A Truncated Mutation of TP53 Promotes Chemoresistance in Tongue Squamous Cell Carcinoma


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
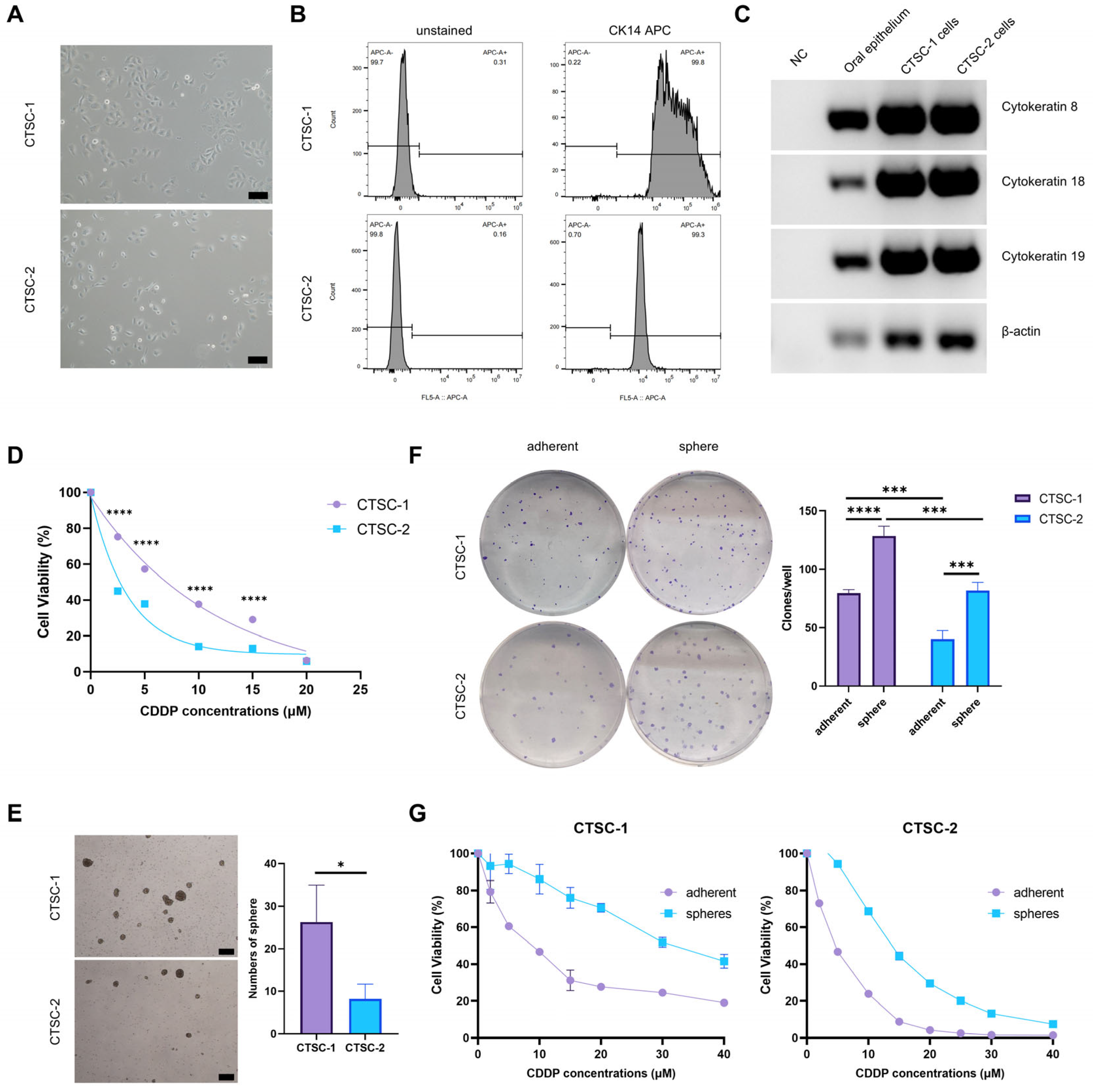
2.1. Establishment of CTSC-1 and CTSC-2
2.2. CTSC-1 and CTSC-2 Have Different Cellular Characteristics
2.3. A Special TP53 Mutation in CTSC-1
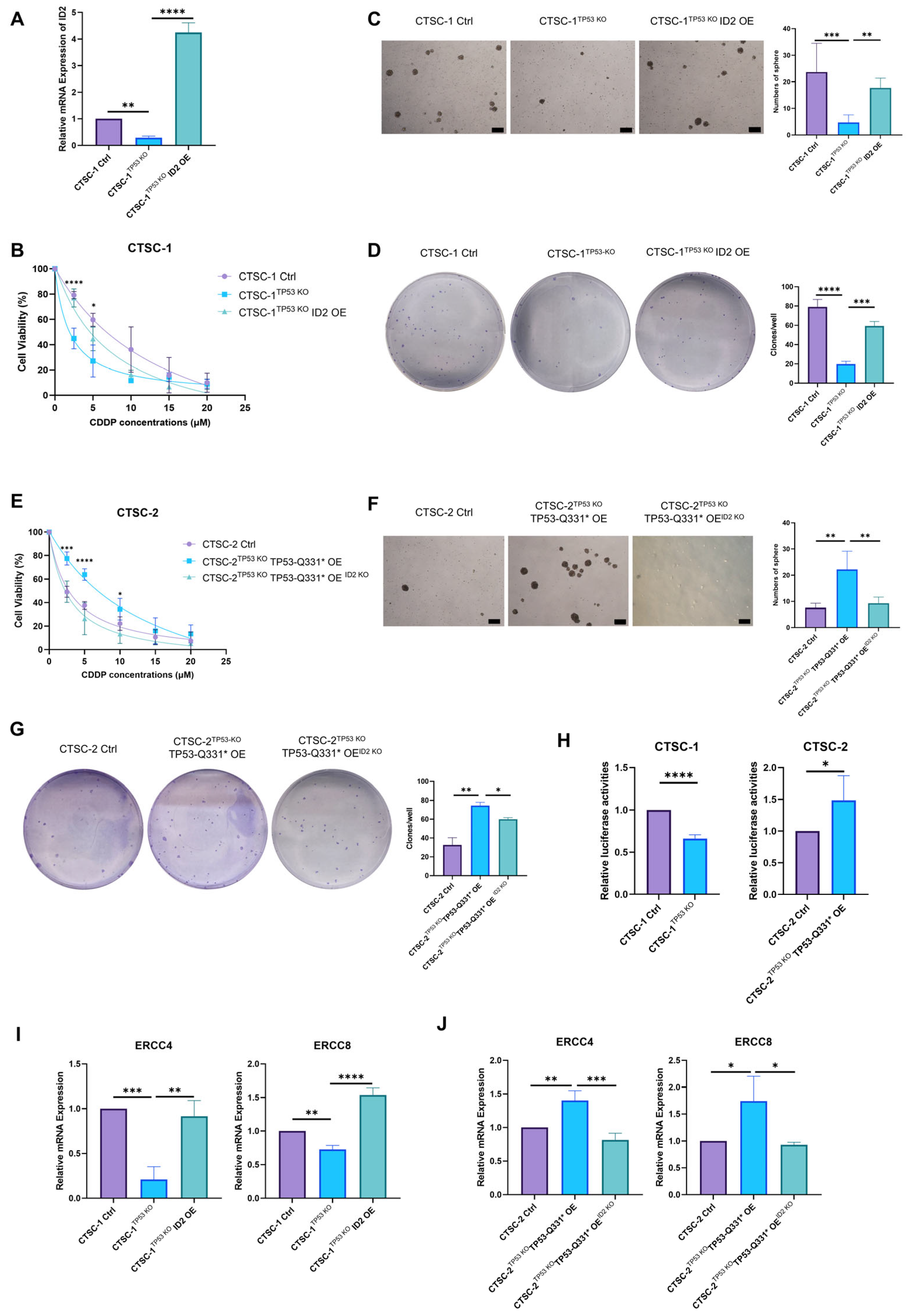
2.4. TP53-Q331* Induces Enhanced Chemoresistance and Stemness
2.5. ID2 Contributes to TP53-Q331*-Mediated Cell Chemoresistance and Stemness
2.6. TP53-Q331*-ID2-ERCC4/8 Axis Mediates Chemoresistance
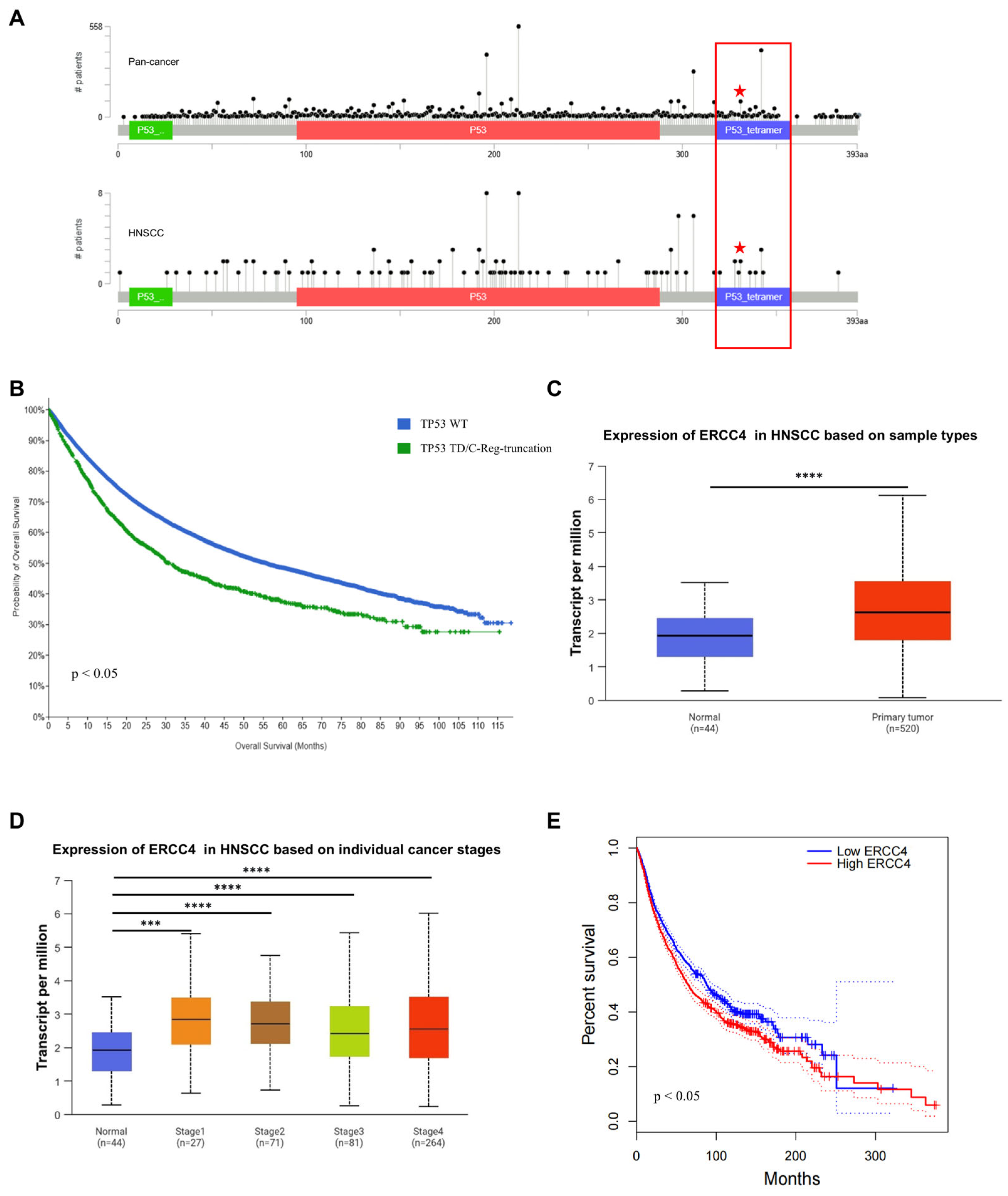
2.7. TP53-Q331* and ERCC4 Might Be Associated with Poor Prognosis
3. Discussion
4. Materials and Methods
4.1. Primary Cells, Cell Lines, and Culture Conditions
4.2. RNA Extraction and RT-PCR
4.3. Cell Viability and Chemotherapeutic Sensitivity Assay
4.4. Neosphere Formation Assay
4.5. In Vivo Tumorigenicity Assay
4.6. Hematoxylin–Eosin (H&E) Staining
4.7. Plasmids and Viral Transduction
4.8. Flow Cytometry Analysis
4.9. Colony Formation ASSAY
4.10. In Vitro Differentiation Assay
4.11. Western Blotting
4.12. RNA Sequencing and Bioinformatic Analysis
4.13. Luciferase Reporter Assay
4.14. Clinical Datasets and Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fuereder, T. Essential news of current guidelines: Head and neck squamous cell carcinoma. memo—Mag. Eur. Med. Oncol. 2022, 15, 278–281. [Google Scholar] [CrossRef]
- Tan, Y.; Wang, Z.; Xu, M.; Li, B.; Huang, Z.; Qin, S.; Nice, E.C.; Tang, J.; Huang, C. Oral squamous cell carcinomas: State of the field and emerging directions. Int. J. Oral Sci. 2023, 15, 44. [Google Scholar] [CrossRef]
- Li, H.; Zhang, Y.; Xu, M.; Yang, D. Current trends of targeted therapy for oral squamous cell carcinoma. J. Cancer Res. Clin. Oncol. 2022, 148, 2169–2186. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, X.; Jia, Y.; Guo, F.; Zhengjun, S.; Shao, Z. Intratumoural heterogeneity and clone evolution of oral squamous cell carcinoma. Mol. Carcinog. 2021, 60, 758–768. [Google Scholar] [CrossRef]
- Chang, Y.-S.; Hsu, H.-T.; Ko, Y.-C.; Yeh, K.-T.; Chang, S.-J.; Lin, C.-Y.; Chang, J.-G. Combined mutational analysis of RAS, BRAF, PIK3CA, and TP53 genes in Taiwanese patients with oral squamous cell carcinoma. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2014, 118, 110–116.e1. [Google Scholar] [CrossRef]
