
Targeting Glucosylceramide Synthase: Innovative Drug Repurposing Strategies for Lysosomal Diseases

 ,
,  ,
,  and
and 
Abstract

1. Introduction
2. Results and Discussion
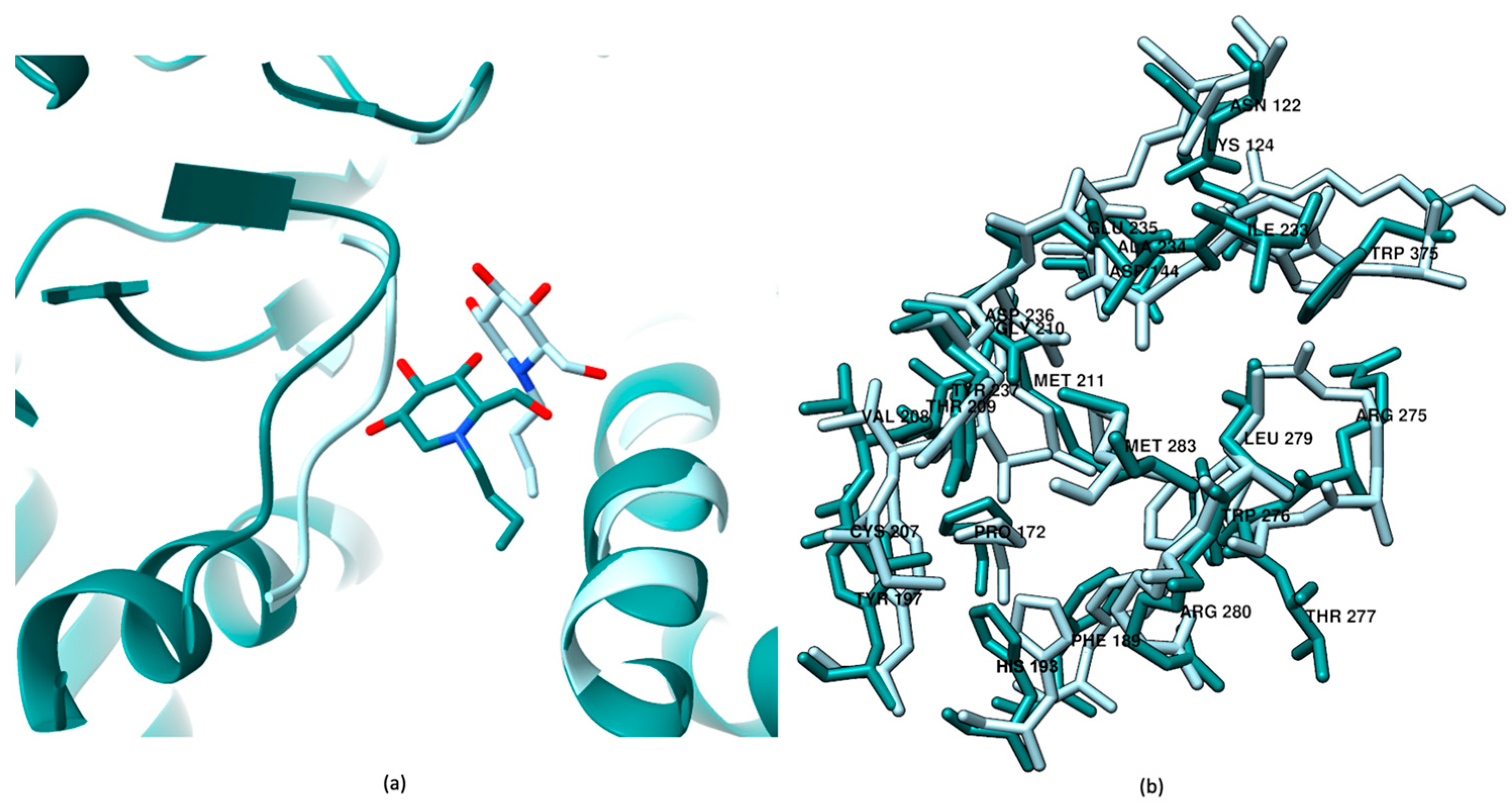
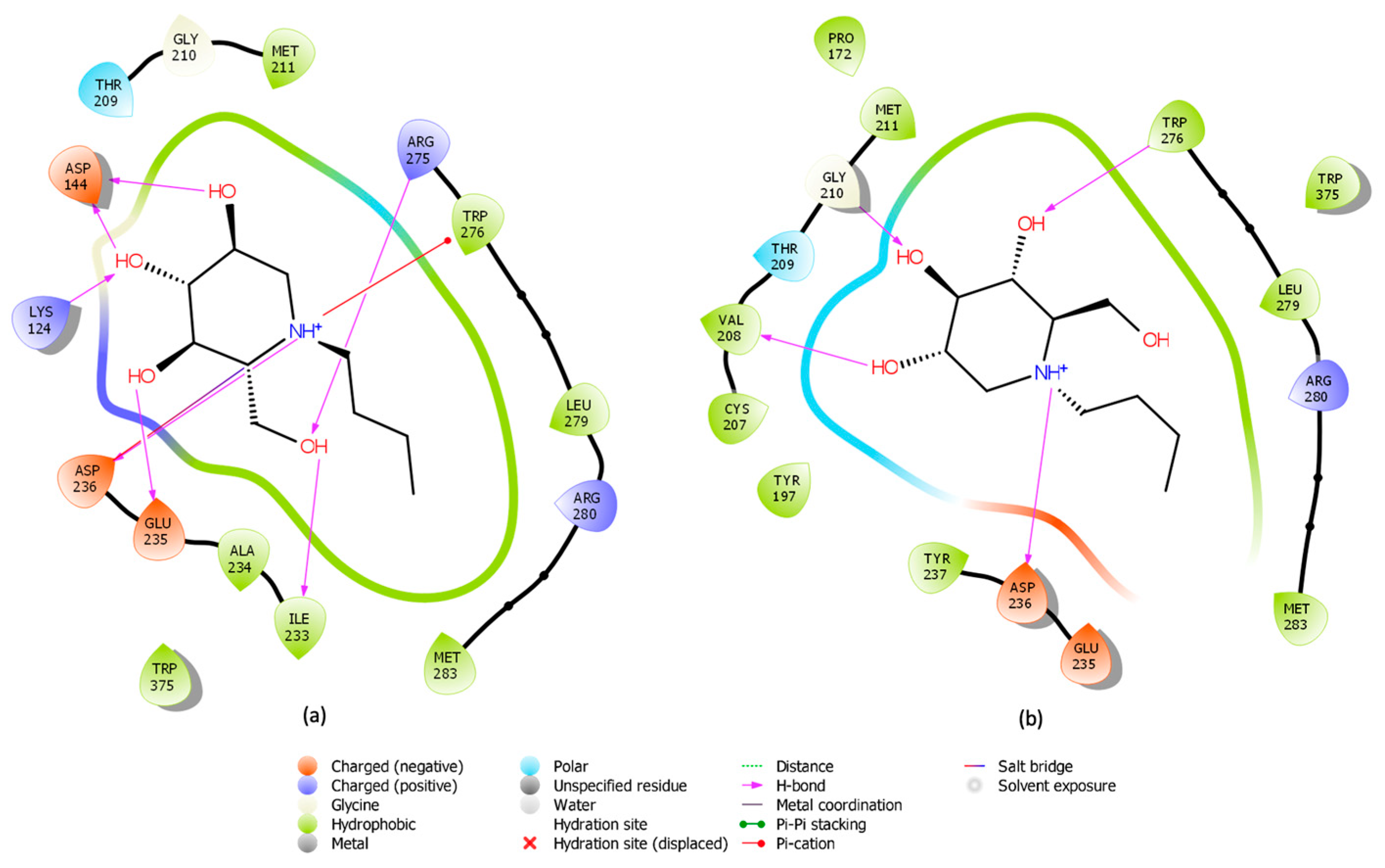
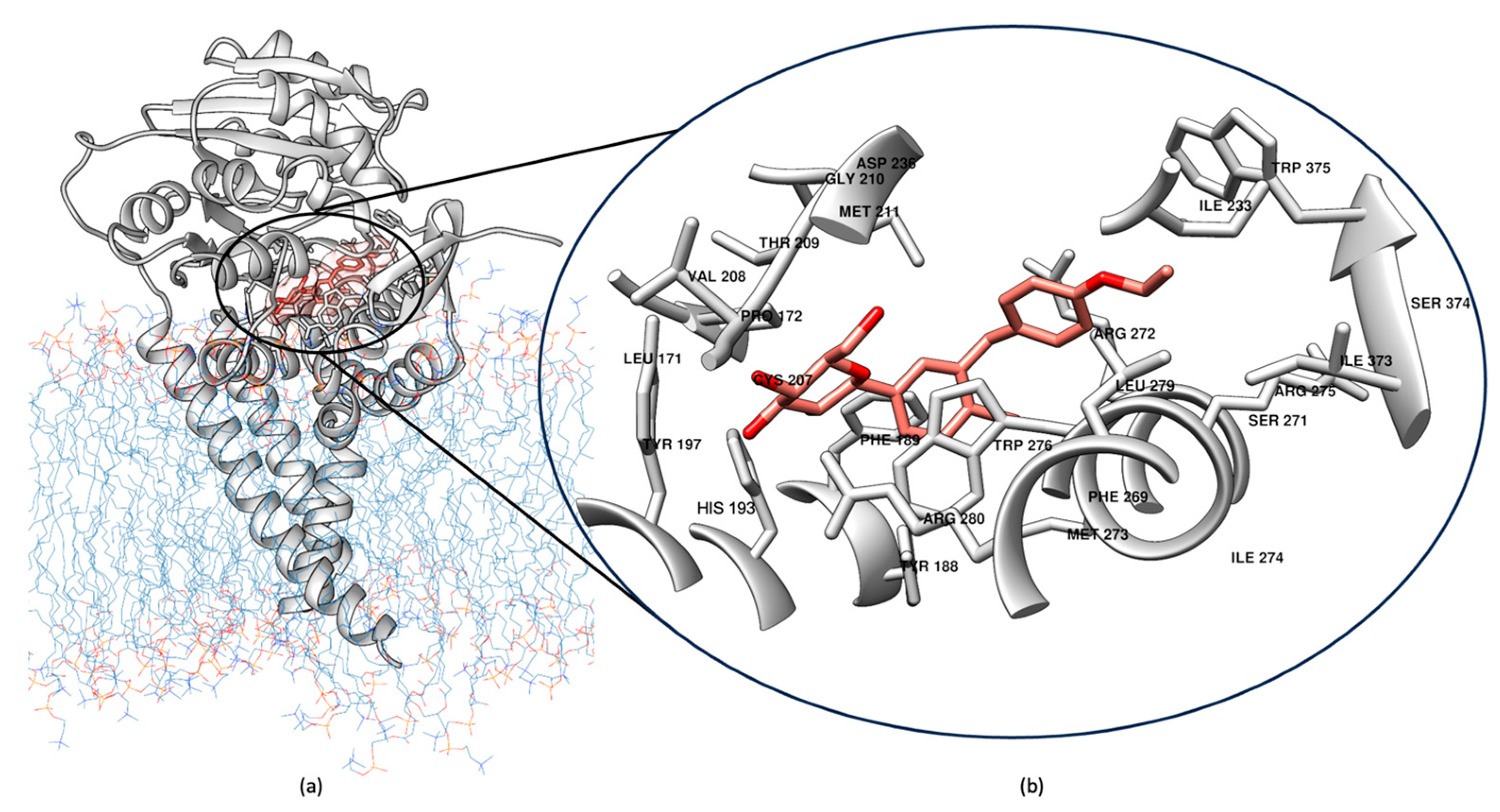
2.1. Induced Fit Docking for Miglustat: A Unique Treatment for NPC as a GCS Inhibitor
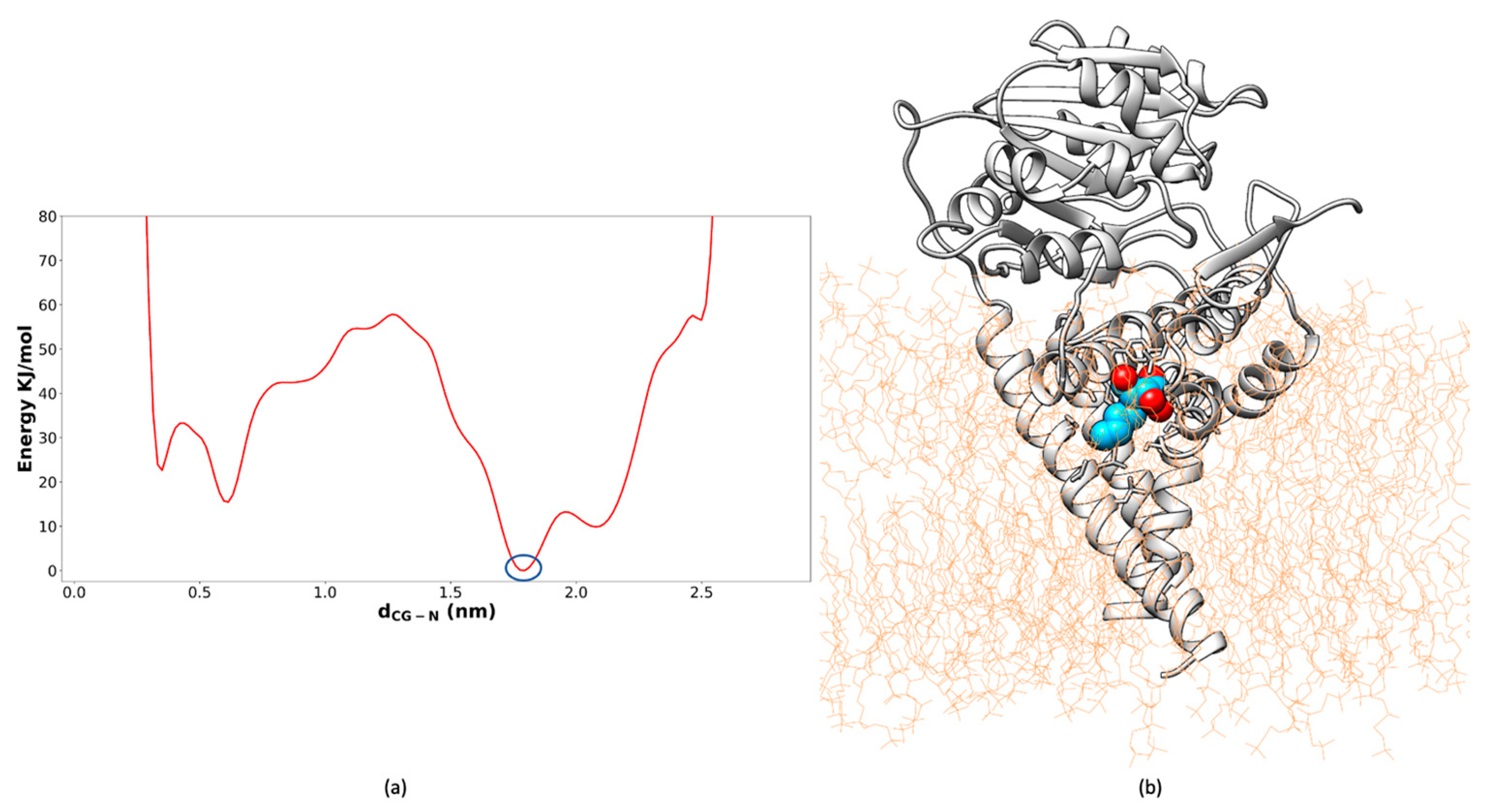
2.2. Well-Tempered FM Analysis of the GCS–Inhibitor Complex
2.3. In Silico Drug Repurposing
2.4. Virtual Screening Protocol
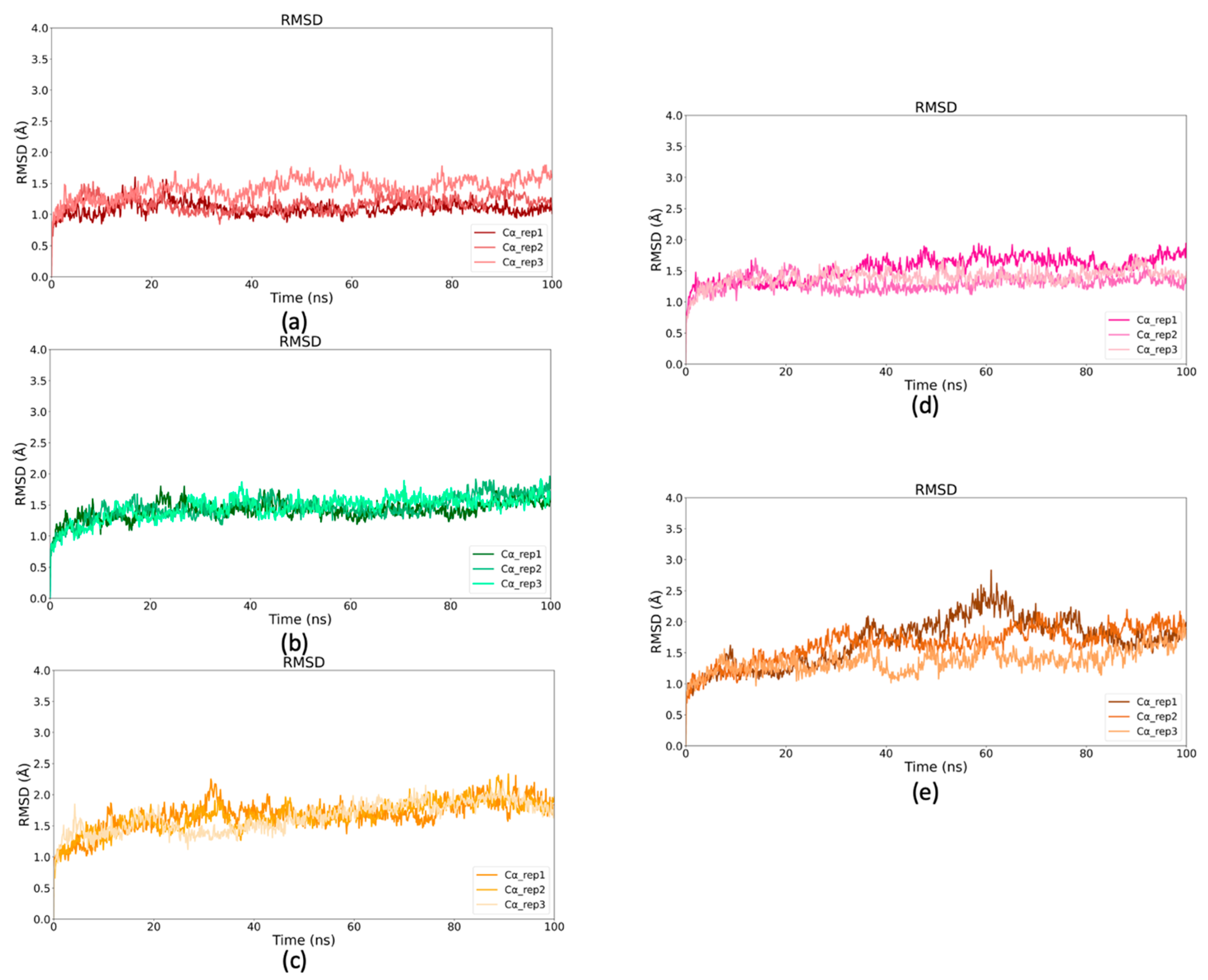
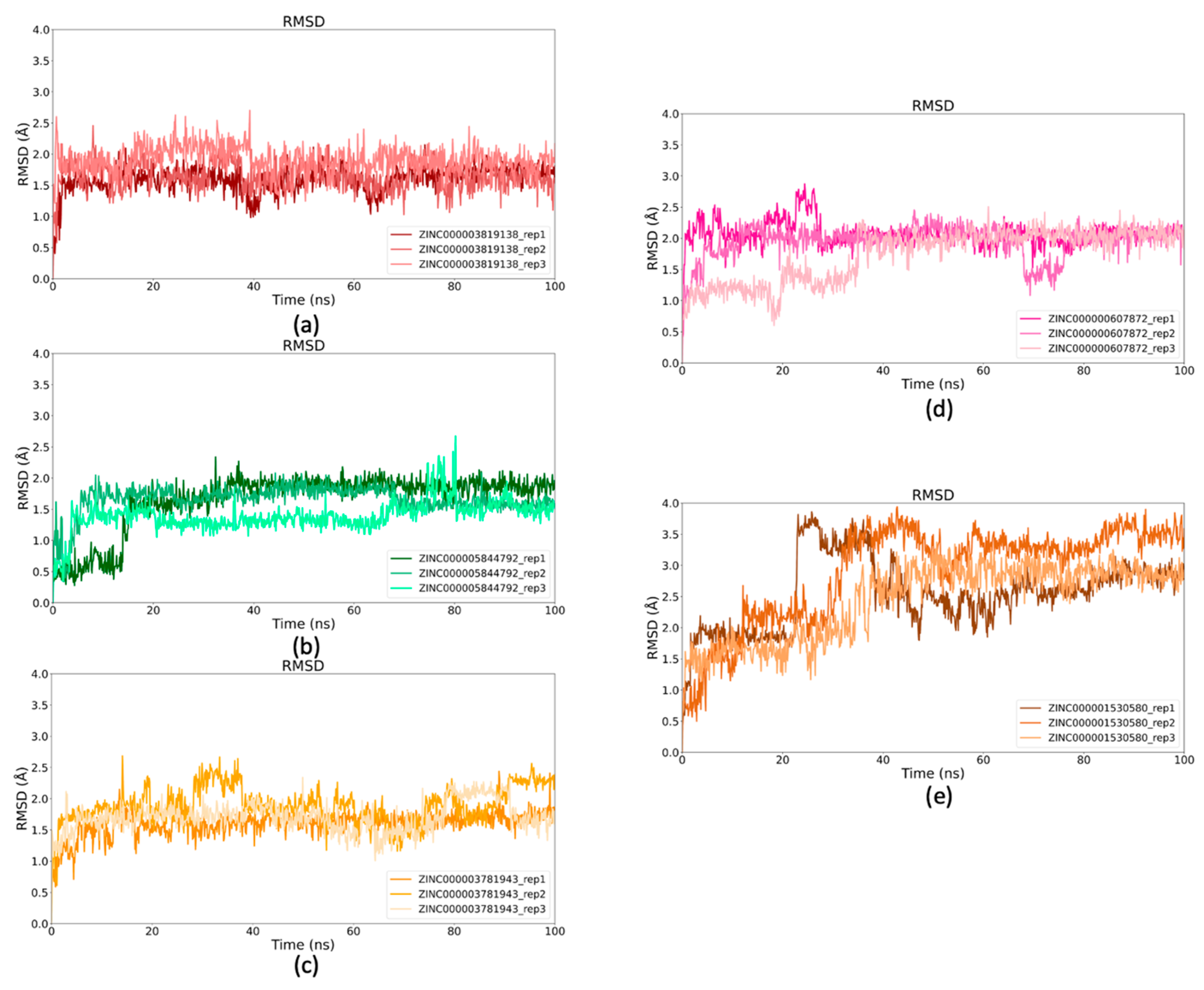
2.5. Binding Stability of Selected Drugs Through MD Simulations
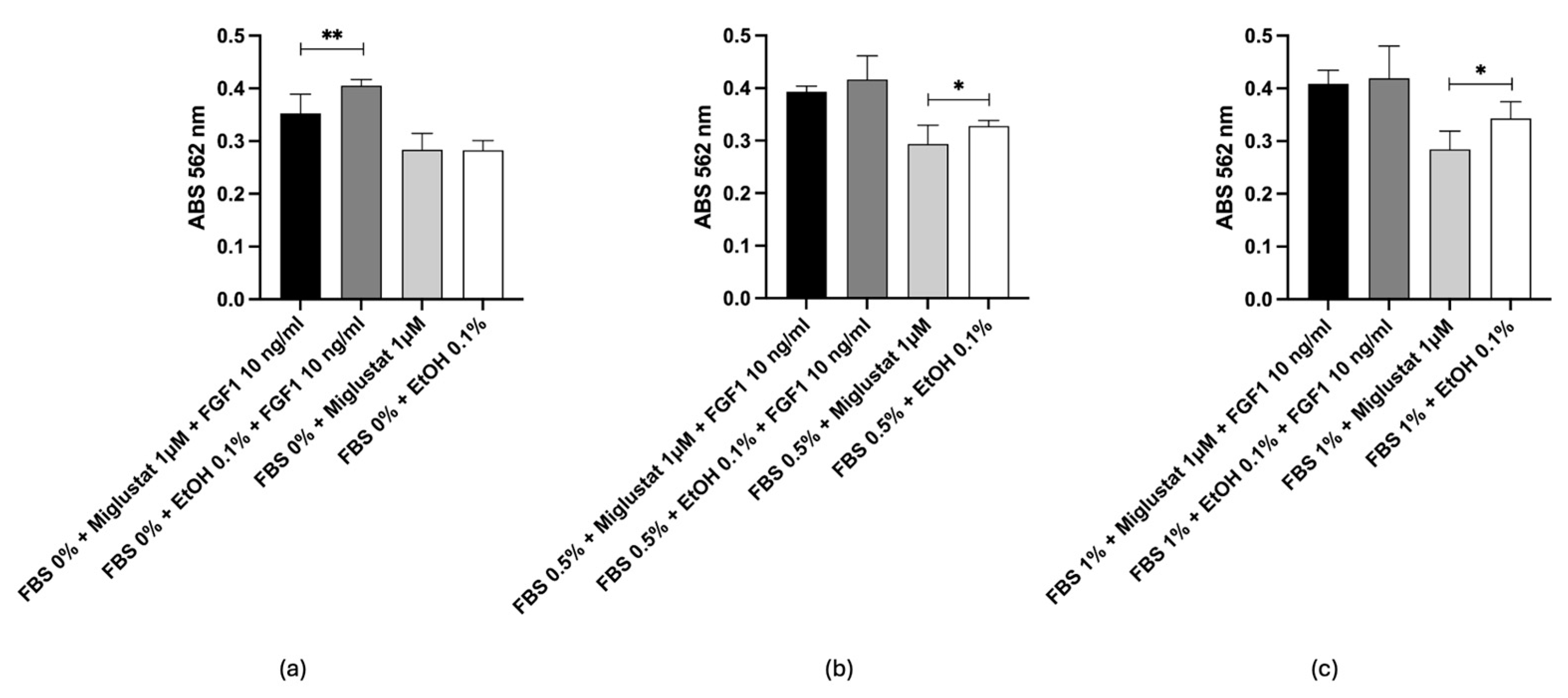
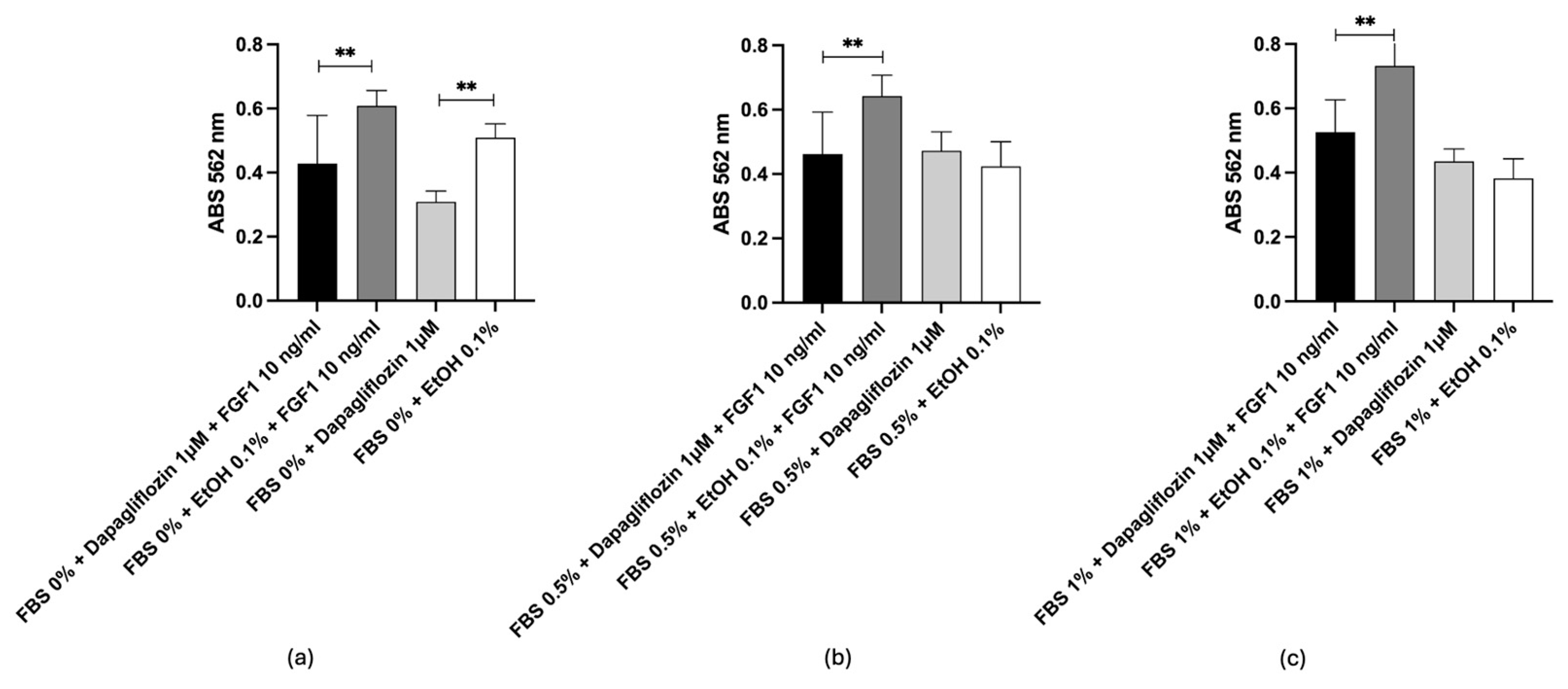
2.6. In Vitro Test
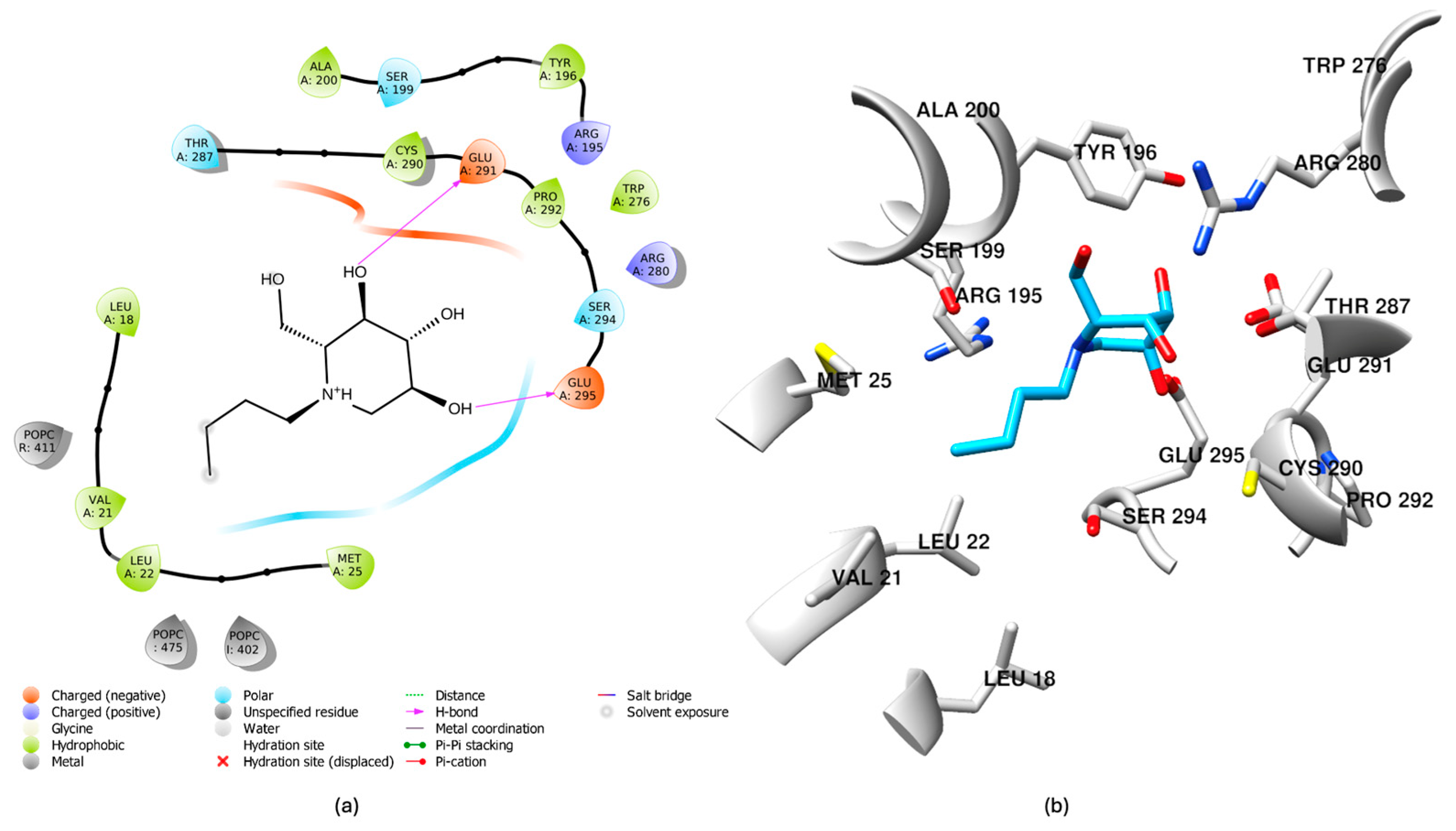
2.7. Comparative Binding Modes of Dapagliflozin and Miglustat
3. Materials and Methods
3.1. Induced Fit Docking for Miglustat, the Only Approved Drug for NPC
3.2. MD Simulation of Miglustat
3.3. Well-Tempered FM Simulations of Miglustat
3.4. Cross-Docking Procedure
3.5. Virtual Screening
3.6. Simulations of Top Ligands
3.7. In Vitro Tests
3.7.1. Materials
3.7.2. Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| LSDs | Lysosomal storage diseases |
| GCS | Glucosylceramide synthase |
| BMT | Bone marrow transplantation |
| ERT | Enzyme replacement therapy |
| SRT | Substrate reduction therapy |
| OCT2 | Organic cation transporter 2 |
| Mn2+ | Manganese ion |
| MD | Molecular dynamics |
| FM | Funnel metadynamics |
| CVs | Collective variables |
| dCG-N | Distance between the carbon atom of the carboxyl group in Asp236 and the nitrogen atom in Miglustat’s piperidine ring |
| Cα atoms | Alpha-carbons |
| H-bonds | Hydrogen bonds |
| SP | Standard precision |
| XP | Extra precision |
| VS | Virtual screening |
| QPPCaco | Predicted apparent Caco-2 cell permeability in nm/sec |
| QPlogBB | Predicted brain/blood partition coefficient |
| QPlogPo/w | Predicted octanol/water partition coefficient |
| CNS | Central nervous system |
| PSA | Polar surface area |
| MM-GBSA | Molecular mechanics/generalized born surface area |
| ΔGbind | Free energy |
| SGLT1 | Sodium-glucose transport protein 1 |
| SGLT2 | Sodium-glucose transport protein 2 |
| RMSD | Root Mean square Deviation |
