
Heme Regulatory Motif of Heme Oxygenase-2 Is Involved in the Interaction with NADPH–Cytochrome P450 Reductase and Regulates Enzymatic Activity
 , , , , and
, , , , and
Abstract
1. Introduction
2. Results
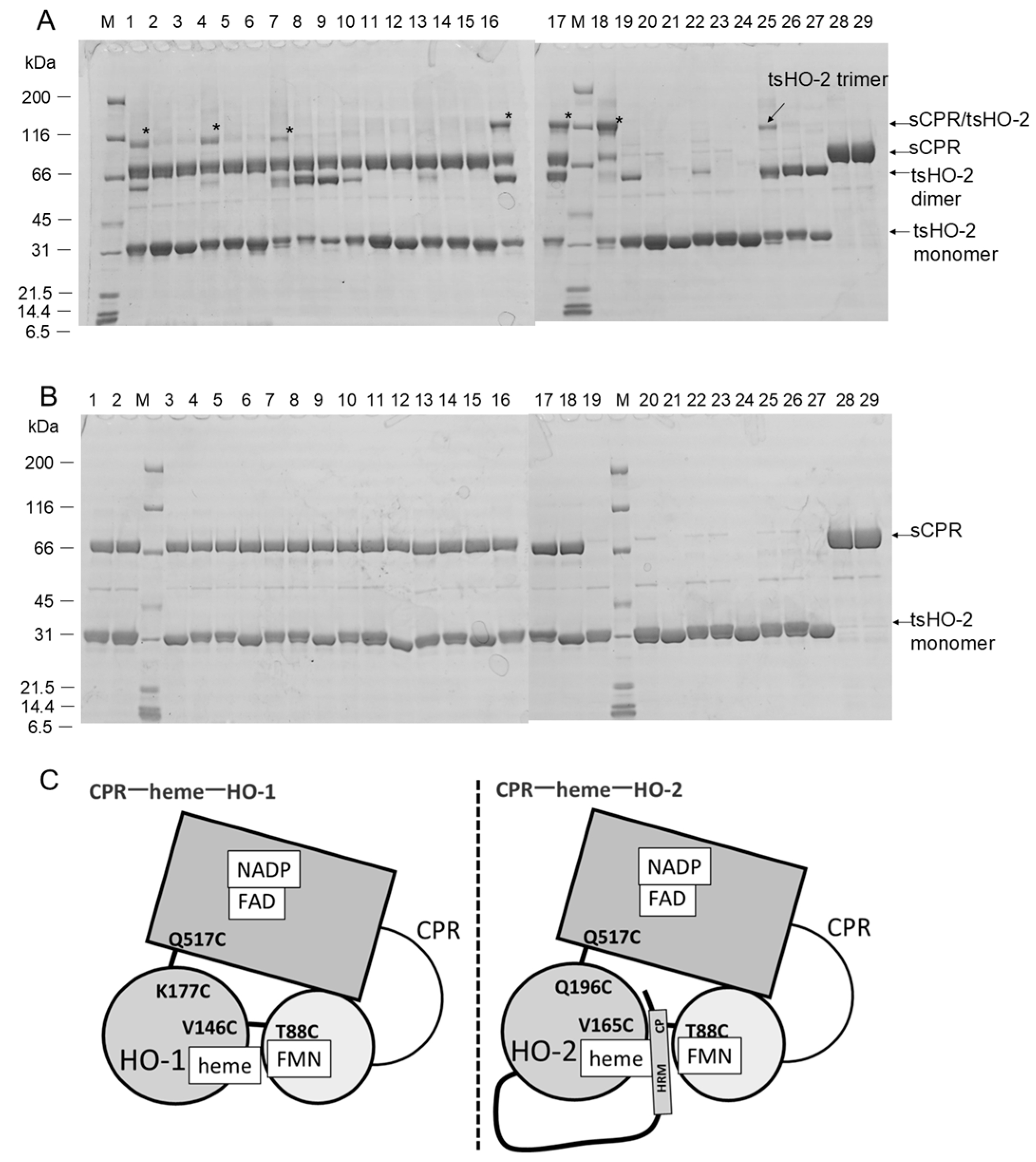
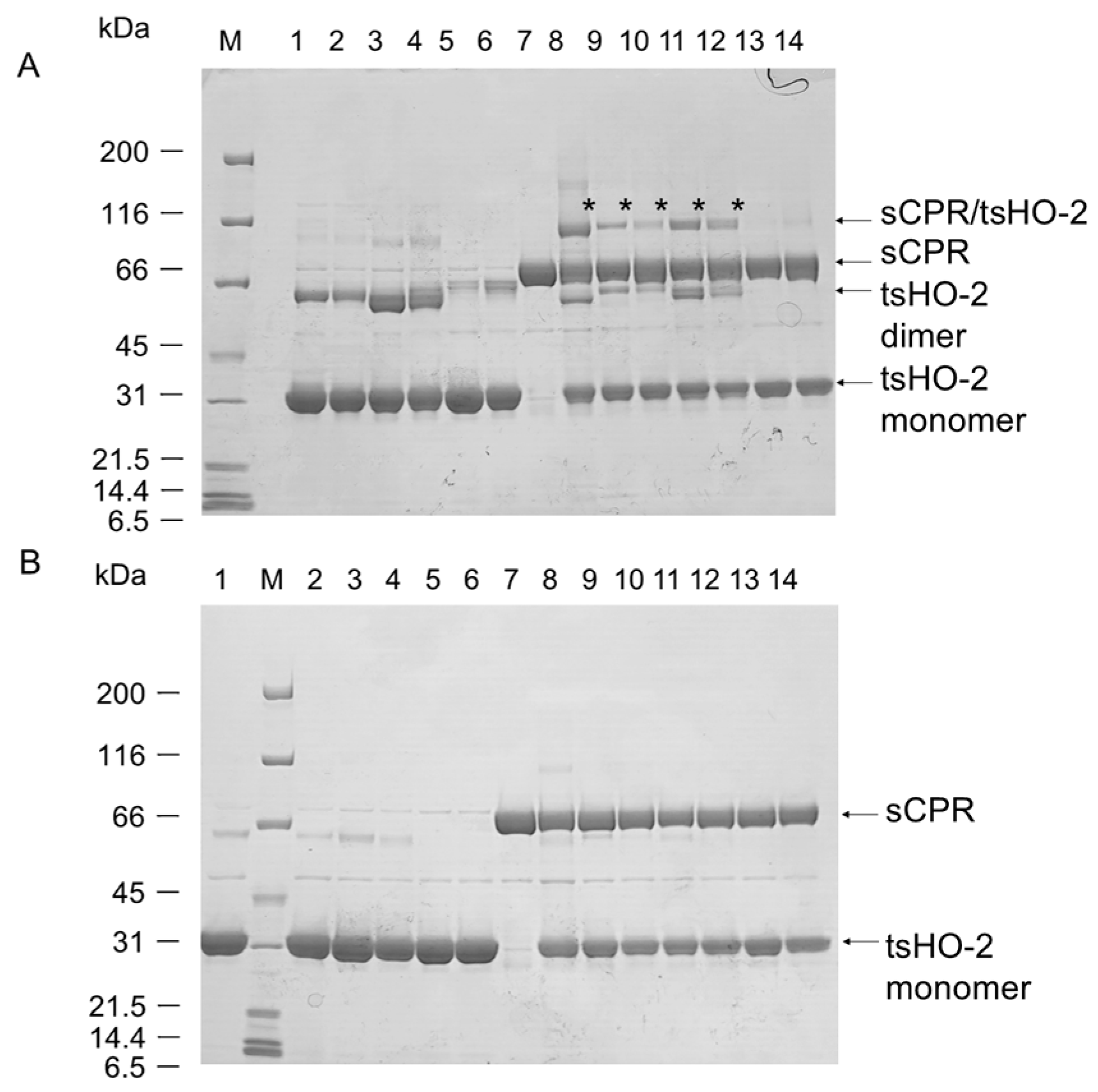
2.1. Crosslinking Experiments
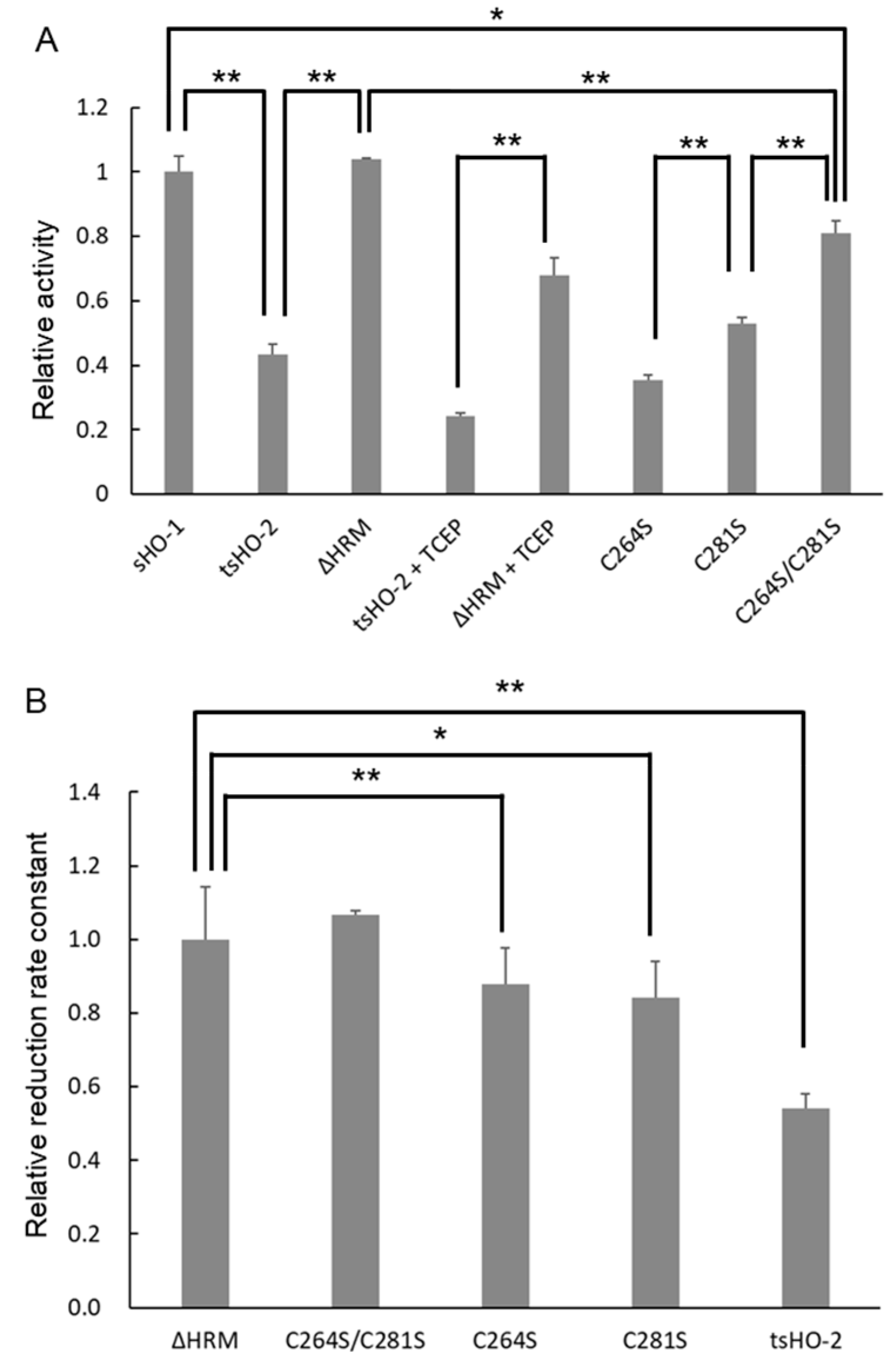
2.2. Enzymatic Assay
2.3. Purification of Heme-Bound tsHO-2 Complex
2.4. Reduction Kinetics
2.5. Single-Turnover Analysis
3. Discussion
4. Materials and Methods
4.1. Protein Expression and Purification
4.2. Crosslinking Experiments
4.3. Enzymatic Assay
4.4. MALDI-TOF MS Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kikuchi, G.; Yoshida, T.; Noguchi, M. Heme oxygenase and heme degradation. Biochem. Biophys. Res. Commun. 2005, 338, 558–567. [Google Scholar] [CrossRef] [PubMed]
- Ortiz de Montellano, P.R.; Wilks, A. Heme oxygenase structure and mechanism. In Advances in Inorgranic Chemistry; Sykes, A.G., Ed.; Academic Press: San Diego, CA, USA, 2001; Volume 51, pp. 359–407. [Google Scholar]
- Tenhunen, R.; Marver, H.S.; Schmid, R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc. Natl. Acad. Sci. USA 1968, 61, 748–755. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S.W.; Alam, J.; Choi, A.M. Heme oxygenase-1/carbon monoxide: From basic science to therapeutic applications. Physiol. Rev. 2006, 86, 583–650. [Google Scholar] [CrossRef]
- Morikawa, T.; Kajimura, M.; Nakamura, T.; Hishiki, T.; Nakanishi, T.; Yukutake, Y.; Nagahata, Y.; Ishikawa, M.; Hattori, K.; Takenouchi, T.; et al. Hypoxic regulation of the cerebral microcirculation is mediated by a carbon monoxide-sensitive hydrogen sulfide pathway. Proc. Natl. Acad. Sci. USA 2012, 109, 1293–1298. [Google Scholar] [CrossRef]
- Ayer, A.; Zarjou, A.; Agarwal, A.; Stocker, R. Heme Oxygenases in Cardiovascular Health and Disease. Physiol. Rev. 2016, 96, 1449–1508. [Google Scholar] [CrossRef]
- Peers, C.; Wyatt, C.N.; Evans, A.M. Mechanisms for acute oxygen sensing in the carotid body. Respir. Physiol. Neurobiol. 2010, 174, 292–298. [Google Scholar] [CrossRef]