- Koo, K.; Mouradov, D.; Angel, C.M.; Iseli, T.A.; Wiesenfeld, D.; McCullough, M.J.; Burgess, A.W.; Sieber, O.M. Genomic Signature of Oral Squamous Cell Carcinomas from Non-Smoking Non-Drinking Patients. Cancers 2021, 13, 1029. [Google Scholar] [CrossRef]
- Dyson, H.J.; Wright, P.E. How does p53 work? Regulation by the intrinsically disordered domains. Trends Biochem. Sci. 2024, 50, 9–17. [Google Scholar] [CrossRef]
- Kulesza, M.; Dansonka-Mieszkowska, A.; Pieńkowska-Grela, B. Repair or perish—The role of p53 protein in a cell’s life. Nowotw. J. Oncol. 2019, 69, 168–178. [Google Scholar] [CrossRef]
- Babamohamadi, M.; Babaei, E.; Ahmed Salih, B.; Babamohammadi, M.; Jalal Azeez, H.; Othman, G. Recent findings on the role of wild-type and mutant p53 in cancer development and therapy. Front. Mol. Biosci. 2022, 9, 903075. [Google Scholar] [CrossRef]
- Ghatak, D.; Das Ghosh, D.; Roychoudhury, S. Cancer Stemness: p53 at the Wheel. Front. Oncol. 2021, 10, 604124. [Google Scholar] [CrossRef]
- Kanapathipillai, M. Treating p53 Mutant Aggregation-Associated Cancer. Cancers 2018, 10, 154. [Google Scholar] [CrossRef] [PubMed]
- Gurpinar, E.; Vousden, K.H. Hitting cancers’ weak spots: Vulnerabilities imposed by p53 mutation. Trends Cell Biol. 2015, 25, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Elfaki, R.M.; Abdelaziz, M.S.; Altayb, H.N.; Munsoor, M.M.; Gameel, A.A. Molecular and in-silico analysis of single nucleotide polymorphism targeting human TP53 gene exon 5-8 in Sudanese esophageal cancer patients. F1000Research 2018, 7, 1741. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, M.C.; Lowe, S.W. Mutant p53: It’s not all one and the same. Cell Death Differ. 2022, 29, 983–987. [Google Scholar] [CrossRef]
- Dobes, P.; Podhorec, J.; Coufal, O.; Jureckova, A.; Petrakova, K.; Vojtesek, B.; Hrstka, R. Influence of mutation type on prognostic and predictive values of TP53 status in primary breast cancer patients. Oncol. Rep. 2014, 32, 1695–1702. [Google Scholar] [CrossRef]
