
Stress-Related LncRNAs and Their Roles in Diabetes and Diabetic Complications

Abstract
1. Introduction
2. Classification and Functional Mechanisms of LncRNAs
3. Stresses and Stress-Associated LncRNAs
3.1. Endoplasmic Reticulum Stress and Major Cellular Signaling Pathways
3.2. Oxidative Stress and Major Cellular Signaling Pathways
3.3. Stresses in Diabetes and Diabetic Complications
4. Stress-Associated LncRNAs Implicated in Diabetes
4.1. LncRNAs Implicated in Diabetes via Regulation of Oxidative Stress
4.2. LncRNAs Implicated in Diabetes via Regulation of ER Stress
5. The Role of LncRNAs in Diabetes and Diabetic Complications
5.1. LncRNAs Related to Pancreatic β Cell Function
5.2. LncRNAs Related to Insulin Resistance
5.3. LncRNAs Related to Diabetic Complications
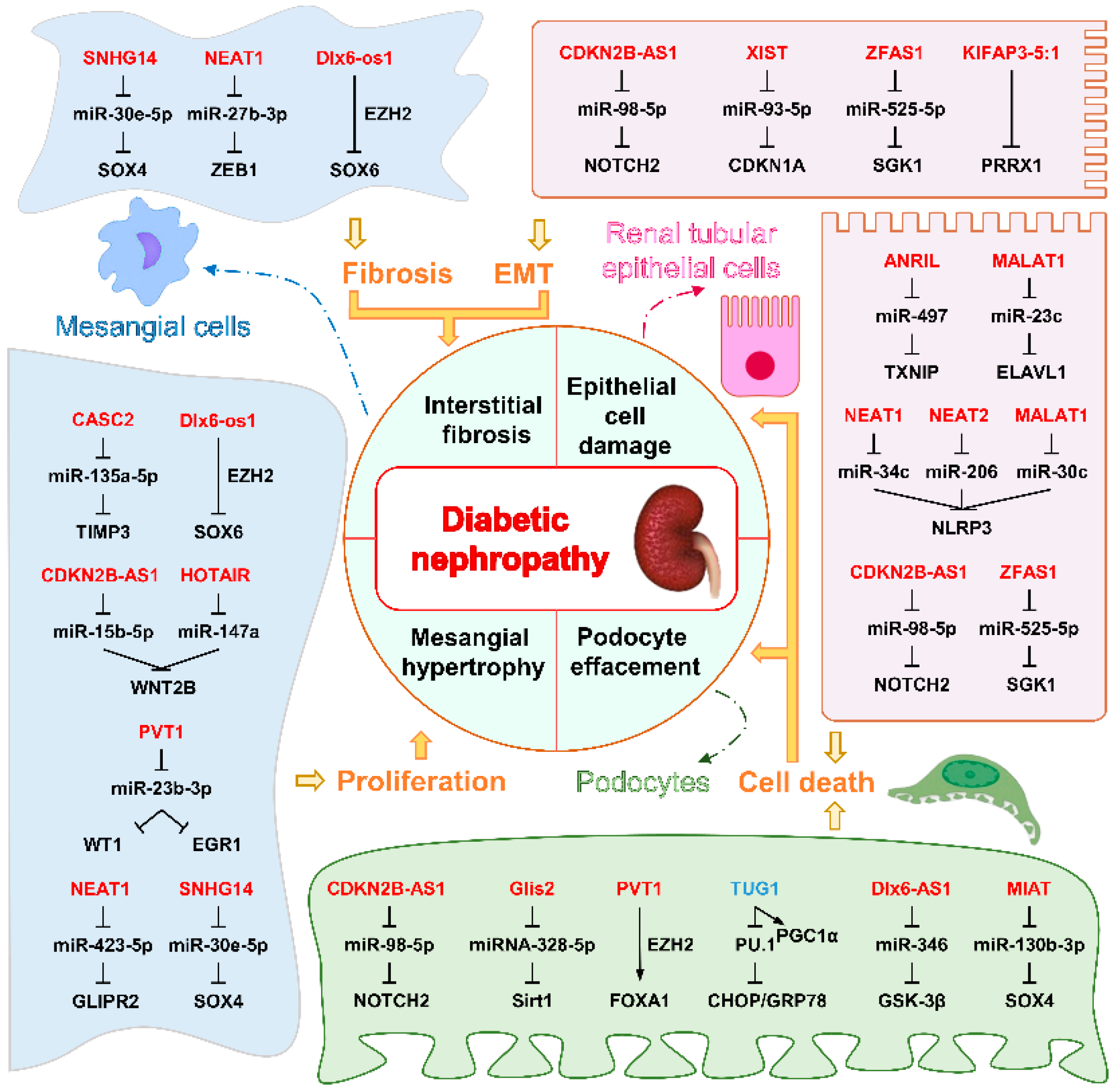
5.3.1. LncRNAs Associated with DN
5.3.2. LncRNAs Associated with DR
6. Conclusions and Further Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ANRIL | Antisense non-coding RNA in the INK4 locus |
| ASMER-1/2 | Adipocyte-specific metabolic-related lncRNA 1/2 |
| betaFaar | β cell function and apoptosis regulator |
| BGLs | Blood glucose levels |
| BSA | Bovine serum albumin |
| CaM | Calmodulin |
| CASC2 | Cancer susceptibility candidate 2 |
| CDKN2B-AS1 | Cyclin-dependent kinase inhibitor 2B antisense RNA 1 |
| ceRNA | Competitive endogenous RNA |
| circRNAs | Circular RNAs |
| DC | Diabetic cataract |
| DCM | Diabetic cardiomyopathy |
| DLX6-AS1 (Dlx6-os1 in mice) | distal-less homeobox 6 (DLX6) antisense RNA 1 |
| DN | Diabetic nephropathy |
| DR | Diabetic retinopathy |
| ECM | Extracellular matrix |
| EGR1 | Early growth response factor 1 |
| EndMT | Endothelial–mesenchymal transition |
| ER | Endoplasmic reticulum |
| EMT | Epithelial–mesenchymal transition |
| ETC | Electron transport chain |
| EZH2 | Zeste homolog 2 |
| ERAD | ER-associated degradation |
| FOXA1 | Forkhead box A1 |
| GAS5 | Growth stabilization specific transcript, |
| GLIPR2 | Glioma pathogenesis related-2 |
| Glis2 | ENSMUST00000122896 |
| Gomafu | also referred to as RNCR2/MIAT |
| HFD | High-fat diet |
| HG | High glucose |
| HGMCs | Human glomerular mesangial cells |
| hnRNPA1 | Heterogeneous nuclear ribonucleoprotein A1 |
| IGF2BP2 | Insulin like growth factor 2 mRNA binding protein 2 |
| IR | Insulin resistance |
| KO | Knockout |
| MALAT1 | Metastasis-associated lung adenocarcinoma transcript 1 |
| MDA | Malondialdehyde |
| MEG3 | Maternally expressed gene 3 |
| MGC | Megacluster of nearly 40 microRNAs and their host long non-coding RNA transcript |
| MME | Membrane metallo-endopeptidase |
| NEAT1 | nuclear paraspeckle assembly transcript 1 |
| NLRP3 | NOD-like receptor thermal protein domain associated protein 3 |
| OS | Oxidative stress |
| PEG | Paternally expressed genes |
