xu, Q.; Zhang, X.; Huang, M.; Dai, X.; Huang, K.; Liu, L. A Signature-Based Classification of Gastric Cancer That Stratifies Tumor Immunity and Predicts Responses to PD-1 Inhibitors. Front. Immunol. 2021, 12, 693314. [Google Scholar] [CrossRef]
- Hou, C.K.J.; Behdinan, K. Dimensionality Reduction in Surrogate Modeling: A Review of Combined Methods. Data Sci. Eng. 2022, 7, 402–427. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.-Y.; Wu, A.-H.; Wang, Y.; Zhang, L.; Chai, H.; Liang, X.-F. Deep Learning-Based Ovarian Cancer Subtypes Identification Using Multi-Omics Data. BioData Min. 2020, 13, 10. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Feng, W.; Yao, Z.; Zhou, X.; Xiao, W. Deep Learning Model as a New Trend in Computer-Aided Diagnosis of Tumor Pathology for Lung Cancer. J. Cancer 2020, 11, 3615–3622. [Google Scholar] [CrossRef] [PubMed]
- Lupat, R.; Perera, R.; Loi, S.; Li, J. Moanna: Multi-Omics Autoencoder-Based Neural Network Algorithm for Predicting Breast Cancer Subtypes. IEEE Access 2023, 11, 10912–10924. [Google Scholar] [CrossRef]
- Park, K.H.; Batbaatar, E.; Piao, Y.; Theera-Umpon, N.; Ryu, K.H. Deep Learning Feature Extraction Approach for Hematopoietic Cancer Subtype Classification. IJERPH 2021, 18, 2197. [Google Scholar] [CrossRef]
- Lin, M.J.; Svensson-Arvelund, J.; Lubitz, G.S.; Marabelle, A.; Melero, I.; Brown, B.D.; Brody, J.D. Cancer Vaccines: The next Immunotherapy Frontier. Nat. Cancer 2022, 3, 911–926. [Google Scholar] [CrossRef]
- Prawiningrum, A.F.; Paramita, R.I.; Panigoro, S.S. Immunoinformatics Approach for Epitope-Based Vaccine Design: Key Steps for Breast Cancer Vaccine. Diagnostics 2022, 12, 2981. [Google Scholar] [CrossRef]
- Kon, E.; Ad-El, N.; Hazan-Halevy, I.; Stotsky-Oterin, L.; Peer, D. Targeting Cancer with mRNA–Lipid Nanoparticles: Key Considerations and Future Prospects. Nat. Rev. Clin. Oncol. 2023, 20, 739–754. [Google Scholar] [CrossRef]
- Thompson, E.D.; Zahurak, M.; Murphy, A.; Cornish, T.; Cuka, N.; Abdelfatah, E.; Yang, S.; Duncan, M.; Ahuja, N.; Taube, J.M.; et al. Patterns of PD-L1 Expression and CD8 T Cell Infiltration in Gastric Adenocarcinomas and Associated Immune Stroma. Gut 2017, 66, 794–801. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.P.; Pedersen, J.; Zhan, Y.; Lew, A.M.; Pearse, M.J.; Wijburg, O.L.C.; Strugnell, R.A. CD8+ T Cells Are Associated with Severe Gastritis in Helicobacter Pylori-Infected Mice in the Absence of CD4+ T Cells. Infect. Immun. 2008, 76, 1289–1297. [Google Scholar] [CrossRef]
- Hou, W.; Yi, C.; Zhu, H. Predictive Biomarkers of Colon Cancer Immunotherapy: Present and Future. Front. Immunol. 2022, 13, 1032314. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Pham, B.; Li, T. Cancer Neoantigens as Potential Targets for Immunotherapy. Clin. Exp. Metastasis 2022, 39, 51–60. [Google Scholar] [CrossRef]
- Lin, R.A.; Lin, J.K.; Lin, S. Mechanisms of Immunogenic Cell Death and Immune Checkpoint Blockade Therapy. Kaohsiung J. Med. Sci. 2021, 37, 448–458. [Google Scholar] [CrossRef]
- Lin, A.; Yan, W.-H. The Emerging Roles of Human Leukocyte Antigen-F in Immune Modulation and Viral Infection. Front. Immunol. 2019, 10, 964. [Google Scholar] [CrossRef] [PubMed]
- Mistretta, B.; Rankothgedera, S.; Castillo, M.; Rao, M.; Holloway, K.; Bhardwaj, A.; El Noafal, M.; Albarracin, C.; El-Zein, R.; Rezaei, H.; et al. Chimeric RNAs Reveal Putative Neoantigen Peptides for Developing Tumor Vaccines for Breast Cancer. Front. Immunol. 2023, 14, 1188831. [Google Scholar] [CrossRef]
- Fiolet, T.; Kherabi, Y.; MacDonald, C.-J.; Ghosn, J.; Peiffer-Smadja, N. Comparing COVID-19 Vaccines for Their Characteristics, Efficacy and Effectiveness against SARS-CoV-2 and Variants of Concern: A Narrative Review. Clin. Microbiol. Infect. 2022, 28, 202–221. [Google Scholar] [CrossRef]