- Petitjean, A.; Mathe, E.; Kato, S.; Ishioka, C.; Tavtigian, S.V.; Hainaut, P.; Olivier, M. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: Lessons from recent developments in the IARC TP53 database. Hum. Mutat. 2007, 28, 622–629. [Google Scholar] [CrossRef]
- Thompson, L.R.; Oliveira, T.G.; Hermann, E.R.; Chowanadisai, W.; Clarke, S.L.; Montgomery, M.R. Distinct TP53 Mutation Types Exhibit Increased Sensitivity to Ferroptosis Independently of Changes in Iron Regulatory Protein Activity. Int. J. Mol. Sci. 2020, 21, 6751. [Google Scholar] [CrossRef]
- Barbato, L.; Bocchetti, M.; Di Biase, A.; Regad, T. Cancer Stem Cells and Targeting Strategies. Cells 2019, 8, 926. [Google Scholar] [CrossRef]
- Gurzu, S.; Silveanu, C.; Fetyko, A.; Butiurca, V.; Kovacs, Z.; Jung, I. Systematic review of the old and new concepts in the epithelial-mesenchymal transition of colorectal cancer. World J. Gastroenterol. 2016, 22, 6764–6775. [Google Scholar] [CrossRef]
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef]
- Phi, L.T.H.; Sari, I.N.; Yang, Y.-G.; Lee, S.-H.; Jun, N.; Kim, K.S.; Lee, Y.K.; Kwon, H.Y. Cancer Stem Cells (CSCs) in Drug Resistance and their Therapeutic Implications in Cancer Treatment. Stem Cells Int. 2018, 2018, 5416923. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Zhan, Y.; Sun, L.; Zhu, W. Cancer Stem Cells and the Tumor Microenvironment in Tumor Drug Resistance. Stem Cell Rev. Rep. 2023, 19, 2141–2154. [Google Scholar] [CrossRef] [PubMed]
- Rezayatmand, H.; Razmkhah, M.; Razeghian-Jahromi, I. Drug resistance in cancer therapy: The Pandora’s Box of cancer stem cells. Stem Cell Res. Ther. 2022, 13, 181. [Google Scholar] [CrossRef]
- Garimella, S.V.; Gampa, S.C.; Chaturvedi, P. Mitochondria in Cancer Stem Cells: From an Innocent Bystander to a Central Player in Therapy Resistance. Stem Cells Cloning Adv. Appl. 2023, 16, 19–41. [Google Scholar] [CrossRef]
- Aramini, B.; Masciale, V.; Grisendi, G.; Bertolini, F.; Maur, M.; Guaitoli, G.; Chrystel, I.; Morandi, U.; Stella, F.; Dominici, M.; et al. Dissecting Tumor Growth: The Role of Cancer Stem Cells in Drug Resistance and Recurrence. Cancers 2022, 14, 976. [Google Scholar] [CrossRef]
- Bose, B.; Bozdag, S. Identifying cell lines across pan-cancer to be used in preclinical research as a proxy for patient tumor samples. Commun. Biol. 2024, 7, 1101. [Google Scholar] [CrossRef]