| PPPP | D-l-threo-1-phenyl-2-hexadecanoylamino-3-pyrrolidino-1-propanol-HCl |
| FBS | Fetal bovine serum |
| FGF1 | Recombinant human fibroblast growth Factor acidic |
| MCE | MedChemExpress |
| EtOH | Ethanol |
| DMEM | Dulbecco’s Modified Eagle Medium |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide |
| PBS | Phosphate buffered saline |
| Abs | Absorbance |
References
- Platt, F.M.; Boland, B.; van der Spoel, A.C. Lysosomal Storage Disorders: The Cellular Impact of Lysosomal Dysfunction. J. Cell Biol. 2012, 199, 723–734. [Google Scholar] [CrossRef]
- Alkhzouz, C.; Miclea, D.; Bucerzan, S.; Lazea, C.; Nascu, I.; Sido, P.G. Early Clinical Signs in Lysosomal Diseases. Med. Pharm. Rep. 2021, 94, S43–S46. [Google Scholar] [CrossRef] [PubMed]
- Scerra, G.; De Pasquale, V.; Scarcella, M.; Caporaso, M.G.; Pavone, L.M.; D’Agostino, M. Lysosomal Positioning Diseases: Beyond Substrate Storage. Open Biol. 2022, 12, 220155. [Google Scholar] [CrossRef] [PubMed]
- Vanier, M.T. Niemann-Pick Disease Type C. Orphanet J. Rare Dis. 2010, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, C.R.; Gahl, W.A. Lysosomal Storage Diseases. Transl. Sci. Rare Dis. 2017, 2, 1–71. [Google Scholar] [CrossRef]
- Marques, A.R.A.; Saftig, P. Lysosomal Storage Disorders—Challenges, Concepts and Avenues for Therapy: Beyond Rare Diseases. J. Cell Sci. 2019, 132, jcs221739. [Google Scholar] [CrossRef]
- Parenti, G.; Andria, G.; Ballabio, A. Lysosomal Storage Diseases: From Pathophysiology to Therapy. Annu. Rev. Med. 2015, 66, 471–486. [Google Scholar] [CrossRef]
- Luzio, J.P.; Pryor, P.R.; Bright, N.A. Lysosomes: Fusion and Function. Nat. Rev. Mol. Cell Biol. 2007, 8, 622–632. [Google Scholar] [CrossRef]
- Patterson, M.C.; Hendriksz, C.J.; Walterfang, M.; Sedel, F.; Vanier, M.T.; Wijburg, F.; NP-C Guidelines Working Group. Recommendations for the Diagnosis and Management of Niemann-Pick Disease Type C: An Update. Mol. Genet. Metab. 2012, 106, 330–344. [Google Scholar] [CrossRef]
- Mengel, E.; Pineda, M.; Hendriksz, C.J.; Walterfang, M.; Torres, J.V.; Kolb, S.A. Differences in Niemann-Pick Disease Type C Symptomatology Observed in Patients of Different Ages. Mol. Genet. Metab. 2017, 120, 180–189. [Google Scholar] [CrossRef]
- Mengel, E.; Klünemann, H.-H.; Lourenço, C.M.; Hendriksz, C.J.; Sedel, F.; Walterfang, M.; Kolb, S.A. Niemann-Pick Disease Type C Symptomatology: An Expert-Based Clinical Description. Orphanet J. Rare Dis. 2013, 8, 166. [Google Scholar] [CrossRef]