- Leffler, C.W.; Parfenova, H.; Jaggar, J.H. Carbon monoxide as an endogenous vascular modulator. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1–H11. [Google Scholar] [CrossRef] [PubMed]
- Schuller, D.J.; Wilks, A.; Ortiz de Montellano, P.R.; Poulos, T.L. Crystal structure of human heme oxygenase-1. Nat. Struct. Biol. 1999, 6, 860–867. [Google Scholar] [CrossRef]
- Sugishima, M.; Omata, Y.; Kakuta, Y.; Sakamoto, H.; Noguchi, M.; Fukuyama, K. Crystal structure of rat heme oxygenase-1 in complex with heme. FEBS Lett. 2000, 471, 61–66. [Google Scholar] [CrossRef]
- Sugishima, M.; Sakamoto, H.; Higashimoto, Y.; Noguchi, M.; Fukuyama, K. Crystal structure of rat heme oxygenase-1 in complex with biliverdin-iron chelate: Conformational change of the distal helix during the heme cleavage reaction. J. Biol. Chem. 2003, 278, 32352–32358. [Google Scholar] [CrossRef]
- Sugishima, M.; Sakamoto, H.; Noguchi, M.; Fukuyama, K. Crystal structures of ferrous and CO-, CN−-, and NO-bound forms of rat heme oxygenase-1 (HO-1) in complex with heme: Structural implications for discrimination between CO and O2 in HO-1. Biochemistry 2003, 42, 9898–9905. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Sugishima, M.; Sakamoto, H.; Higashimoto, Y.; Shimokawa, C.; Fukuyama, K.; Palmer, G.; Noguchi, M. Crystal structure of rat haem oxygenase-1 in complex with ferrous verdohaem: Presence of a hydrogen-bond network on the distal side. Biochem. J. 2009, 419, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Sugishima, M.; Moffat, K.; Noguchi, M. Discrimination between CO and O2 in heme oxygenase: Comparison of static structures and dynamic conformation changes following CO photolysis. Biochemistry 2012, 51, 8554–8562. [Google Scholar] [CrossRef] [PubMed]
- Unno, M.; Matsui, T.; Chu, G.C.; Couture, M.; Yoshida, T.; Rousseau, D.L.; Olson, J.S.; Ikeda-Saito, M. Crystal structure of the dioxygen-bound heme oxygenase from Corynebacterium diphtheriae: Implications for heme oxygenase function. J. Biol. Chem. 2004, 279, 21055–21061. [Google Scholar] [CrossRef]