- Al Delbany, D.; Ghosh, M.S.; Krivec, N.; Huyghebaert, A.; Regin, M.; Duong, M.C.; Lei, Y.; Sermon, K.; Olsen, C.; Spits, C. De Novo Cancer Mutations Frequently Associate with Recurrent Chromosomal Abnormalities during Long-Term Human Pluripotent Stem Cell Culture. Cells 2024, 13, 1395. [Google Scholar] [CrossRef]
- Zhang, H.; Wu, H.; Zheng, J.; Yu, P.; Xu, L.; Jiang, P.; Gao, J.; Wang, H.; Zhang, Y. Transforming growth factor beta1 signal is crucial for dedifferentiation of cancer cells to cancer stem cells in osteosarcoma. Stem Cells 2013, 31, 433–446. [Google Scholar] [CrossRef]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H.J. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef]
- Cummings, M.; Higginbottom, K.; McGurk, C.J.; Wong, O.G.-W.; Köberle, B.; Oliver, R.T.D.; Masters, J.R. XPA versus ERCC1 as chemosensitising agents to cisplatin and mitomycin C in prostate cancer cells: Role of ERCC1 in homologous recombination repair. Biochem. Pharmacol. 2006, 72, 166–175. [Google Scholar] [CrossRef]
- Keller, J.R.; Ji, M.; Li, H.; Kozlov, S.V.; Tessarollo, L.; Klarmann, K.D. Dynamic Expression of Id2 Determines Cell Fate Decisions in Multipotent Hematopoietic Cells. Blood 2011, 118, 2369. [Google Scholar] [CrossRef]
- Paolella, B.R.; Havrda, M.C.; Mantani, A.; Wray, C.M.; Zhang, Z.; Israel, M.A. p53 Directly Represses Id2 to Inhibit the Proliferation of Neural Progenitor Cells. Stem Cells 2011, 29, 1090–1101. [Google Scholar] [CrossRef]
- Yan, W.; Liu, G.; Scoumanne, A.; Chen, X. Suppression of Inhibitor of Differentiation 2, a Target of Mutant p53, Is Required for Gain-of-Function Mutations. Cancer Res. 2008, 68, 6789–6796. [Google Scholar] [CrossRef] [PubMed]
- Vessoni, A.T.; Filippi-Chiela, E.C.; Menck, C.F.; Lenz, G. Autophagy and genomic integrity. Cell Death Differ. 2013, 20, 1444–1454. [Google Scholar] [CrossRef] [PubMed]
- Palomera-Sanchez, Z.; Zurita, M. Open, repair and close again: Chromatin dynamics and the response to UV-induced DNA damage. DNA Repair 2011, 10, 119–125. [Google Scholar] [CrossRef]
- Jee, J.; Fong, C.; Pichotta, K.; Tran, T.N.; Luthra, A.; Waters, M.; Fu, C.; Altoe, M.; Liu, S.-Y.; Maron, S.B.; et al. Automated real-world data integration improves cancer outcome prediction. Nature 2024, 636, 728–736. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.V.S.K.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef]