- Cox, T.M. Gaucher Disease: Understanding the Molecular Pathogenesis of Sphingolipidoses. J. Inherit. Metab. Dis. 2001, 24 (Suppl. S2), 106–121; discussion 87–88. [Google Scholar] [CrossRef] [PubMed]
- Ferraz, M.J.; Kallemeijn, W.W.; Mirzaian, M.; Herrera Moro, D.; Marques, A.; Wisse, P.; Boot, R.G.; Willems, L.I.; Overkleeft, H.S.; Aerts, J.M. Gaucher Disease and Fabry Disease: New Markers and Insights in Pathophysiology for Two Distinct Glycosphingolipidoses. Biochim. Biophys. Acta 2014, 1841, 811–825. [Google Scholar] [CrossRef]
- Stern, G. Niemann-Pick’s and Gaucher’s Diseases. Park. Relat. Disord. 2014, 20 (Suppl. S1), S143–S146. [Google Scholar] [CrossRef]
- Elstein, D.; Abrahamov, A.; Hadas-Halpern, I.; Zimran, A. Gaucher’s Disease. Lancet 2001, 358, 324–327. [Google Scholar] [CrossRef] [PubMed]
- Abed Rabbo, M.; Khodour, Y.; Kaguni, L.S.; Stiban, J. Sphingolipid Lysosomal Storage Diseases: From Bench to Bedside. Lipids Health Dis. 2021, 20, 44. [Google Scholar] [CrossRef] [PubMed]
- Bernardes, T.P.; Foresto, R.D.; Kirsztajn, G.M. Fabry Disease: Genetics, Pathology, and Treatment. Rev. Assoc. Med. Bras. 2020, 66 (Suppl. S1), s10–s16. [Google Scholar] [CrossRef]
- Newton, J.; Milstien, S.; Spiegel, S. Niemann-Pick Type C Disease: The Atypical Sphingolipidosis. Adv. Biol. Regul. 2018, 70, 82–88. [Google Scholar] [CrossRef]
- Biffi, A. Hematopoietic Stem Cell Gene Therapy for Storage Disease: Current and New Indications. Mol. Ther. 2017, 25, 1155–1162. [Google Scholar] [CrossRef]
- Kim, S.U. Lysosomal Storage Diseases: Stem Cell-Based Cell- and Gene-Therapy. Cell Transplant. 2014. epub ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Shaimardanova, A.A.; Chulpanova, D.S.; Mullagulova, A.I.; Afawi, Z.; Gamirova, R.G.; Solovyeva, V.V.; Rizvanov, A.A. Gene and Cell Therapy for Epilepsy: A Mini Review. Front. Mol. Neurosci. 2022, 15, 868531. [Google Scholar] [CrossRef] [PubMed]
- Valayannopoulos, V. Enzyme Replacement Therapy and Substrate Reduction Therapy in Lysosomal Storage Disorders with Neurological Expression. In Handbook of Clinical Neurology; Elsevier: Philadelphia, PA, USA, 2013; Volume 113, pp. 1851–1857. [Google Scholar]
