
Comparative Chloroplast Genomics Reveals Intrageneric Divergence in Salix


Abstract
1. Introduction
2. Results
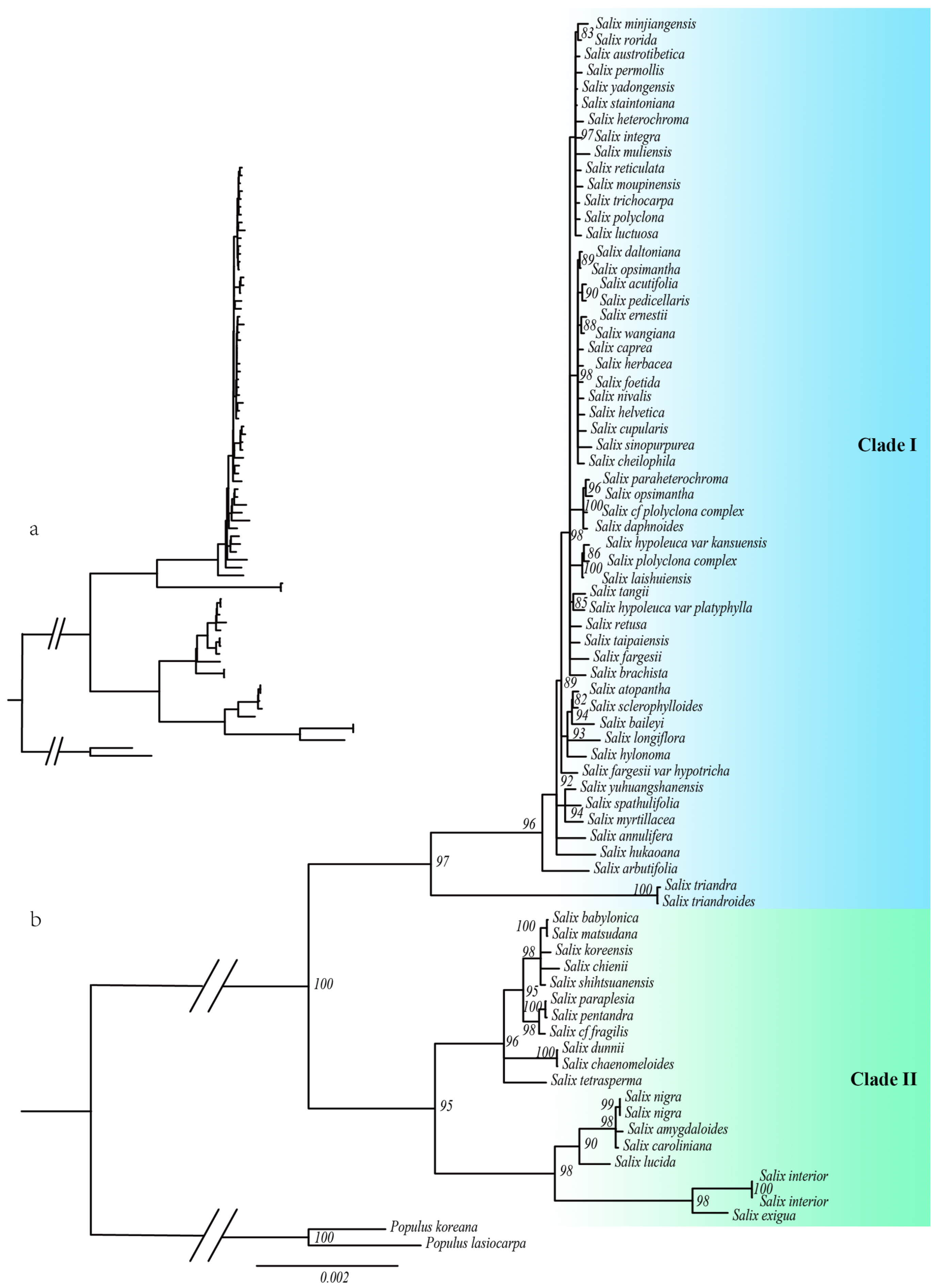
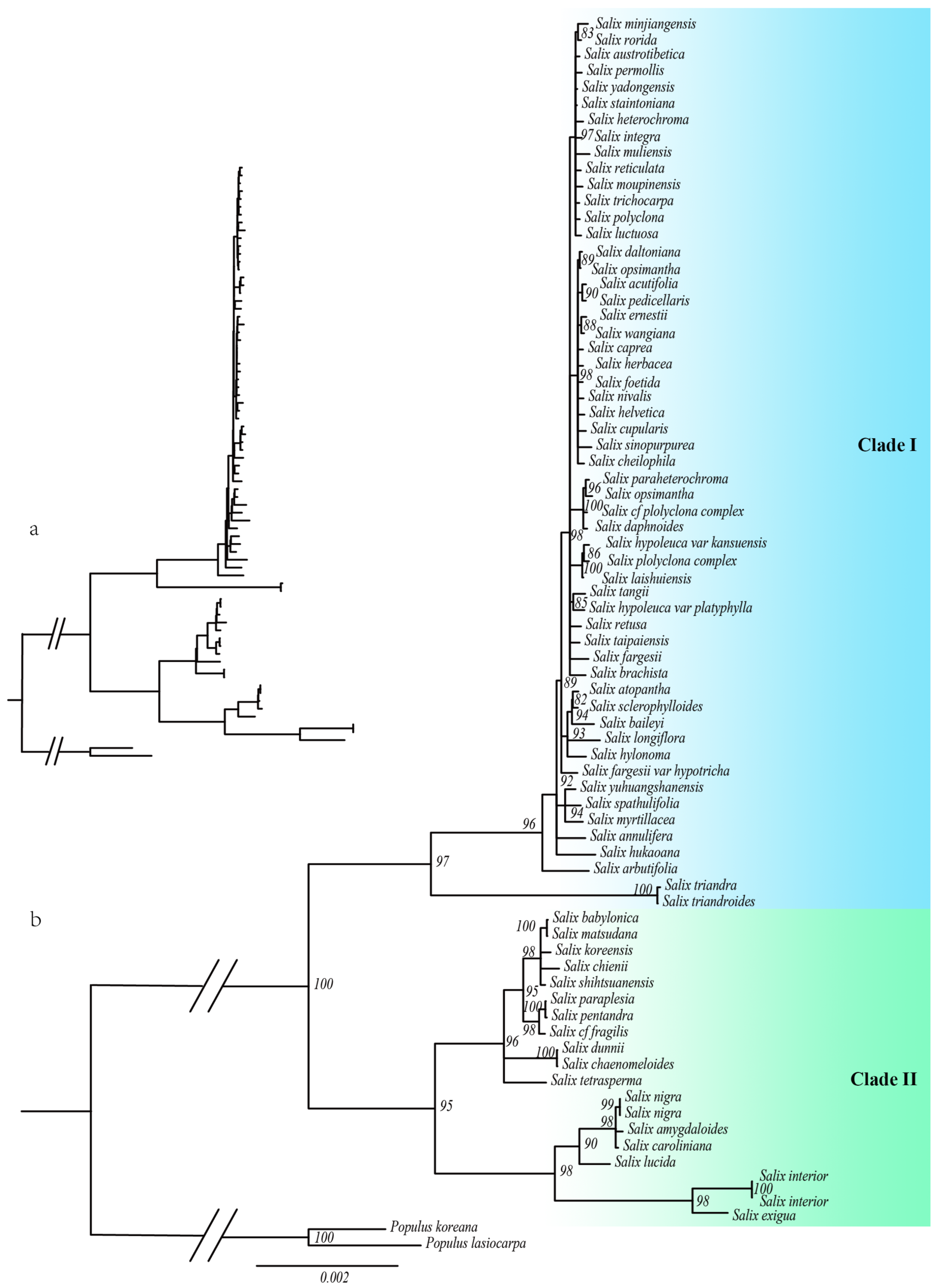
2.1. Phylogenetic Relationships in Salix
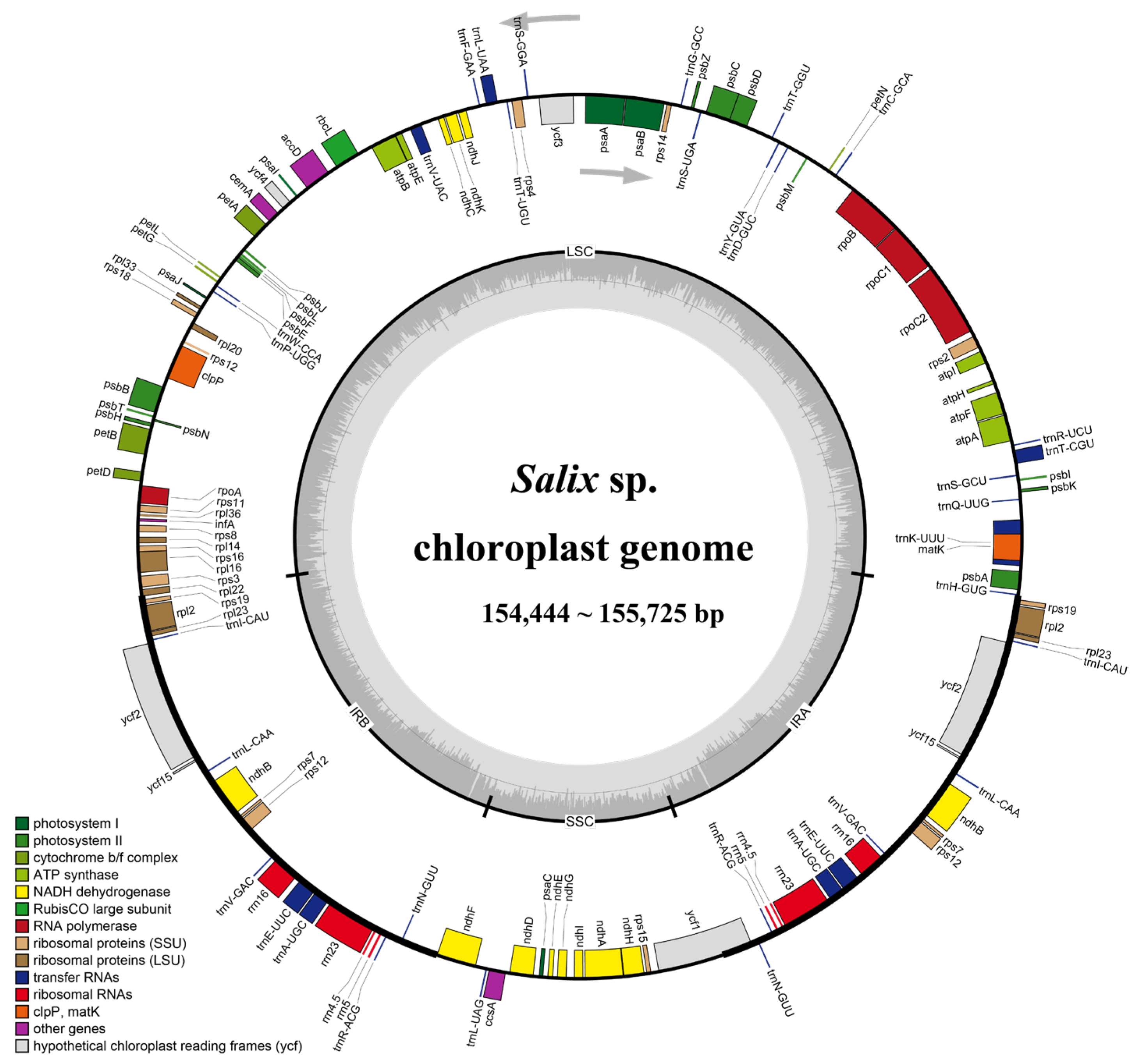
2.2. Chloroplast Genome Features
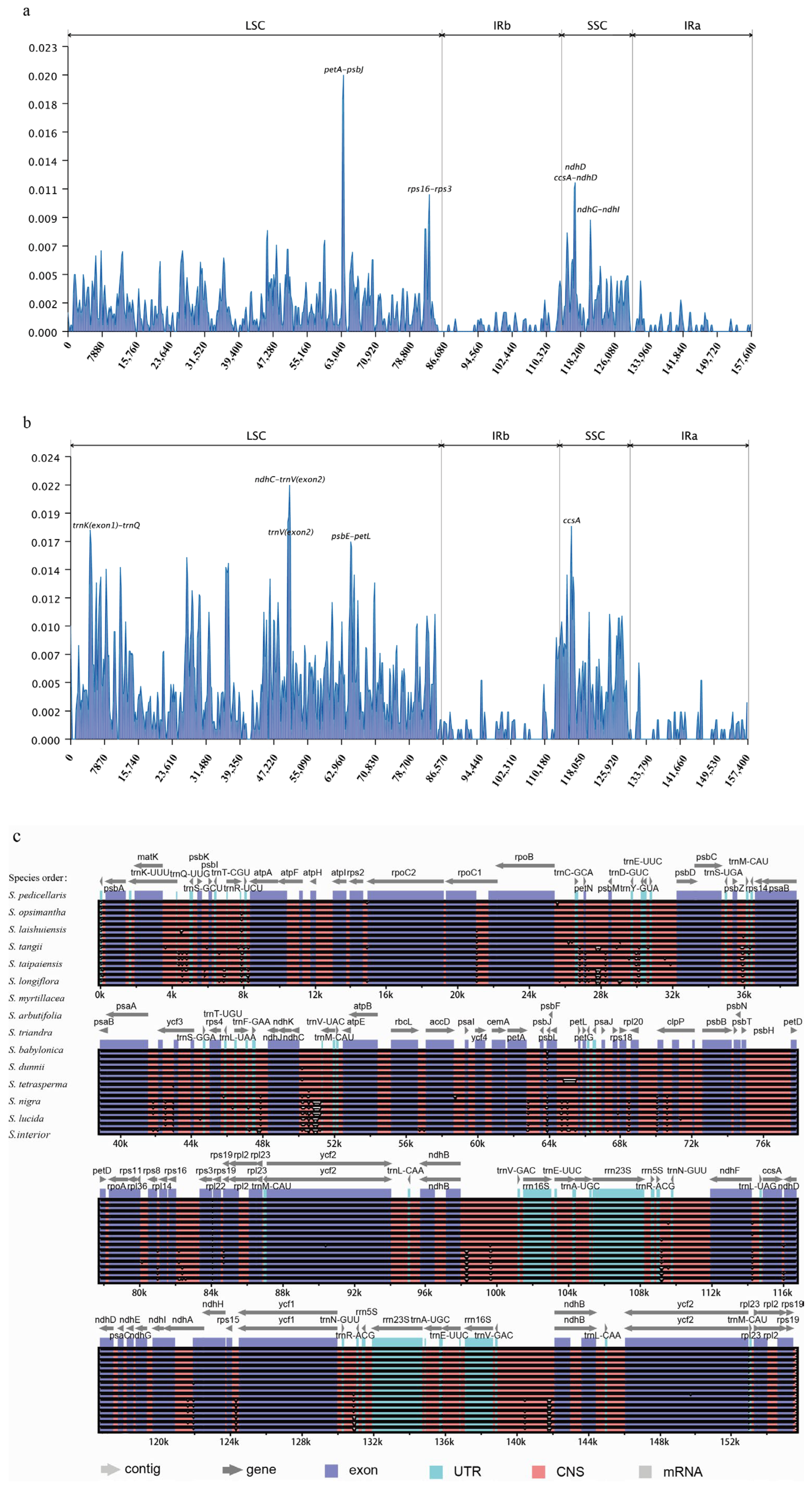
2.3. Comparative Analysis of Salix Chloroplast Genome
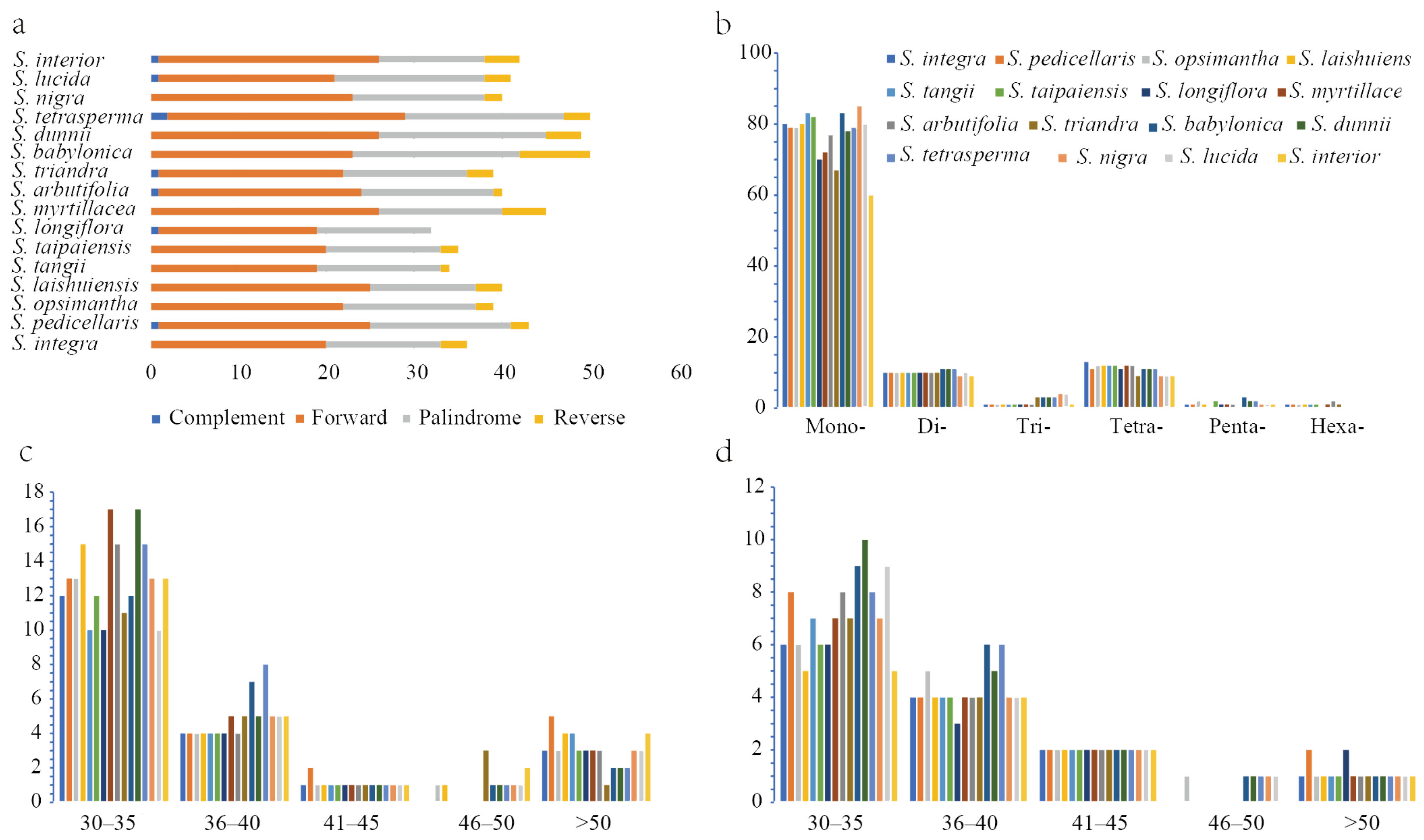
2.4. Result of Chloroplast Genome Repeat Sequences
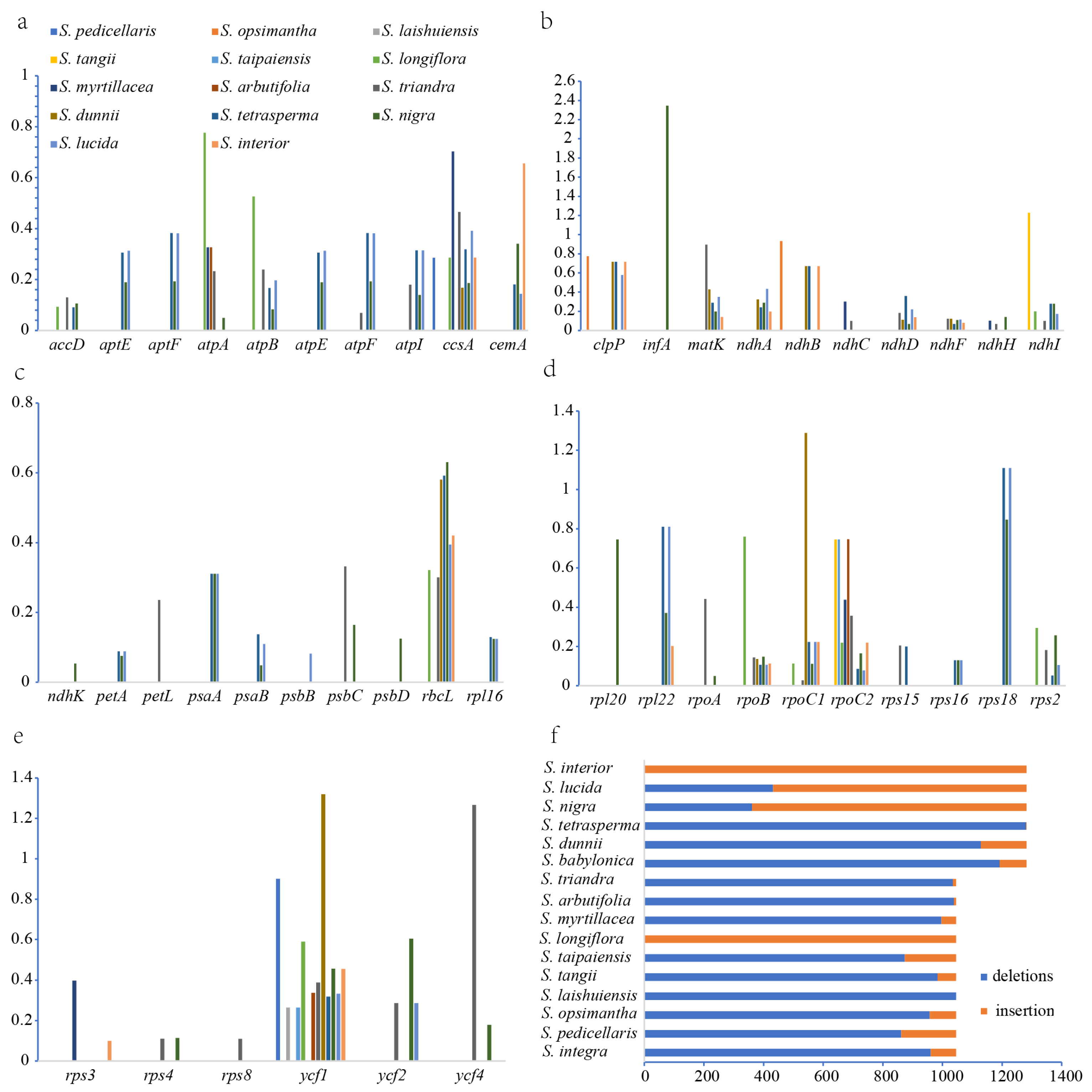
2.5. Result of InDel and Selection Pressure
3. Discussion
3.1. Phylogenetic Relationships Among the Salix
3.2. Chloroplast Genome Differences Between Two Lineages of Salix
4. Materials and Methods
4.1. Plant Material, DNA Extraction, and Genome Sequencing
4.2. Chloroplast Genome Assembly and Annotation
4.3. Phylogenetic Analysis
4.4. Chloroplast Genome Structural Comparison
4.5. Analysis of Repeat Sequences and SSRs
4.6. Analysis of Gene Selection Pressure
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tawfeek, N.; Mahmoud, M.F.; Hamdan, D.I.; Sobeh, M.; Farrag, N.; Wink, M.; El-Shazly, A.M. Phytochemistry, Pharmacology and Medicinal Uses of Plants of the Genus Salix: An Updated Review. Front. Pharmacol. 2021, 12, 593856. [Google Scholar] [CrossRef] [PubMed]
- Keoleian, G.A.; Volk, T.A. Renewable Energy from Willow Biomass Crops: Life Cycle Energy, Environmental and Economic Performance. Crit. Rev. Plant Sci. 2005, 24, 385–406. [Google Scholar] [CrossRef]
- Myers-Smith, I.H.; Forbes, B.C.; Wilmking, M. Shrub Encroachment in Arctic and Alpine Tundra: Dynamics, Impacts and Research Priorities. Environ. Res. Lett. 2011, 6, 1–15. [Google Scholar] [CrossRef]