- Ning, J.; Jiao, Y.; Xie, X.; Deng, X.; Zhang, Y.; Yang, Y.; Zhao, C.; Wang, H.; Gu, K. miR-138-5p modulates the expression of excision repair cross-complementing proteins ERCC1 and ERCC4, and regulates the sensitivity of gastric cancer cells to cisplatin. Oncol. Rep. 2018, 41, 1131–1139. [Google Scholar] [CrossRef]
- Kashofer, K.; Winter, E.; Halbwedl, I.; Thueringer, A.; Kreiner, M.; Sauer, S.; Regauer, S. HPV-negative penile squamous cell carcinoma: Disruptive mutations in the TP53 gene are common. Mod. Pathol. 2017, 30, 1013–1020. [Google Scholar] [CrossRef]
- Alsofyani, A.A.; Dallol, A.; Farraj, S.A.; Alsiary, R.A.; Samkari, A.; Alhaj-Hussain, B.T.; Khan, J.A.; Al-Maghrabi, J.; Al-Khayyat, S.S.; Alkhatabi, H.; et al. Molecular characterisation in tongue squamous cell carcinoma reveals key variants potentially linked to clinical outcomes. Cancer Biomark. 2020, 28, 213–220. [Google Scholar] [CrossRef]
- Ono, S.; Hirose, K.; Sukegawa, S.; Obata, K.; Masui, M.; Hasegawa, K.; Fujimura, A.; Shimada, K.; Nakamura, S.; Teramoto, A.; et al. Squamous cell carcinoma initially occurring on the tongue dorsum: A case series report with molecular analysis. Diagn. Pathol. 2024, 19, 63. [Google Scholar] [CrossRef] [PubMed]
- Hyodo, T.; Kuribayashi, N.; Fukumoto, C.; Komiyama, Y.; Shiraishi, R.; Kamimura, R.; Sawatani, Y.; Yaguchi, E.; Hasegawa, T.; Izumi, S.; et al. The mutational spectrum in whole exon of p53 in oral squamous cell carcinoma and its clinical implications. Sci. Rep. 2022, 12, 21695. [Google Scholar] [CrossRef] [PubMed]
- Lapke, N.; Lu, Y.-J.; Liao, C.-T.; Lee, L.-Y.; Lin, C.-Y.; Wang, H.-M.; Ng, S.-H.; Chen, S.-J.; Yen, T.-C. Missense mutations in the TP53 DNA-binding domain predict outcomes in patients with advanced oral cavity squamous cell carcinoma. Oncotarget 2016, 7, 44194–44210. [Google Scholar] [CrossRef] [PubMed]
- van Kempen, P.M.W.; Verdam, F.J.; de Poel, E.; Braunius, W.W.; de Weger, R.A.; van Es, R.J.J.; Grolman, W.; Willems, S.M. TP53 Y220C Is a Hotspot Mutation in Oropharyngeal Squamous Cell Carcinoma. Pathobiology 2015, 82, 21–27. [Google Scholar] [CrossRef]