- Diaz, G.A.; Jones, S.A.; Scarpa, M.; Mengel, K.E.; Giugliani, R.; Guffon, N.; Batsu, I.; Fraser, P.A.; Li, J.; Zhang, Q.; et al. One-Year Results of a Clinical Trial of Olipudase Alfa Enzyme Replacement Therapy in Pediatric Patients with Acid Sphingomyelinase Deficiency. Genet. Med. 2021, 23, 1543–1550. [Google Scholar] [CrossRef]
- Marshall, J.; Nietupski, J.B.; Park, H.; Cao, J.; Bangari, D.S.; Silvescu, C.; Wilper, T.; Randall, K.; Tietz, D.; Wang, B.; et al. Substrate Reduction Therapy for Sandhoff Disease through Inhibition of Glucosylceramide Synthase Activity. Mol. Ther. 2019, 27, 1495–1506. [Google Scholar] [CrossRef] [PubMed]
- Hughes, D.A.; Deegan, P.; Giraldo, P.; Göker-Alpan, Ö.; Lau, H.; Lukina, E.; Revel-Vilk, S.; Scarpa, M.; Botha, J.; Gadir, N.; et al. Switching between Enzyme Replacement Therapies and Substrate Reduction Therapies in Patients with Gaucher Disease: Data from the Gaucher Outcome Survey (GOS). J. Clin. Med. 2022, 11, 5158. [Google Scholar] [CrossRef]
- Coutinho, M.; Santos, J.; Alves, S. Less Is More: Substrate Reduction Therapy for Lysosomal Storage Disorders. Int. J. Mol. Sci. 2016, 17, 1065. [Google Scholar] [CrossRef]
- Weidemann, F.; Jovanovic, A.; Herrmann, K.; Vardarli, I. Chaperone Therapy in Fabry Disease. Int. J. Mol. Sci. 2022, 23, 1887. [Google Scholar] [CrossRef] [PubMed]
- Chiricozzi, E.; Niemir, N.; Aureli, M.; Magini, A.; Loberto, N.; Prinetti, A.; Bassi, R.; Polchi, A.; Emiliani, C.; Caillaud, C.; et al. Chaperone Therapy for GM2 Gangliosidosis: Effects of Pyrimethamine on β-Hexosaminidase Activity in Sandhoff Fibroblasts. Mol. Neurobiol. 2014, 50, 159–167. [Google Scholar] [CrossRef]
- Suzuki, Y. Chemical Chaperone Therapy for GM1-Gangliosidosis. Cell. Mol. Life Sci. 2008, 65, 351–353. [Google Scholar] [CrossRef]
- Shaimardanova, A.A.; Solovyeva, V.V.; Issa, S.S.; Rizvanov, A.A. Gene Therapy of Sphingolipid Metabolic Disorders. Int. J. Mol. Sci. 2023, 24, 3627. [Google Scholar] [CrossRef]
- Issa, S.S.; Shaimardanova, A.A.; Valiullin, V.V.; Rizvanov, A.A.; Solovyeva, V.V. Mesenchymal Stem Cell-Based Therapy for Lysosomal Storage Diseases and Other Neurodegenerative Disorders. Front. Pharmacol. 2022, 13, 859516. [Google Scholar] [CrossRef]
- Shaimardanova, A.; Solovyeva, V.; Chulpanova, D.; James, V.; Kitaeva, K.; Rizvanov, A. Extracellular Vesicles in the Diagnosis and Treatment of Central Nervous System Diseases. Neural Regen. Res. 2020, 15, 586. [Google Scholar] [CrossRef] [PubMed]