- Mohsin, M.; Salam, M.M.A.; Nawrot, N.; Kaipiainen, E.; Lane, D.J.; Wojciechowska, E.; Kinnunen, N.; Heimonen, M.; Tervahauta, A.; Peräniemi, S.; et al. Phytoextraction and Recovery of Rare Earth Elements Using Willow (Salix spp.). Sci. Total Environ. 2022, 809, 152209. [Google Scholar] [CrossRef]
- Dorn, R.D. A Synopsis of American Salix. Can. J. Bot. 1976, 54, 2769–2789. [Google Scholar] [CrossRef]
- Argus, G.W. Infrageneric Classification of Salix (Salicaceae) in the New World. Syst. Bot. Monogr. 1997, 52, 1–121. [Google Scholar] [CrossRef]
- Argus, G.W. Salix. In Flora of North America Editorial Committee; Flora of North America North of Mexico, Ed.; Oxford University: New York, NY, USA, 2010; Volume 7, pp. 23–51. [Google Scholar]
- Meucci, S.; Schulte, L.; Zimmermann, H.H.; Stoof-Leichsenring, K.R.; Epp, L.; Eidesen, P.B.; Herzschuh, U. Holocene Chloroplast Genetic Variation of Shrubs (Alnus Alnobetula, Betula Nana, Salix sp.) at the Siberian Tundra-Taiga Ecotone Inferred from Modern Chloroplast Genome Assembly and Sedimentary Ancient DNA Analyses. Ecol. Evol. 2021, 11, 2173–2193. [Google Scholar] [CrossRef]
- Wu, J.; Nyman, T.; Wang, D.C.; Argus, G.W.; Yang, Y.P.; Chen, J.H. Phylogeny of Salix Subgenus Salix s.l. (Salicaceae): Delimitation, Biogeography, and Reticulate Evolution. BMC Evol. Biol. 2015, 15, 31. [Google Scholar] [CrossRef]
- Zhang, L.; Xi, Z.; Wang, M.; Guo, X.; Ma, T. Plastome phylogeny and lineage diversification of Salicaceae with focus on poplars and willows. Ecol. Evol. 2018, 8, 7817–7823. [Google Scholar] [CrossRef]