- Lindemann, A.; Takahashi, H.; Patel, A.A.; Osman, A.A.; Myers, J.N. Targeting the DNA Damage Response in OSCC with TP53 Mutations. J. Dent. Res. 2018, 97, 635–644. [Google Scholar] [CrossRef]
- Jovic, S.; Kozomara, R.; Stosic, S.; Jovandic, S.; Zeljic, K.; Supic, G. Pathogenic TP53 mutations influence chemotherapy response and survival rate of HPV-negative oral carcinomas. Vojnosanit. Pregl. 2022, 79, 1063–1072. [Google Scholar] [CrossRef]
- Galuppini, F.; Salmaso, R.; Valentini, E.; Lanza, C.; Maretto, I.; Nitti, D.; Rugge, M.; Fassan, M. Adenosquamous gallbladder carcinoma: Multigene hotspot mutational profiling reveals a monoclonal origin of the two components. Pathol.—Res. Pract. 2017, 213, 1010–1013. [Google Scholar] [CrossRef]
- Li, X.; Chen, X.; Wen, L.; Wang, Y.; Chen, B.; Xue, Y.; Guo, L.; Liao, N. Impact of TP53 mutations in breast cancer: Clinicopathological features and prognosisImpact of TP53 mutations in breast. Thorac. Cancer 2020, 11, 1861–1868. [Google Scholar] [CrossRef]
- Méndez, O.; Villanueva, J. Challenges and opportunities for cell line secretomes in cancer proteomics. Proteom.—Clin. Appl. 2015, 9, 348–357. [Google Scholar] [CrossRef]
- Fusenig, N.E.; Capes-Davis, A.; Bianchini, F.; Sundell, S.; Lichter, P. The need for a worldwide consensus for cell line authentication: Experience implementing a mandatory requirement at the International Journal of Cancer. PLoS Biol. 2017, 15, e2001438. [Google Scholar] [CrossRef]
- Liu, X.; Mondal, A.M. Conditional cell reprogramming for modeling host-virus interactions and human viral diseases. J. Med. Virol. 2020, 92, 2440–2452. [Google Scholar] [CrossRef]
- Hsieh, J.-W.A.; Chang, P.; Kuang, L.-Y.; Hsing, Y.-I.C.; Chen, P.-Y. Rice transformation treatments leave specific epigenome changes beyond tissue culture. Plant Physiol. 2023, 193, 1297–1312. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Feng, Y.; Dai, X.; Huang, L.; Li, J.; Tao, P.; Crabbe, M.J.C.; Zhang, T.; Qiao, Q. Dynamic Changes of DNA Methylation During Wild Strawberry (Fragaria nilgerrensis) Tissue Culture. Front. Plant Sci. 2021, 12, 765383. [Google Scholar] [CrossRef]