- Israr, J.; Alam, S.; Kumar, A. Drug Repurposing for Rare Diseases. Prog. Mol. Biol. Transl. Sci. 2024, 207, 231–247. [Google Scholar] [CrossRef] [PubMed]
- Jonker, A.H.; O’Connor, D.; Cavaller-Bellaubi, M.; Fetro, C.; Gogou, M.; ’t Hoen, P.A.C.; de Kort, M.; Stone, H.; Valentine, N.; Pasmooij, A.M.G. Drug Repurposing for Rare: Progress and Opportunities for the Rare Disease Community. Front. Med. 2024, 11, 1352803. [Google Scholar] [CrossRef]
- Roessler, H.I.; Knoers, N.V.A.M.; van Haelst, M.M.; van Haaften, G. Drug Repurposing for Rare Diseases. Trends Pharmacol. Sci. 2021, 42, 255–267. [Google Scholar] [CrossRef]
- Hughes, D.A.; Pastores, G.M. Gaucher Disease. In GeneReviews® [Internet]; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Stirnemann, J.; Belmatoug, N.; Camou, F.; Serratrice, C.; Froissart, R.; Caillaud, C.; Levade, T.; Astudillo, L.; Serratrice, J.; Brassier, A.; et al. A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. Int. J. Mol. Sci. 2017, 18, 441. [Google Scholar] [CrossRef]
- Pineda, M.; Walterfang, M.; Patterson, M.C. Miglustat in Niemann-Pick Disease Type C Patients: A Review. Orphanet J. Rare Dis. 2018, 13, 140. [Google Scholar] [CrossRef] [PubMed]
- Sitarska, D.; Tylki-Szymańska, A.; Ługowska, A. Treatment Trials in Niemann-Pick Type C Disease. Metab. Brain Dis. 2021, 36, 2215–2221. [Google Scholar] [CrossRef]
- Canini, G.; Lo Cascio, E.; Della Longa, S.; Cecconi, F.; Arcovito, A. Human Glucosylceramide Synthase at Work as Provided by “In Silico” Molecular Docking, Molecular Dynamics, and Metadynamics. ACS Omega 2023, 8, 8755–8765. [Google Scholar] [CrossRef]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein−Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Dasmahapatra, U.; Kumar, C.K.; Das, S.; Subramanian, P.T.; Murali, P.; Isaac, A.E.; Ramanathan, K.; Mm, B.; Chanda, K. In-Silico Molecular Modelling, MM/GBSA Binding Free Energy and Molecular Dynamics Simulation Study of Novel Pyrido Fused Imidazo[4,5-c]Quinolines as Potential Anti-Tumor Agents. Front. Chem. 2022, 10, 991369. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA Methods to Estimate Ligand-Binding Affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Shukla, G.S.; Radin, N.S. Glucosylceramide Synthase of Mouse Kidney: Further Characterization with an Improved Assay Method. Arch. Biochem. Biophys. 1990, 283, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Kaufman, B.; Roseman, S. Enzymatic Synthesis of Ceramide-Glucose and Ceramide-Lactose by Glycosyltransferases from Embryonic Chicken Brain. J. Biol. Chem. 1968, 243, 5802–5804. [Google Scholar] [CrossRef]