- Gulyaev, S.; Cai, X.J.; Guo, F.Y.; Kikuchi, S.; Applequist, W.L.; Zhang, Z.X.; Hörandl, E.; He, L. The phylogeny of Salix revealed by whole genome re-sequencing suggests different sex-determination systems in major groups of the genus. Ann. Bot. 2022, 129, 485–498. [Google Scholar] [CrossRef]
- Zhou, X.; Sheng, S.; Xu, Q.; Lu, R.; Chen, C.; Peng, H.; Feng, C. Structure and features of the complete chloroplast genome of Salix triandroides (Salicaceae). Biotechnol. Biotechnol. Equip. 2022, 36, 148–158. [Google Scholar] [CrossRef]
- Jansen, R.K.; Wojciechowski, M.F.; Sanniyasi, E.; Lee, S.B.; Daniell, H. Complete plastid genome sequence of the chickpea (Cicer arietinum) and the phylogenetic distribution of rps12 and clpP intron losses among legumes (Leguminosae). Mol. Phylogenet. Evol. 2008, 48, 1204–1217. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.J.; Dhingra, A.; Soltis, P.S.; Shaw, R.; Farmerie, W.G.; Folta, K.M.; Soltis, D.E. Rapid and accurate pyrosequencing of angiosperm plastid genomes. BMC Plant Biol. 2006, 6, 17. [Google Scholar] [CrossRef] [PubMed]
- Wicke, S.; Schneeweiss, G.M.; dePamphilis, C.W.; Müller, K.F.; Quandt, D. The Evolution of the Plastid Chromosome in Land Plants: Gene Content, Gene Order, Gene Function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef]
- Fajri, H.; Sunandar, A.R.I.; Qurbaniah, M. Phylogenetic Analysis of Wild Bananas (Musa spp.) in West Kalimantan, Indonesia, Based on Maturase K (Matk) Genes. Biodiversitas J. Biol. Divers. 2024, 25, 8. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, K.; Wang, Y.; He, J.; Wu, Y.; Zhang, Z. Complete Chloroplast Genomes of Three Salix Species: Genome Structures and Phylogenetic Analysis. Forests 2021, 12, 12. [Google Scholar] [CrossRef]