- Unno, M.; Matsui, T.; Ikeda-Saito, M. Structure and catalytic mechanism of heme oxygenase. Nat. Prod. Rep. 2007, 24, 553–570. [Google Scholar] [CrossRef]
- Unno, M.; Matsui, T.; Ikeda-Saito, M. Crystallographic studies of heme oxygenase complexed with an unstable reaction intermediate, verdoheme. J. Inorg. Biochem. 2012, 113, 102–109. [Google Scholar] [CrossRef]
- Unno, M.; Ardevol, A.; Rovira, C.; Ikeda-Saito, M. Structures of the substrate-free and product-bound forms of HmuO, a heme oxygenase from corynebacterium diphtheriae: X-ray crystallography and molecular dynamics investigation. J. Biol. Chem. 2013, 288, 34443–34458. [Google Scholar] [CrossRef]
- Bianchetti, C.M.; Yi, L.; Ragsdale, S.W.; Phillips, G.N., Jr. Comparison of apo- and heme-bound crystal structures of a truncated human heme oxygenase-2. J. Biol. Chem. 2007, 282, 37624–37631. [Google Scholar] [CrossRef]
- Pandey, A.V.; Fluck, C.E. NADPH P450 oxidoreductase: Structure, function, and pathology of diseases. Pharmacol. Ther. 2013, 138, 229–254. [Google Scholar] [CrossRef]
- Iyanagi, T.; Xia, C.; Kim, J.J. NADPH-cytochrome P450 oxidoreductase: Prototypic member of the diflavin reductase family. Arch. Biochem. Biophys. 2012, 528, 72–89. [Google Scholar] [CrossRef]
- Wang, M.; Roberts, D.L.; Paschke, R.; Shea, T.M.; Masters, B.S.; Kim, J.J. Three-dimensional structure of NADPH-cytochrome P450 reductase: Prototype for FMN- and FAD-containing enzymes. Proc. Natl. Acad. Sci. USA 1997, 94, 8411–8416. [Google Scholar] [CrossRef] [PubMed]
- Iijima, M.; Ohnuki, J.; Sato, T.; Sugishima, M.; Takano, M. Coupling of Redox and Structural States in Cytochrome P450 Reductase Studied by Molecular Dynamics Simulation. Sci. Rep. 2019, 9, 9341. [Google Scholar] [CrossRef] [PubMed]