- Balasubramanian, A.; Sundrud, M.S. ATP-Dependent Transporters: Emerging Players at the Crossroads of Immunity and Metabolism. Front. Immunol. 2023, 14, 1286696. [Google Scholar] [CrossRef]
- Logesh, R.; Prasad, S.R.; Chipurupalli, S.; Robinson, N.; Mohankumar, S.K. Natural Tyrosinase Enzyme Inhibitors: A Path from Melanin to Melanoma and Its Reported Pharmacological Activities. Biochim. Et. Biophys. Acta (BBA) Rev. Cancer 2023, 1878, 188968. [Google Scholar] [CrossRef]
- Zhu, K.-L.; Su, F.; Yang, J.-R.; Xiao, R.-W.; Wu, R.-Y.; Cao, M.-Y.; Ling, X.-L.; Zhang, T. TP53 to Mediate Immune Escape in Tumor Microenvironment: An Overview of the Research Progress. Mol. Biol. Rep. 2024, 51, 205. [Google Scholar] [CrossRef]
- Jiang, X.; Wu, M.; Xu, X.; Zhang, L.; Huang, Y.; Xu, Z.; He, K.; Wang, H.; Wang, H.; Teng, L. COL12A1, a Novel Potential Prognostic Factor and Therapeutic Target in Gastric Cancer. Mol. Med. Rep. 2019. [Google Scholar] [CrossRef] [PubMed]
- Gamage, D.G.; Gunaratne, A.; Periyannan, G.R.; Russell, T.G. Applicability of Instability Index for In Vitro Protein Stability Prediction. PPL 2019, 26, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An Enhanced Web Server for Large-Scale Expression Profiling and Interactive Analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef]
- Yoshihara, K.; Shahmoradgoli, M.; Martínez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Treviño, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring Tumour Purity and Stromal and Immune Cell Admixture from Expression Data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef] [PubMed]
- Mayakonda, A.; Lin, D.-C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and Comprehensive Analysis of Somatic Variants in Cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef]
- Wei, S.; Sun, Q.; Chen, J.; Li, X.; Hu, Z. Bioinformatics Analyses for the Identification of Tumor Antigens and Immune Subtypes of Gastric Adenocarcinoma. Front. Genet. 2022, 13, 1068112. [Google Scholar] [CrossRef]
- Li, T.; Fu, J.; Zeng, Z.; Cohen, D.; Li, J.; Chen, Q.; Li, B.; Liu, X.S. TIMER2.0 for Analysis of Tumor-Infiltrating Immune Cells. Nucleic Acids Res. 2020, 48, W509–W514. [Google Scholar] [CrossRef]
- Charoentong, P.; Finotello, F.; Angelova, M.; Mayer, C.; Efremova, M.; Rieder, D.; Hackl, H.; Trajanoski, Z. Pan-Cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017, 18, 248–262. [Google Scholar] [CrossRef]
- Wu, J.; Zhao, W.; Zhou, B.; Su, Z.; Gu, X.; Zhou, Z.; Chen, S. TSNAdb: A Database for Tumor-Specific Neoantigens from Immunogenomics Data Analysis. Genom. Proteom. Bioinform. 2018, 16, 276–282. [Google Scholar] [CrossRef]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Open Source Drug Discovery Consortium; Raghava, G.P.S. In Silico Approach for Predicting Toxicity of Peptides and Proteins. PLoS ONE 2013, 8, e73957. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, I.; Bangov, I.; Flower, D.R.; Doytchinova, I. AllerTOP v.2—a Server for in Silico Prediction of Allergens. J. Mol. Model. 2014, 20, 2278. [Google Scholar] [CrossRef]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A Server for Prediction of Protective Antigens, Tumour Antigens and Subunit Vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef]
- Ahammad, I.; Lira, S.S. Designing a Novel mRNA Vaccine against SARS-CoV-2: An Immunoinformatics Approach. Int. J. Biol. Macromol. 2020, 162, 820–837. [Google Scholar] [CrossRef]