- Lu, D.; Huang, H.; Zhang, L.; Hao, L.; Zhang, G. Complete chloroplast genomes of three sand-fixing Salix shrubs from Northwest China: Comparative and phylogenetic analysis and interspecific identification. Trees 2023, 37, 849–861. [Google Scholar] [CrossRef]
- Zhou, J.; Jiao, Z.; Guo, J.; Wang, B.S.; Zheng, J. Complete Chloroplast Genome Sequencing of Five Salix Species and Its Application in the Phylogeny and Taxonomy of the Genus. Mitochondrial DNA B Resour. 2021, 6, 2348–2352. [Google Scholar] [CrossRef]
- Azuma, T.; Kajita, T.; Yokoyama, J.; Ohashi, H. Phylogenetic Relationships of Salix (Salicaceae) Based on Rbcl Quence Data. Am. J. Bot. 2000, 87, 67–75. [Google Scholar] [CrossRef]
- Hardig, T.M.; Anttila, C.K.; Brunsfeld, S.J. A Phylogenetic Analysis of Salix (Salicaceae) Based on Matk and Ribosomal DNA Sequence Data. J. Bot. 2010, 2010, 197696. [Google Scholar]
- Ogutcen, E.; Ferreira, P.d.L.; Wagner, N.D.; Marinček, P.; Leong, J.V.; Aubona, G.; Cavender-Bares, J.; Michálek, J.; Schroeder, L.; Sedio, B.E.; et al. Phylogenetic Insights into the Salicaceae: The Evolution of Willows and Beyond. Mol. Phylogenetics Evol. 2024, 199, 108161. [Google Scholar] [CrossRef]
- Chen, K.-Y.; Wang, J.-D.; Xiang, R.-Q.; Yang, X.-D.; Yun, Q.-Z.; Huang, Y.; Sun, H.; Chen, J.-H. Backbone Phylogeny of Salix Based on Genome Skimming Data. Plant Divers. 2024. [Google Scholar] [CrossRef]