- de Peppo, G.M.; Svensson, S.; Lennerås, M.; Synnergren, J.; Stenberg, J.; Strehl, R.; Hyllner, J.; Thomsen, P.; Karlsson, C. Human Embryonic Mesodermal Progenitors Highly Resemble Human Mesenchymal Stem Cells and Display High Potential for Tissue Engineering Applications. Tissue Eng. Part A 2010, 16, 2161–2182. [Google Scholar] [CrossRef] [PubMed]
- Hughes, P.; Marshall, D.; Reid, Y.; Parkes, H.; Gelber, C. The Costs of using Unauthenticated, Over-Passaged Cell Lines: How Much more Data do we Need? BioTechniques 2018, 43, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Andreani, N.A.; Renzi, S.; Piovani, G.; Ajmone Marsan, P.; Bomba, L.; Villa, R.; Ferrari, M.; Dotti, S. Potential neoplastic evolution of Vero cells: In vivo and in vitro characterization. Cytotechnology 2017, 69, 741–750. [Google Scholar] [CrossRef]
- Andus, I.; Prall, F.; Linnebacher, M.; Linnebacher, C.S. Establishment, characterization, and drug screening of low-passage patient individual non-small cell lung cancer in vitro models including the rare pleomorphic subentity. Front. Oncol. 2023, 13, 1089681. [Google Scholar] [CrossRef]
- Hirsch, D.; Seyfried, S.; Staib, T.; Fiedler, D.; Sauer, C.; Ried, T.; Witt, S.; Rueckert, F.; Gaiser, T. Newly established gastrointestinal cancer cell lines retain the genomic and immunophenotypic landscape of their parental cancers. Sci. Rep. 2020, 10, 17895. [Google Scholar] [CrossRef]
- Lythe, G.; Jin, W.; Penington, C.J.; McCue, S.W.; Simpson, M.J. A computational modelling framework to quantify the effects of passaging cell lines. PLoS ONE 2017, 12, e0181941. [Google Scholar] [CrossRef]
- Wang, T.; Zhang, J.; Liao, J.; Zhang, F.; Zhou, G. Donor genetic backgrounds contribute to the functional heterogeneity of stem cells and clinical outcomes. Stem Cells Transl. Med. 2020, 9, 1495–1499. [Google Scholar] [CrossRef]
- Cring, M.R.; Sheffield, V.C. Gene therapy and gene correction: Targets, progress, and challenges for treating human diseases. Gene Ther. 2020, 29, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Sarkar, T.; Gangopadhyay, D. Gene therapy and its application in dermatology. Indian J. Dermatol. 2020, 65, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Zamyatnin, A.A. Special issue: Genome editing and gene therapy. Biochemistry 2016, 81, 651–652. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Li, G.; Li, P.; Wang, H.; Fang, X.; He, T.; Li, J. Twenty years of Gendicine® rAd-p53 cancer gene therapy: The first-in-class human cancer gene therapy in the era of personalized oncology. Genes Dis. 2024, 11, 101155. [Google Scholar] [CrossRef]