| PRKCB2 | Protein kinase C beta 2 |
| PVT1 | Plasmacytoma variant translocation 1 |
| ROS | Reactive oxygen species |
| RTN1 | Reticulon-1 |
| SGK1 | Serum- and glucocorticoid-regulated kinase 1, Sirt1, Sirtuin1 |
| SHGL | Suppressor of hepatic gluconeogenesis and lipogenesis in mice, B4GALT1-AS1 in human |
| SNHG14 | Small nucleolar host gene 14 |
| SOX6/4 | SRY-related high-mobility-group box 6/4 |
| SREBP-2 | Sterol regulatory element binding protein 2 |
| STZ | Streptozotocin |
| TRAF3IP2 | Tumor necrosis factor receptor-associated factor 3 interacting protein 2 |
| TUG1 | Taurine upregulated gene 1 |
| TUNAR | TCL1 upstream neural differentiation-associated RNA |
| TXNIP | Thioredoxin-interacting protein |
| T1DM | Type 1 diabetes mellitus |
| T2DM | Type 2 diabetes mellitus |
| UPR | Unfolded protein response |
| WAT | White adipose tissue |
| WNT2B | Wingless-type family member 2B |
| WT1 | Wilms tumor protein 1 |
| XIST | X inactive specific transcript |
| ZEB1 | zinc finger E-box binding homeobox 1 |
| ZFAS1 | ZNFX1 antisense RNA 1 |
References
- GBD 2021 Diabetes Collaborators. Global, regional, and national burden of diabetes from 1990 to 2021, with projections of prevalence to 2050: A systematic analysis for the Global Burden of Disease Study 2021. Lancet 2023, 402, 203–234. [Google Scholar] [CrossRef] [PubMed]
- Palazzo, A.F.; Lee, E.S. Non-coding RNA: What is functional and what is junk? Front. Genet. 2015, 6, 2. [Google Scholar] [CrossRef]
- Mudge, J.M.; Carbonell-Sala, S.; Diekhans, M.; Martinez, J.G.; Hunt, T.; Jungreis, I.; Loveland, J.E.; Arnan, C.; Barnes, I.; Bennett, R.; et al. GENCODE 2025: Reference gene annotation for human and mouse. Nucleic Acids Res. 2024, 53, D966–D975. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, W.; Zhu, W.; Dong, J.; Cheng, Y.; Yin, Z.; Shen, F. Mechanisms and Functions of Long Non-Coding RNAs at Multiple Regulatory Levels. Int. J. Mol. Sci. 2019, 20, 5573. [Google Scholar] [CrossRef]
- Lemmer, I.L.; Willemsen, N.; Hilal, N.; Bartelt, A. A guide to understanding endoplasmic reticulum stress in metabolic disorders. Mol. Metab. 2021, 47, 101169. [Google Scholar] [CrossRef]
- Raut, S.K.; Khullar, M. Oxidative stress in metabolic diseases: Current scenario and therapeutic relevance. Mol. Cell. Biochem. 2023, 478, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Vijayalalitha, R.; Archita, T.; Juanitaa, G.R.; Jayasuriya, R.; Amin, K.N.; Ramkumar, K.M. Role of Long Non-Coding RNA in Regulating ER Stress Response to the Progression of Diabetic Complications. Curr. Gene Ther. 2023, 23, 96–110. [Google Scholar] [CrossRef]
- Hussein, R.M. Long non-coding RNAs: The hidden players in diabetes mellitus-related complications. Diabetes Metab. Syndr. 2023, 17, 102872. [Google Scholar] [CrossRef]