- Hamdane, D.; Xia, C.; Im, S.C.; Zhang, H.; Kim, J.J.; Waskell, L. Structure and function of an NADPH-cytochrome P450 oxidoreductase in an open conformation capable of reducing cytochrome P450. J. Biol. Chem. 2009, 284, 11374–11384. [Google Scholar] [CrossRef]
- Sugishima, M.; Sato, H.; Higashimoto, Y.; Harada, J.; Wada, K.; Fukuyama, K.; Noguchi, M. Structural basis for the electron transfer from an open form of NADPH-cytochrome P450 oxidoreductase to heme oxygenase. Proc. Natl. Acad. Sci. USA 2014, 111, 2524–2529. [Google Scholar] [CrossRef]
- Sugishima, M.; Sato, H.; Wada, K.; Yamamoto, K. Crystal structure of a NADPH-cytochrome P450 oxidoreductase (CYPOR) and heme oxygenase 1 fusion protein implies a conformational change in CYPOR upon NADPH/NADP(+) binding. FEBS Lett. 2019, 593, 868–875. [Google Scholar] [CrossRef] [PubMed]
- Sugishima, M.; Taira, J.; Sagara, T.; Nakao, R.; Sato, H.; Noguchi, M.; Fukuyama, K.; Yamamoto, K.; Yasunaga, T.; Sakamoto, H. Conformational Equilibrium of NADPH-Cytochrome P450 Oxidoreductase Is Essential for Heme Oxygenase Reaction. Antioxidants 2020, 9, 673. [Google Scholar] [CrossRef]
- Higashimoto, Y.; Sakamoto, H.; Hayashi, S.; Sugishima, M.; Fukuyama, K.; Palmer, G.; Noguchi, M. Involvement of NADPH in the interaction between heme oxygenase-1 and cytochrome P450 reductase. J. Biol. Chem. 2005, 280, 729–737. [Google Scholar] [CrossRef]
- Spencer, A.L.; Bagai, I.; Becker, D.F.; Zuiderweg, E.R.; Ragsdale, S.W. Protein/protein interactions in the mammalian heme degradation pathway: Heme oxygenase-2, cytochrome P450 reductase, and biliverdin reductase. J. Biol. Chem. 2014, 289, 29836–29858. [Google Scholar] [CrossRef]
- Yi, L.; Jenkins, P.M.; Leichert, L.I.; Jakob, U.; Martens, J.R.; Ragsdale, S.W. Heme regulatory motifs in heme oxygenase-2 form a thiol/disulfide redox switch that responds to the cellular redox state. J. Biol. Chem. 2009, 284, 20556–20561. [Google Scholar] [CrossRef]
- Yi, L.; Ragsdale, S.W. Evidence that the heme regulatory motifs in heme oxygenase-2 serve as a thiol/disulfide redox switch regulating heme binding. J. Biol. Chem. 2007, 282, 21056–21067. [Google Scholar] [CrossRef]