- Marks, D.L.; Dominguez, M.; Wu, K.; Pagano, R.E. Identification of Active Site Residues in Glucosylceramide Synthase. J. Biol. Chem. 2001, 276, 26492–26498. [Google Scholar] [CrossRef] [PubMed]
- Wraith, J.E.; Imrie, J. New Therapies in the Management of Niemann-Pick Type C Disease: Clinical Utility of Miglustat. Ther. Clin. Risk Manag. 2009, 5, 877–887. [Google Scholar] [CrossRef]
- Butters, T.D.; van den Broek, L.A.G.M.; Fleet, G.W.J.; Krulle, T.M.; Wormald, M.R.; Dwek, R.A.; Platt, F.M. Molecular Requirements of Imino Sugars for the Selective Control of N-Linked Glycosylation and Glycosphingolipid Biosynthesis. Tetrahedron Asymmetry 2000, 11, 113–124. [Google Scholar] [CrossRef]
- Wishart, D.S. DrugBank: A Comprehensive Resource for in Silico Drug Discovery and Exploration. Nucleic Acids Res. 2006, 34, D668–D672. [Google Scholar] [CrossRef]
- Wu, K.; Marks, D.L.; Watanabe, R.; Paul, P.; Rajan, N.; Pagano, R.E. Histidine-193 of Rat Glucosylceramide Synthase Resides in a UDP-Glucose- and Inhibitor (D-Threo-1-Phenyl-2-Decanoylamino-3-Morpholinopropan-1-Ol)-Binding Region: A Biochemical and Mutational Study. Biochem. J. 1999, 341 Pt 2, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Padda, I.S.; Mahtani, A.U.; Parmar, M. Sodium-Glucose Transport Protein 2 (SGLT2) Inhibitors. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Sha, S.; Polidori, D.; Farrell, K.; Ghosh, A.; Natarajan, J.; Vaccaro, N.; Pinheiro, J.; Rothenberg, P.; Plum-Mörschel, L. Pharmacodynamic Differences between Canagliflozin and Dapagliflozin: Results of a Randomized, Double-Blind, Crossover Study. Diabetes Obes. Metab. 2015, 17, 188–197. [Google Scholar] [CrossRef]
- Li, R.; Manela, J.; Kong, Y.; Ladisch, S. Cellular Gangliosides Promote Growth Factor-Induced Proliferation of Fibroblasts. J. Biol. Chem. 2000, 275, 34213–34223. [Google Scholar] [CrossRef]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel Procedure for Modeling Ligand/Receptor Induced Fit Effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Friesner, R.A.; Xiang, Z.; Honig, B. On the Role of the Crystal Environment in Determining