- Mallet, J. Hybridization as an Invasion of the Genome. Trends Ecol. Evol. 2005, 20, 229–237. [Google Scholar] [CrossRef]
- Seehausen, O. Hybridization and Adaptive Radiation. Trends Ecol. Evol. 2004, 19, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Petit, R.J.; Vendramin, G.G. Plant Phylogeography Based on Organelle Genes: An Introduction. In Phylogeography of Southern European Refugia; Springer: Berlin/Heidelberg, Germany, 2007; pp. 23–97. [Google Scholar]
- Fontaine, M.C.; Pease, J.B.; Steele, A.; Waterhouse, R.M.; Neafsey, D.E.; Sharakhov, I.V.; Jiang, X.; Hall, A.B.; Catteruccia, F.; Kakani, E.; et al. Mosquito Genomics. Extensive Introgression in a Malaria Vector Species Complex Revealed by Phylogenomics. Science 2015, 347, 1258524. [Google Scholar] [CrossRef] [PubMed]
- Birky, C.W., Jr. The Inheritance of Genes in Mitochondria and Chloroplasts: Laws, Mechanisms, and Models. Annu. Rev. Genet. 2001, 35, 125–148. [Google Scholar] [CrossRef]
- Palmer, J.D.; Thompson, W.F. Rearrangements in the Chloroplast Genomes of Mung Bean and Pea. Proc. Natl. Acad. Sci. USA 1981, 78, 5533–5537. [Google Scholar] [CrossRef]
- Guisinger, M.M.; Kuehl, J.V.; Boore, J.L.; Jansen, R.K. Extreme Reconfiguration of Plastid Genomes in the Angiosperm Family Geraniaceae: Rearrangements, Repeats, and Codon Usage. Mol. Biol. Evol. 2011, 28, 583–600. [Google Scholar] [CrossRef]
- Song, Y.; Chen, Y.; Lv, J.; Xu, J.; Zhu, S.; Li, M.; Chen, N. Development of Chloroplast Genomic Resources for Oryza Species Discrimination. Front. Plant Sci. 2017, 8, 1854. [Google Scholar] [CrossRef]