- Fleischhacker, A.S.; Gunawan, A.L.; Kochert, B.A.; Liu, L.; Wales, T.E.; Borowy, M.C.; Engen, J.R.; Ragsdale, S.W. The heme-regulatory motifs of heme oxygenase-2 contribute to the transfer of heme to the catalytic site for degradation. J. Biol. Chem. 2020, 295, 5177–5191. [Google Scholar] [CrossRef] [PubMed]
- Fleischhacker, A.S.; Carter, E.L.; Ragsdale, S.W. Redox Regulation of Heme Oxygenase-2 and the Transcription Factor, Rev-Erb, Through Heme Regulatory Motifs. Antioxid. Redox Signal. 2018, 29, 1841–1857. [Google Scholar] [CrossRef] [PubMed]
- Fleischhacker, A.S.; Sharma, A.; Choi, M.; Spencer, A.M.; Bagai, I.; Hoffman, B.M.; Ragsdale, S.W. The C-terminal heme regulatory motifs of heme oxygenase-2 are redox-regulated heme binding sites. Biochemistry 2015, 54, 2709–2718. [Google Scholar] [CrossRef]
- Bagai, I.; Sarangi, R.; Fleischhacker, A.S.; Sharma, A.; Hoffman, B.M.; Zuiderweg, E.R.; Ragsdale, S.W. Spectroscopic studies reveal that the heme regulatory motifs of heme oxygenase-2 are dynamically disordered and exhibit redox-dependent interaction with heme. Biochemistry 2015, 54, 2693–2708. [Google Scholar] [CrossRef]
- Lathrop, J.T.; Timko, M.P. Regulation by heme of mitochondrial protein transport through a conserved amino acid motif. Science 1993, 259, 522–525. [Google Scholar] [CrossRef]
- Shimizu, T.; Alzbeta, L.; Václav, M.; Markéta, M. Heme: Emergent roles of heme in signal transduction, functional regulation and as catalytic centres. Chem. Soc. Rev. 2019, 48, 5619–5808. [Google Scholar] [CrossRef] [PubMed]
- Kochert, B.A.; Fleischhacker, A.S.; Wales, T.E.; Becker, D.F.; Engen, J.R.; Ragsdale, S.W. Dynamic and structural differences between heme oxygenase-1 and -2 are due to differences in their C-terminal regions. J. Biol. Chem. 2019, 294, 8259–8272. [Google Scholar] [CrossRef]
- McCoubrey, W.K., Jr.; Huang, T.J.; Maines, M.D. Heme oxygenase-2 is a hemoprotein and binds heme through heme regulatory motifs that are not involved in heme catalysis. J. Biol. Chem. 1997, 272, 12568–12574. [Google Scholar] [CrossRef]