Protein Side-Chain Conformations. J. Mol. Biol. 2002, 320, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, M.P.; Kaminski, G.A.; Friesner, R.A.; Rapp, C.S. Force Field Validation Using Protein Side Chain Prediction. J. Phys. Chem. B 2002, 106, 11673–11680. [Google Scholar] [CrossRef]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem Substance and Compound Databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2023 Update. Nucleic Acids Res. 2023, 51, D1373–D1380. [Google Scholar] [CrossRef] [PubMed]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Khan, H.M.; MacKerell, A.D.; Reuter, N. Cation-π Interactions between Methylated Ammonium Groups and Tryptophan in the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2019, 15, 7–12. [Google Scholar] [CrossRef]
- Brooks, B.R.; Brooks, C.L.; Mackerell, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The Biomolecular Simulation Program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed]
- Wataha, J.C.; Hanks, C.T.; Craig, R.G. In Vitro Synergistic, Antagonistic, and Duration of Exposure Effects of Metal Cations on Eukaryotic Cells. J. Biomed. Mater. Res. 1992, 26, 1297–1309. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drugs | Zinc Code | Docking Score | Interactions | Residue |
|---|---|---|---|---|
| Labetalol | ZINC416 | −11.769 | H-bonds/ Salt bridge/ π-stacking | Asp144, Ile233, Glu235, Asp236, Arg275/Asp144, Glu235, Asp236, Arg275/Trp276 |
| Pitavastatin | ZINC1534965 | −11.305 | H-bonds/ Salt bridge/ π-stacking | Lys124, Glu235, Asp236, Arg275, Trp375/Trp276 |
| Macimorelin | ZINC1554197 | −11.081 | H-bonds/ Salt bridge/ π-stacking | Asp144, Ile233, Glu235, Asp236, Trp276/Asp144, Glu235, Asp236/Trp276 |
| Pravastatin | ZINC3798763 | −10.946 | H-bonds/ Salt bridge | Gly210, Glu235, Asp236, Trp276, Arg280/Lys124 |
| Ertugliflozin | ZINC68197809 | −10.525 | H-bonds/ π-stacking | His193, Glu291/Trp276 |
| Canagliflozin | ZINC43207238 | −10.244 | H-bonds | His193, Thr209, Gly210 |
| Nebivolol | ZINC5844792 | −10.153 | H-bonds/ Salt bridge/π-stacking | Asp144, Asp236/Asp144, Glu235, Asp236/Trp276 |
| Dapagliflozin | ZINC3819138 | −10.138 | H-bonds | His193, Thr209, Val208, Asp236, Arg275 |
| Benazepril | ZINC3781943 | −10.456 | H-bonds/ π-stacking | Asp236, Glu235, Gly210/Trp375 |
| Floctafenine | ZINC607872 | −9.241 | H-bonds/ π-stacking | His193, Glu291/Trp276 |