- Yang, Q.; Fang, D.; Chen, J.; Hu, S.; Chen, N.; Jiang, J.; Zeng, M.; Luo, M. LncRNAs associated with oxidative stress in diabetic wound healing: Regulatory mechanisms and application prospects. Theranostics 2023, 13, 3655–3674. [Google Scholar] [CrossRef]
- Yang, Y.; Cheng, H. Emerging Roles of ncRNAs in Type 2 Diabetes Mellitus: From Mechanisms to Drug Discovery. Biomolecules 2024, 14, 1364. [Google Scholar] [CrossRef]
- Qin, T.; Li, J.; Zhang, K.Q. Structure, Regulation, and Function of Linear and Circular Long Non-Coding RNAs. Front. Genet. 2020, 11, 150. [Google Scholar] [CrossRef] [PubMed]
- Ponting, C.P.; Oliver, P.L.; Reik, W. Evolution and functions of long noncoding RNAs. Cell 2009, 136, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Mattick, J.S.; Amaral, P.P.; Carninci, P.; Carpenter, S.; Chang, H.Y.; Chen, L.L.; Chen, R.; Dean, C.; Dinger, M.E.; Fitzgerald, K.A.; et al. Long non-coding RNAs: Definitions, functions, challenges and recommendations. Nat. Rev. Mol. Cell Biol. 2023, 24, 430–447. [Google Scholar] [CrossRef]
- Grammatikakis, I.; Lal, A. Significance of lncRNA abundance to function. Mamm. Genome 2022, 33, 271–280. [Google Scholar] [CrossRef]
- Meola, N.; Domanski, M.; Karadoulama, E.; Chen, Y.; Gentil, C.; Pultz, D.; Vitting-Seerup, K.; Lykke-Andersen, S.; Andersen, J.S.; Sandelin, A.; et al. Identification of a Nuclear Exosome Decay Pathway for Processed Transcripts. Mol. Cell 2016, 64, 520–533. [Google Scholar] [CrossRef]
- Tan, K.; Stupack, D.G.; Wilkinson, M.F. Nonsense-mediated RNA decay: An emerging modulator of malignancy. Nat. Rev. Cancer 2022, 22, 437–451. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhu, P.; Lu, T.; Du, Y.; Wang, Y.; He, L.; Ye, B.; Liu, B.; Yang, L.; Wang, J.; et al. The long non-coding RNA LncHDAC2 drives the self-renewal of liver cancer stem cells via activation of Hedgehog signaling. J. Hepatol. 2019, 70, 918–929. [Google Scholar] [CrossRef]
- Wang, Y.; Zhu, P.; Luo, J.; Wang, J.; Liu, Z.; Wu, W.; Du, Y.; Ye, B.; Wang, D.; He, L.; et al. LncRNA HAND2-AS1 promotes liver cancer stem cell self-renewal via BMP signaling. EMBO J. 2019, 38, e101110. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, Z.; Trottier, J.; Barbier, O.; Wang, L. Long noncoding RNA MEG3 induces cholestatic liver injury by interaction with PTBP1 to facilitate shp mRNA decay. Hepatology 2017, 65, 604–615. [Google Scholar] [CrossRef]
- Zhang, H.; Xing, Z.; Mani, S.K.; Bancel, B.; Durantel, D.; Zoulim, F.; Tran, E.J.; Merle, P.; Andrisani, O. RNA helicase DEAD box protein 5 regulates Polycomb repressive complex 2/Hox transcript antisense intergenic RNA function in hepatitis B virus infection and hepatocarcinogenesis. Hepatology 2016, 64, 1033–1048. [Google Scholar] [CrossRef]