- Lyu, D.; Sun, S.; Shan, X.; Wang, W. Inbreeding Evaluation Using Microsatellite Confirmed Inbreeding Depression in Growth in the Fenneropenaeus Chinensis Natural Population. Front. Genet. 2023, 14, 1077814. [Google Scholar] [CrossRef]
- Köhler, M.; Reginato, M.; Souza-Chies, T.T.; Majure, L.C. Insights into Chloroplast Genome Evolution across Opuntioideae (Cactaceae) Reveals Robust yet Sometimes Conflicting Phylogenetic Topologies. Front. Plant Sci. 2020, 11, 729. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.; Lickey, E.B.; Beck, J.T.; Farmer, S.B.; Liu, W.; Miller, J.; Siripun, K.C.; Winder, C.T.; Schilling, E.E.; Small, R.L. The Tortoise and the Hare Ii: Relative Utility of 21 Noncoding Chloroplast DNA Sequences for Phylogenetic Analysis. Am. J. Bot. 2005, 92, 142–166. [Google Scholar] [CrossRef] [PubMed]
- Wright, S.I.; Gaut, B.S. Molecular Population Genetics and the Search for Adaptive Evolution in Plants. Mol. Biol. Evol. 2005, 22, 506–519. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.S.W.; Yang, Z.; Goldman, N.; Nielsen, R. Accuracy and Power of Statistical Methods for Detecting Adaptive Evolution in Protein Coding Sequences and for Identifying Positively Selected Sites. Genetics 2004, 168, 1041–1051. [Google Scholar] [CrossRef]
- Clark, R.M.; Schweikert, G.; Toomajian, C.; Ossowski, S.; Zeller, G.; Shinn, P.; Warthmann, N.; Hu, T.T.; Fu, G.; Hinds, D.A.; et al. Common Sequence Polymorphisms Shaping Genetic Diversity in Arabidopsis thaliana. Science 2007, 317, 338–342. [Google Scholar] [CrossRef]
- Li, J.; Wang, S.; Yu, J.; Wang, L.; Zhou, S. A Modified Ctab Protocol for Plant DNA Extraction. Acta Bot. 2013, 48, 72. [Google Scholar]