- Ogura, M.; Endo, R.; Ishikawa, H.; Takeda, Y.; Uchida, T.; Iwai, K.; Kobayashi, K.; Ishimori, K. Redox-dependent axial ligand replacement and its functional significance in heme-bound iron regulatory proteins. J. Inorg. Biochem. 2018, 182, 238–248. [Google Scholar] [CrossRef]
- Sugishima, M.; Sakamoto, H.; Higashimoto, Y.; Omata, Y.; Hayashi, S.; Noguchi, M.; Fukuyama, K. Crystal structure of rat heme oxygenase-1 in complex with heme bound to azide. Implication for regiospecific hydroxylation of heme at the alpha-meso carbon. J. Biol. Chem. 2002, 277, 45086–45090. [Google Scholar] [CrossRef]
- Hayashi, S.; Omata, Y.; Sakamoto, H.; Hara, T.; Noguchi, M. Purification and characterization of a soluble form of rat liver NADPH-cytochrome P-450 reductase highly expressed in Escherichia coli. Protein Expr. Purif. 2003, 29, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Sato, M.; Yoshida, T.; Shimizu, H.; Miyatake, H.; Adachi, S.; Shiro, Y.; Kikuchi, A. Crystallization and preliminary X-ray diffraction analysis of a rat biliverdin reductase. Acta Crystallogr. D Biol. Crystallogr. 2000, 56 Pt 9, 1180–1182. [Google Scholar] [CrossRef]
- Noguchi, M.; Yoshida, T.; Kikuchi, G. Purification and properties of biliverdin reductases from pig spleen and rat liver. J. Biochem. 1979, 86, 833–848. [Google Scholar] [CrossRef] [PubMed]
- Takemoto, M.; Sakamoto, H.; Higashimoto, Y.; Taira, J. Complex Formation of Heme Oxygenase-2 with Heme Is Competitively Inhibited by the Cytosolic Domain of Caveolin-1. Biochemistry 2021, 60, 2300–2308. [Google Scholar] [CrossRef]
- Yoshida, T.; Takahashi, S.; Kikuchi, G. Partial purification and reconstitution of the heme oxygenase system from pig spleen microsomes. J. Biochem. 1974, 75, 1187–1191. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Kikuchi, G. Purification and properties of heme oxygenase from pig spleen microsomes. J. Biol. Chem. 1978, 253, 4224–4229. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Enzymatic Activity | Apparent Reduction Rate Constant | ||||