- Yan, C.; Li, J.; Feng, S.; Li, Y.; Tan, L. Long noncoding RNA Gomafu upregulates Foxo1 expression to promote hepatic insulin resistance by sponging miR-139-5p. Cell Death Dis. 2018, 9, 289. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Zheng, Y.; Sun, Y.; Lai, M.; Qiu, J.; Gui, F.; Zeng, Q.; Liu, F. Suppressing long noncoding RNA OGRU ameliorates diabetic retinopathy by inhibition of oxidative stress and inflammation via miR-320/USP14 axis. Free Radic. Biol. Med. 2021, 169, 361–381. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chang, Y.; Liu, Y.; Liu, B.; Zhen, J.; Li, X.; Lin, J.; Yu, Q.; Lv, Z.; Wang, R. Inhibition of the lncRNA MIAT prevents podocyte injury and mitotic catastrophe in diabetic nephropathy. Mol. Ther. Nucleic Acids 2022, 28, 136–153. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, X.; Zhou, J.; Hu, J.; Zhang, D.; Liu, J.; Qiao, Y.; Zhan, Q. Long noncoding RNA HULC modulates the phosphorylation of YB-1 through serving as a scaffold of extracellular signal-regulated kinase and YB-1 to enhance hepatocarcinogenesis. Hepatology 2017, 65, 1612–1627. [Google Scholar] [CrossRef]
- Wang, W.T.; Ye, H.; Wei, P.P.; Han, B.W.; He, B.; Chen, Z.H.; Chen, Y.Q. LncRNAs H19 and HULC, activated by oxidative stress, promote cell migration and invasion in cholangiocarcinoma through a ceRNA manner. J. Hematol. Oncol. 2016, 9, 117. [Google Scholar] [CrossRef]
- Chen, X.; Shi, C.; He, M.; Xiong, S.; Xia, X. Endoplasmic reticulum stress: Molecular mechanism and therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 352. [Google Scholar] [CrossRef]
- Afzal, S.; Abdul Manap, A.S.; Attiq, A.; Albokhadaim, I.; Kandeel, M.; Alhojaily, S.M. From imbalance to impairment: The central role of reactive oxygen species in oxidative stress-induced disorders and therapeutic exploration. Front. Pharmacol. 2023, 14, 1269581. [Google Scholar] [CrossRef]
- Wei, Y.; Giunta, S.; Xia, S. Hypoxia in Aging and Aging-Related Diseases: Mechanism and Therapeutic Strategies. Int. J. Mol. Sci. 2022, 23, 8165. [Google Scholar] [CrossRef]
- Wang, J.; Takeuchi, T.; Tanaka, S.; Kubo, S.K.; Kayo, T.; Lu, D.; Takata, K.; Koizumi, A.; Izumi, T. A mutation in the insulin 2 gene induces diabetes with severe pancreatic beta-cell dysfunction in the Mody mouse. J. Clin. Investig. 1999, 103, 27–37. [Google Scholar] [CrossRef]
- Ni, L.; Yuan, C.; Wu, X. Endoplasmic Reticulum Stress in Diabetic Nephrology: Regulation, Pathological Role, and Therapeutic Potential. Oxid. Med. Cell Longev. 2021, 2021, 7277966. [Google Scholar] [CrossRef]
- Caturano, A.; D’Angelo, M.; Mormone, A.; Russo, V.; Mollica, M.P.; Salvatore, T.; Galiero, R.; Rinaldi, L.; Vetrano, E.; Marfella, R.; et al. Oxidative Stress in Type 2 Diabetes: Impacts from Pathogenesis to Lifestyle Modifications. Curr. Issues Mol. Biol. 2023, 45, 6651–6666. [Google Scholar] [CrossRef] [PubMed]