- Jin, J.-J.; Yu, W.-B.; Yang, J.-B.; Song, Y.; Depamphilis, C.W.; Yi, T.-S.; Li, D.-Z. Getorganelle: A Fast and Versatile Toolkit for Accurate De Novo Assembly of Organelle Genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An Integrated and Extendable Desktop Software Platform for the Organization and Analysis of Sequence Data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Qu, X.; Moore, M.J.; Li, D.-Z.; Yi, T. Pga: A Software Package for Rapid, Accurate, and Flexible Batch Annotation of Plastomes. Plant Methods 2019, 15, 50. [Google Scholar] [CrossRef]
- Greiner, S.; Lehwark, P.; Bock, R. Organellargenomedraw (Ogdraw) Version 1.3.1: Expanded Toolkit for the Graphical Visualization of Organellar Genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. Mafft Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. Trimal: A Tool for Automated Alignment Trimming in Large-Scale Phylogenetic Analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, J.; Zhao, X.; Wang, J.; Wong, G.K.; Yu, J. Kaks_Calculator: Calculating Ka and Ks through Model Selection and Model Averaging. Genom. Proteom. Bioinform. 2006, 4, 259–263. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Species Length (bp) | GC (%) | Number of Genes | ||||
|---|---|---|---|---|---|---|---|
| Coding Genes | tRNA | rRNA | Total | ||||
| Clade I | S. integra | 155,607 | 36.69 | 88 | 35 | 8 | 131 |
| S. pedicellaris | 155,508 | 36.7 | 88 | 35 | 8 | 131 | |
| S. opsimantha | 155,603 | 36.68 | 88 | 35 | 8 | 131 | |
| S. laishuiensis | 155,692 | 36.68 | 88 | 35 | 8 | 131 | |
| S. tangii | 155,630 | 36.69 | 88 | 35 | 8 | 131 | |