|---|---|---|---|---|---|
| (min−1) | Relative to sHO-1 (%) | (min−1) | Relative to ΔHRM (%) | ||
| sHO-1 | 13.8 ± 0.69 | 100 ± 4.99 | 122 ± 3.8 [27] | 115 ± 3.1 | |
| tsHO-2 | 6.01 ± 0.43 | 43.5 ± 3.09 | 57.5 ± 2.30 | 54.1 ± 4.0 | |
| ΔHRM | 14.3 ± 0.069 | 104 ± 0.50 | 106 ± 15.3 | 100 ± 14.4 | |
| C264S | 4.91 ± 0.22 | 35.5 ± 1.57 | 93.2 ± 9.24 | 87.7 ± 9.9 | |
| C281S | 7.32 ± 0.27 | 53.0 ± 1.96 | 89.6 ± 8.81 | 84.3 ± 9.8 | |
| C264S/C281S | 11.2 ± 0.56 | 80.8 ± 4.08 | 113.2 ± 1.40 | 107 ± 1.2 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sugishima, M.; Kusumoto, T.; Sato, H.; Sakamoto, H.; Higashimoto, Y.; Yamamoto, K.; Taira, J. Heme Regulatory Motif of Heme Oxygenase-2 Is Involved in the Interaction with NADPH–Cytochrome P450 Reductase and Regulates Enzymatic Activity. Int. J. Mol. Sci. 2025, 26, 2318. https://doi.org/10.3390/ijms26052318
Sugishima M, Kusumoto T, Sato H, Sakamoto H, Higashimoto Y, Yamamoto K, Taira J. Heme Regulatory Motif of Heme Oxygenase-2 Is Involved in the Interaction with NADPH–Cytochrome P450 Reductase and Regulates Enzymatic Activity. International Journal of Molecular Sciences. 2025; 26(5):2318. https://doi.org/10.3390/ijms26052318
Chicago/Turabian StyleSugishima, Masakazu, Tomoichiro Kusumoto, Hideaki Sato, Hiroshi Sakamoto, Yuichiro Higashimoto, Ken Yamamoto, and Junichi Taira. 2025. "Heme Regulatory Motif of Heme Oxygenase-2 Is Involved in the Interaction with NADPH–Cytochrome P450 Reductase and Regulates Enzymatic Activity" International Journal of Molecular Sciences 26, no. 5: 2318. https://doi.org/10.3390/ijms26052318
APA StyleSugishima, M., Kusumoto, T., Sato, H., Sakamoto, H., Higashimoto, Y., Yamamoto, K., & Taira, J. (2025). Heme Regulatory Motif of Heme Oxygenase-2 Is Involved in the Interaction with NADPH–Cytochrome P450 Reductase and Regulates Enzymatic Activity. International Journal of Molecular Sciences, 26(5), 2318. https://doi.org/10.3390/ijms26052318