- Garg, S.S.; Gupta, J. Polyol pathway and redox balance in diabetes. Pharmacol. Res. 2022, 182, 106326. [Google Scholar] [CrossRef]
- Bhatti, J.S.; Sehrawat, A.; Mishra, J.; Sidhu, I.S.; Navik, U.; Khullar, N.; Kumar, S.; Bhatti, G.K.; Reddy, P.H. Oxidative stress in the pathophysiology of type 2 diabetes and related complications: Current therapeutics strategies and future perspectives. Free Radic. Biol. Med. 2022, 184, 114–134. [Google Scholar] [CrossRef]
- Cai, R.; Jiang, J. LncRNA ANRIL Silencing Alleviates High Glucose-Induced Inflammation, Oxidative Stress, and Apoptosis via Upregulation of MME in Podocytes. Inflammation 2020, 43, 2147–2155. [Google Scholar] [CrossRef]
- Wang, G.; Wu, B.; Zhang, B.; Wang, K.; Wang, H. LncRNA CTBP1-AS2 alleviates high glucose-induced oxidative stress, ECM accumulation, and inflammation in diabetic nephropathy via miR-155-5p/FOXO1 axis. Biochem. Biophys. Res. Commun. 2020, 532, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Li, L.; Xiao, F.; Fu, J. LncRNA FLG-AS1 Mitigates Diabetic Retinopathy by Regulating Retinal Epithelial Cell Inflammation, Oxidative Stress, and Apoptosis via miR-380-3p/SOCS6 Axis. Inflammation 2022, 45, 1936–1949. [Google Scholar] [CrossRef]
- Jiang, L.; Wang, C.; Shen, X. LncRNA GAS5 suppresses ER stress-induced apoptosis and inflammation by regulating SERCA2b in HG-treated retinal epithelial cell. Mol. Med. Rep. 2020, 22, 1072–1080. [Google Scholar] [CrossRef]
- Luo, R.; Xiao, F.; Wang, P.; Hu, Y.X. lncRNA H19 sponging miR-93 to regulate inflammation in retinal epithelial cells under hyperglycemia via XBP1s. Inflamm. Res. 2020, 69, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Geng, J.; Li, X.; Wan, J.; Liu, J.; Zhou, Z.; Liu, X. Long Noncoding RNA LINC01619 Regulates MicroRNA-27a/Forkhead Box Protein O1 and Endoplasmic Reticulum Stress-Mediated Podocyte Injury in Diabetic Nephropathy. Antioxid. Redox Signal. 2018, 29, 355–376. [Google Scholar] [CrossRef]
- Kato, M.; Wang, M.; Chen, Z.; Bhatt, K.; Oh, H.J.; Lanting, L.; Deshpande, S.; Jia, Y.; Lai, J.Y.C.; O’Connor, C.L.; et al. An endoplasmic reticulum stress-regulated lncRNA hosting a microRNA megacluster induces early features of diabetic nephropathy. Nat. Comm. 2016, 7, 12864. [Google Scholar] [CrossRef]
- Huang, Q.; Qiu, T.; Chen, H.; Tian, T.; Wang, D.; Lu, C. Silencing LncRNA SNHG14 alleviates renal tubular injury via the miR-483-5p/HDAC4 axis in diabetic kidney disease. Hormones 2024. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Yi, P.; Wang, N.; Song, M.; Li, W.; Zheng, Y. LncRNA TUG1/miR-29c-3p/SIRT1 axis regulates endoplasmic reticulum stress-mediated renal epithelial cells injury in diabetic nephropathy model in vitro. PLoS ONE 2021, 16, e0252761. [Google Scholar] [CrossRef] [PubMed]