| S. taipaiensis | 155,520 | 36.7 | 88 | 35 | 8 | 131 | |
| S. longiflora | 154,646 | 36.78 | 88 | 35 | 8 | 131 | |
| S. myrtillacea | 155,643 | 36.68 | 88 | 35 | 8 | 131 | |
| S. arbutifolia | 155,684 | 36.68 | 88 | 35 | 8 | 131 | |
| S. triandra | 155,681 | 36.65 | 88 | 35 | 8 | 131 | |
| Clade II | S. babylonica | 155,725 | 36.64 | 88 | 35 | 8 | 131 |
| S. dunnii | 155,636 | 36.65 | 88 | 35 | 8 | 131 | |
| S. tetrasperma | 155,573 | 36.66 | 88 | 35 | 8 | 131 | |
| S. nigra | 154,805 | 36.76 | 88 | 35 | 8 | 131 | |
| S. lucida | 154,876 | 36.76 | 88 | 35 | 8 | 131 | |
| S. interior | 154,444 | 36.78 | 88 | 35 | 8 | 131 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, F.; Zhou, L.; Wei, X.; Shang, C.; Zhang, Z. Comparative Chloroplast Genomics Reveals Intrageneric Divergence in Salix. Int. J. Mol. Sci. 2025, 26, 2248. https://doi.org/10.3390/ijms26052248
Yuan F, Zhou L, Wei X, Shang C, Zhang Z. Comparative Chloroplast Genomics Reveals Intrageneric Divergence in Salix. International Journal of Molecular Sciences. 2025; 26(5):2248. https://doi.org/10.3390/ijms26052248
Chicago/Turabian StyleYuan, Fulin, Liwei Zhou, Xueya Wei, Ce Shang, and Zhixiang Zhang. 2025. "Comparative Chloroplast Genomics Reveals Intrageneric Divergence in Salix" International Journal of Molecular Sciences 26, no. 5: 2248. https://doi.org/10.3390/ijms26052248
APA StyleYuan, F., Zhou, L., Wei, X., Shang, C., & Zhang, Z. (2025). Comparative Chloroplast Genomics Reveals Intrageneric Divergence in Salix. International Journal of Molecular Sciences, 26(5), 2248. https://doi.org/10.3390/ijms26052248