| Carvedilol | ZINC1530580 | −9.142 | H-bonds/ Salt bridge/ π-stacking | Asp144, Asp 236/Asp144, Glu235, Asp236/Trp276 |
| Drugs | Zinc Code | HB Donor | H-Bonds Donor Acceptor | Rule of Five | QPlogPo/w | Molecular Weight |
|---|---|---|---|---|---|---|
| Nebivolol | ZINC5844792 | 3 | 6.4 | 0 | 3.826 | 405.441 |
| Canagliflozin | ZINC43207238 | 4 | 8.5 | 0 | 3.249 | 444.517 |
| Dapagliflozin | ZINC3819138 | 4 | 9.250 | 0 | 2.236 | 408.878 |
| Carvedilol | ZINC1530580 | 3 | 5.450 | 0 | 4.208 | 406.480 |
| Ertugliflozin | ZINC68197809 | 4 | 9.050 | 0 | 2.458 | 436.888 |
| Floctafenine | ZINC607872 | 2 | 5.9 | 0 | 3.971 | 406.361 |
| Benazepril | ZINC3781943 | 2 | 8.5 | 0 | 1.715 | 424.496 |
| Pravastatin | ZINC3798763 | 3 | 8.1 | 0 | 3.351 | 424.533 |
| Pitavastatin | ZINC1534965 | 2 | 5.4 | 0 | 4.858 | 421.467 |
| Labetalol | ZINC416 | 4 | 5.450 | 0 | 2.874 | 328.410 |
| Drugs | Zinc Code | MM-GBSA (ΔGbind) |
|---|---|---|
| Nebivolol | ZINC5844792 | −67.8 |
| Canagliflozin | ZINC43207238 | −64.5 |
| Dapagliflozin | ZINC3819138 | −63.93 |
| Carvedilol | ZINC1530580 | −63.51 |
| Ertugliflozin | ZINC68197809 | −57.18 |
| Floctafenine | ZINC607872 | −53.58 |
| Benazepril | ZINC3781943 | −39.39 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Canini, G.; Mazzinelli, E.; Nocca, G.; Lattanzi, W.; Arcovito, A. Targeting Glucosylceramide Synthase: Innovative Drug Repurposing Strategies for Lysosomal Diseases. Int. J. Mol. Sci. 2025, 26, 2195. https://doi.org/10.3390/ijms26052195
Canini G, Mazzinelli E, Nocca G, Lattanzi W, Arcovito A. Targeting Glucosylceramide Synthase: Innovative Drug Repurposing Strategies for Lysosomal Diseases. International Journal of Molecular Sciences. 2025; 26(5):2195. https://doi.org/10.3390/ijms26052195
Chicago/Turabian StyleCanini, Giorgia, Elena Mazzinelli, Giuseppina Nocca, Wanda Lattanzi, and Alessandro Arcovito. 2025. "Targeting Glucosylceramide Synthase: Innovative Drug Repurposing Strategies for Lysosomal Diseases" International Journal of Molecular Sciences 26, no. 5: 2195. https://doi.org/10.3390/ijms26052195
APA StyleCanini, G., Mazzinelli, E., Nocca, G., Lattanzi, W., & Arcovito, A. (2025). Targeting Glucosylceramide Synthase: Innovative Drug Repurposing Strategies for Lysosomal Diseases. International Journal of Molecular Sciences, 26(5), 2195. https://doi.org/10.3390/ijms26052195