- Geng, Z.; Dong, B.; Lv, W.; Wang, Z.; Wang, X.; Huang, Y.; Wang, Y.; Xu, L. LncRNA ZFAS1 regulates the proliferation, oxidative stress, fibrosis, and inflammation of high glucose-induced human mesangial cells via the miR-588/ROCK1 axis. Diabetol. Metab. Syndr. 2022, 14, 21. [Google Scholar] [CrossRef]
- Dai, W.; Lee, D. Interfering with long chain noncoding RNA ANRIL expression reduces heart failure in rats with diabetes by inhibiting myocardial oxidative stress. J. Cell Biochem. 2019, 120, 18446–18456. [Google Scholar] [CrossRef]
- Xie, C.; Wu, W.; Tang, A.; Luo, N.; Tan, Y. lncRNA GAS5/miR-452-5p Reduces Oxidative Stress and Pyroptosis of High-Glucose-Stimulated Renal Tubular Cells. Diabetes Metab. Syndr. Obes. 2019, 12, 2609–2617. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Wang, X.; Guo, S.; Xiao, L.; Liang, C.; Wang, Z.; Li, Y.; Liu, Y.; Yao, R.; Liu, Y.; et al. LncRNA HOTAIR functions as a competing endogenous RNA to upregulate SIRT1 by sponging miR-34a in diabetic cardiomyopathy. J. Cell Physiol. 2019, 234, 4944–4958. [Google Scholar] [CrossRef]
- Gong, W.; Zhu, G.; Li, J.; Yang, X. LncRNA MALAT1 promotes the apoptosis and oxidative stress of human lens epithelial cells via p38MAPK pathway in diabetic cataract. Diabetes Res. Clin. Pract. 2018, 144, 314–321. [Google Scholar] [CrossRef]
- Kato, M.; Abdollahi, M.; Omori, K.; Malek, V.; Lanting, L.; Kandeel, F.; Rawson, J.; Tsark, W.; Zhang, L.; Wang, M.; et al. Lowering an ER stress-regulated long noncoding RNA protects mice from diabetes and isolated pancreatic islets from cell death. Mol. Ther. Nucleic Acids 2024, 35, 102252. [Google Scholar] [CrossRef]
- Meng, D.; Wu, L.; Li, Z.; Ma, X.; Zhao, S.; Zhao, D.; Qin, G. LncRNA TUG1 ameliorates diabetic nephropathy via inhibition of PU.1/RTN1 signaling pathway. J. Leukoc. Biol. 2022, 111, 553–562. [Google Scholar] [CrossRef]
- Barnett, R. Type 1 diabetes. Lancet 2018, 391, 195. [Google Scholar] [CrossRef]
- Wang, J.; Wang, H. Oxidative Stress in Pancreatic Beta Cell Regeneration. Oxid. Med. Cell Longev. 2017, 2017, 1930261. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yang, X.; Zhang, J. Bridges between mitochondrial oxidative stress, ER stress and mTOR signaling in pancreatic beta cells. Cell. Signal. 2016, 28, 1099–1104. [Google Scholar] [CrossRef]
- Ding, G.L.; Wang, F.F.; Shu, J.; Tian, S.; Jiang, Y.; Zhang, D.; Wang, N.; Luo, Q.; Zhang, Y.; Jin, F.; et al. Transgenerational glucose intolerance with Igf2/H19 epigenetic alterations in mouse islet induced by intrauterine hyperglycemia. Diabetes 2012, 61, 1133–1142. [Google Scholar] [CrossRef]
- Fadista, J.; Vikman, P.; Laakso, E.O.; Mollet, I.G.; Esguerra, J.L.; Taneera, J.; Storm, P.; Osmark, P.; Ladenvall, C.; Prasad, R.B.; et al. Global genomic and transcriptomic analysis of human pancreatic islets reveals novel genes influencing glucose metabolism. Proc. Natl. Acad. Sci. USA 2014, 111, 13924–13929. [Google Scholar] [CrossRef]