- Hu, J.; Cao, J.; Topatana, W.; Juengpanich, S.; Li, S.; Zhang, B.; Shen, J.; Cai, L.; Cai, X.; Chen, M. Targeting mutant p53 for cancer therapy: Direct and indirect strategies. J. Hematol. Oncol. 2021, 14, 157. [Google Scholar] [CrossRef]
- Hassin, O.; Oren, M. Drugging p53 in cancer: One protein, many targets. Nat. Rev. Drug Discov. 2022, 22, 127–144. [Google Scholar] [CrossRef]
- Roschke, A.V.; Glebov, O.K.; Lababidi, S.; Gehlhaus, K.S.; Weinstein, J.N.; Kirsch, I.R. Chromosomal instability is associated with higher expression of genes implicated in epithelial-mesenchymal transition, cancer invasiveness, and metastasis and with lower expression of genes involved in cell cycle checkpoints, DNA repair, and chromatin maintenance. Neoplasia 2008, 10, 1222–1230. [Google Scholar] [CrossRef]
- Yoshioka, K.I.; Matsuno, Y.; Hyodo, M.; Fujimori, H. Genomic-Destabilization-Associated Mutagenesis and Clonal Evolution of Cells with Mutations in Tumor-Suppressor Genes. Cancers 2019, 11, 1643. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, X.; Zheng, J.; Lu, X.; Zhang, Y. A Truncated Mutation of TP53 Promotes Chemoresistance in Tongue Squamous Cell Carcinoma. Int. J. Mol. Sci. 2025, 26, 2353. https://doi.org/10.3390/ijms26052353
Du X, Zheng J, Lu X, Zhang Y. A Truncated Mutation of TP53 Promotes Chemoresistance in Tongue Squamous Cell Carcinoma. International Journal of Molecular Sciences. 2025; 26(5):2353. https://doi.org/10.3390/ijms26052353
Chicago/Turabian StyleDu, Xiaoyun, Junheng Zheng, Xiangwan Lu, and Yan Zhang. 2025. "A Truncated Mutation of TP53 Promotes Chemoresistance in Tongue Squamous Cell Carcinoma" International Journal of Molecular Sciences 26, no. 5: 2353. https://doi.org/10.3390/ijms26052353
APA StyleDu, X., Zheng, J., Lu, X., & Zhang, Y. (2025). A Truncated Mutation of TP53 Promotes Chemoresistance in Tongue Squamous Cell Carcinoma. International Journal of Molecular Sciences, 26(5), 2353. https://doi.org/10.3390/ijms26052353