- Ramalingam, P.S.; Arumugam, S. Reverse Vaccinology and Immunoinformatics Approaches to Design Multi-Epitope Based Vaccine against Oncogenic KRAS. Med. Oncol. 2023, 40, 283. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Shin, S.J.; Lee, M.H.; Lee, M.-G.; Kang, T.H.; Park, W.S.; Soh, B.Y.; Park, J.H.; Shin, Y.K.; Kim, H.W.; et al. A Potential Protein Adjuvant Derived from Mycobacterium Tuberculosis Rv0652 Enhances Dendritic Cells-Based Tumor Immunotherapy. PLoS ONE 2014, 9, e104351. [Google Scholar] [CrossRef] [PubMed]
- Basmenj, E.R.; Arastonejad, M.; Mamizadeh, M.; Alem, M.; KhalatbariLimaki, M.; Ghiabi, S.; Khamesipour, A.; Majidiani, H.; Shams, M.; Irannejad, H. Engineering and Design of Promising T-Cell-Based Multi-Epitope Vaccine Candidates against Leishmaniasis. Sci. Rep. 2023, 13, 19421. [Google Scholar] [CrossRef]
- Nguyen, T.L.; Nguyen, T.B.; Kim, H. Computational Identification of B and T-Cell Epitopes for Designing a Multi-Epitope Vaccine against SARS-CoV-2 Spike Glycoprotein. J. Struct. Biol. 2025, 108177. [Google Scholar] [CrossRef]
- Shetty, S.; Dash, S.; Kumar, A.; Vishwanath, S.; Kini, S.G.; Brand, A. Immunoinformatics Design of a Multi-Epitope Vaccine for Chlamydia Trachomatis Major Outer Membrane Proteins. Sci. Rep. 2024, 14, 29919. [Google Scholar] [CrossRef]
- Grote, A.; Hiller, K.; Scheer, M.; Munch, R.; Nortemann, B.; Hempel, D.C.; Jahn, D. JCat: A Novel Tool to Adapt Codon Usage of a Target Gene to Its Potential Expression Host. Nucleic Acids Res. 2005, 33, W526–W531. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ACC | Precision | Recall | F1 | |
|---|---|---|---|---|
| Subtype1 | - | 0.958 | 0.984 | 0.978 |
| Subtype2 | - | 0.999 | 0.966 | 0.983 |
| Subtype3 | - | 0.998 | 0.953 | 0.963 |
| Subtype4 | - | 0.933 | 0.999 | 0.965 |
| Overall | 0.976 ± 0.001 | 0.979 ± 0.002 | 0.975 ± 0.002 | 0.976 ± 0.002 |
| Gene | CTL Epitopes | wild_Affinity | mut_Affinity |
|---|---|---|---|
| TP53 | TTIHTNTMY | 49,635.09 | 466.97 |
| TP53 | HMTEVVRRY | 34,089.59 | 231.98 |
| COL12A1 | AAIKKIPYK | 10,552.19 | 81.46 |
| COL12A1 | PVPGKVHKY | 1781.34 | 464.04 |
| COL12A1 | LAAIKKIPY | 10,552.19 | 81.46 |
| COL12A1 | RTVRSSISR | 2040.02 | 223.93 |
| COL12A1 | KQIAYTPSL | 2471.24 | 391.95 |
| COL12A1 | TAQETTRPM | 34,063.04 | 191.8 |
| COL12A1 | MRMVHLERL | 658.66 | 234.55 |
| Feature | Property |
|---|---|
| Number of amino acids | 239 |
| Molecular weight | 25,851.25 |
| Theoretical PI | 8.87 |
| Chemical formula | C1159H1903N307O339S9 |
| Total number of atoms | 3717 |
| Total number of negatively charged residues (Asp + Glu) | 30 |
| Total number of positively charged residues (Arg + Lys) | 34 |
| Estimated half-life (mammalian reticulocytes, in vitro) | 30 h |
| Instability index (II) | 30.89 |
| Aliphatic index | 96.07 |
| Grand average of hydropathicity (GRAVY) | −0.010 |
| Nucleotide length | 780 bp |
| GC content | 69.20% |
| CAI | 0.96 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lan, H.; Zhao, J.; Yuan, L.; Li, M.; Pu, X.; Guo, Y. Deep Clustering-Based Immunotherapy Prediction for Gastric Cancer mRNA Vaccine Development. Int. J. Mol. Sci. 2025, 26, 2453. https://doi.org/10.3390/ijms26062453
Lan H, Zhao J, Yuan L, Li M, Pu X, Guo Y. Deep Clustering-Based Immunotherapy Prediction for Gastric Cancer mRNA Vaccine Development. International Journal of Molecular Sciences. 2025; 26(6):2453. https://doi.org/10.3390/ijms26062453
Chicago/Turabian StyleLan, Hao, Jinyi Zhao, Linxi Yuan, Menglong Li, Xuemei Pu, and Yanzhi Guo. 2025. "Deep Clustering-Based Immunotherapy Prediction for Gastric Cancer mRNA Vaccine Development" International Journal of Molecular Sciences 26, no. 6: 2453. https://doi.org/10.3390/ijms26062453
APA StyleLan, H., Zhao, J., Yuan, L., Li, M., Pu, X., & Guo, Y. (2025). Deep Clustering-Based Immunotherapy Prediction for Gastric Cancer mRNA Vaccine Development. International Journal of Molecular Sciences, 26(6), 2453. https://doi.org/10.3390/ijms26062453