- Arnes, L.; Akerman, I.; Balderes, D.A.; Ferrer, J.; Sussel, L. betalinc1 encodes a long noncoding RNA that regulates islet beta-cell formation and function. Genes. Dev. 2016, 30, 502–507. [Google Scholar] [CrossRef] [PubMed]
- Moran, I.; Akerman, I.; van de Bunt, M.; Xie, R.; Benazra, M.; Nammo, T.; Arnes, L.; Nakic, N.; Garcia-Hurtado, J.; Rodriguez-Segui, S.; et al. Human beta cell transcriptome analysis uncovers lncRNAs that are tissue-specific, dynamically regulated, and abnormally expressed in type 2 diabetes. Cell Metab. 2012, 16, 435–448. [Google Scholar] [CrossRef] [PubMed]
- Voight, B.F.; Scott, L.J.; Steinthorsdottir, V.; Morris, A.P.; Dina, C.; Welch, R.P.; Zeggini, E.; Huth, C.; Aulchenko, Y.S.; Thorleifsson, G.; et al. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat. Genet. 2010, 42, 579–589. [Google Scholar] [CrossRef]
- Akerman, I.; Tu, Z.; Beucher, A.; Rolando, D.M.Y.; Sauty-Colace, C.; Benazra, M.; Nakic, N.; Yang, J.; Wang, H.; Pasquali, L.; et al. Human Pancreatic beta Cell lncRNAs Control Cell-Specific Regulatory Networks. Cell Metab. 2017, 25, 400–411. [Google Scholar] [CrossRef]
- Zhang, F.; Yang, Y.; Chen, X.; Liu, Y.; Hu, Q.; Huang, B.; Liu, Y.; Pan, Y.; Zhang, Y.; Liu, D.; et al. The long non-coding RNA betaFaar regulates islet beta-cell function and survival during obesity in mice. Nat. Commun. 2021, 12, 3997. [Google Scholar] [CrossRef]
- Zhou, A.X.; Mondal, T.; Tabish, A.M.; Abadpour, S.; Ericson, E.; Smith, D.M.; Knoll, R.; Scholz, H.; Kanduri, C.; Tyrberg, B.; et al. The long noncoding RNA TUNAR modulates Wnt signaling and regulates human beta-cell proliferation. Am. J. Physiol. Endocrinol. Metab. 2021, 320, E846–E857. [Google Scholar] [CrossRef]
- Xu, Y.; Mao, S.; Fan, H.; Wan, J.; Wang, L.; Zhang, M.; Zhu, S.; Yuan, J.; Lu, Y.; Wang, Z.; et al. LINC MIR503HG Controls SC-beta Cell Differentiation and Insulin Production by Targeting CDH1 and HES1. Adv. Sci. 2024, 11, e2305631. [Google Scholar] [CrossRef] [PubMed]
- Khetan, S.; Kales, S.; Kursawe, R.; Jillette, A.; Ulirsch, J.C.; Reilly, S.K.; Ucar, D.; Tewhey, R.; Stitzel, M.L. Functional characterization of T2D-associated SNP effects on baseline and ER stress-responsive beta cell transcriptional activation. Nat. Commun. 2021, 12, 5242. [Google Scholar] [CrossRef]
- de Goede, O.M.; Nachun, D.C.; Ferraro, N.M.; Gloudemans, M.J.; Rao, A.S.; Smail, C.; Eulalio, T.Y.; Aguet, F.; Ng, B.; Xu, J.; et al. Population-scale tissue transcriptomics maps long non-coding RNAs to complex disease. Cell 2021, 184, 2633–2648.e19. [Google Scholar] [CrossRef]
- Guo, W.H.; Guo, Q.; Liu, Y.L.; Yan, D.D.; Jin, L.; Zhang, R.; Yan, J.; Luo, X.H.; Yang, M. Mutated lncRNA increase the risk of type 2 diabetes by promoting beta cell dysfunction and insulin resistance. Cell Death Dis. 2022, 13, 904. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.E.; Newgard, C.B. Mechanisms controlling pancreatic islet cell function in insulin secretion. Nat. Rev. Mol. Cell Biol. 2021, 22, 142–158. [Google Scholar] [CrossRef]
- Gao, H.; Kerr, A.; Jiao, H.; Hon, C.C.; Ryden, M.; Dahlman, I.; Arner, P. Long Non-Coding RNAs Associated with Metabolic Traits in Human White Adipose Tissue. EBioMedicine 2018, 30, 248–260. [Google Scholar] [CrossRef] [PubMed]
- Kerr, A.G.; Wang, Z.; Wang, N.; Kwok, K.H.M.; Jalkanen, J.; Ludzki, A.; Lecoutre, S.; Langin, D.; Bergo, M.O.; Dahlman, I.; et al. The long noncoding RNA ADIPINT regulates human adipocyte metabolism via pyruvate carboxylase. Nat. Commun. 2022, 13, 2958. [Google Scholar] [CrossRef]
- Lu, Y.; Qie, D.; Yang, F.; Wu, J. LncRNA MEG3 aggravates adipocyte inflammation and insulin resistance by targeting IGF2BP2 to activate TLR4/NF-kappaB signaling pathway. Int. Immunopharmacol. 2023, 121, 110467. [Google Scholar] [CrossRef]
- Daneshmoghadam, J.; Omidifar, A.; Akbari Dilmaghani, N.; Karimi, Z.; Emamgholipour, S.; Shanaki, M. The gene expression of long non-coding RNAs (lncRNAs): MEG3 and H19 in adipose tissues from obese women and its association with insulin resistance and obesity indices. J. Clin. Lab. Anal. 2021, 35, e23741. [Google Scholar] [CrossRef]
- Chen, D.L.; Shen, D.Y.; Han, C.K.; Tian, Y. LncRNA MEG3 aggravates palmitate-induced insulin resistance by regulating miR-185-5p/Egr2 axis in hepatic cells. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 5456–5467. [Google Scholar] [CrossRef]
- Zhu, X.; Wu, Y.B.; Zhou, J.; Kang, D.M. Upregulation of lncRNA MEG3 promotes hepatic insulin resistance via increasing FoxO1 expression. Biochem. Biophys. Res. Commun. 2016, 469, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Li, H.; Wu, Y.; Zhou, J.; Yang, G.; Wang, W. lncRNA MEG3 promotes hepatic insulin resistance by serving as a competing endogenous RNA of miR-214 to regulate ATF4 expression. Int. J. Mol. Med. 2019, 43, 345–357. [Google Scholar] [CrossRef]
- Eissmann, M.; Gutschner, T.; Hammerle, M.; Gunther, S.; Caudron-Herger, M.; Gross, M.; Schirmacher, P.; Rippe, K.; Braun, T.; Zornig, M.; et al. Loss of the abundant nuclear non-coding RNA MALAT1 is compatible with life and development. RNA Biol. 2012, 9, 1076–1087. [Google Scholar] [CrossRef] [PubMed]
- Miyagawa, R.; Tano, K.; Mizuno, R.; Nakamura, Y.; Ijiri, K.; Rakwal, R.; Shibato, J.; Masuo, Y.; Mayeda, A.; Hirose, T.; et al. Identification of cis- and trans-acting factors involved in the localization of MALAT-1 noncoding RNA to nuclear speckles. RNA 2012, 18, 738–751. [Google Scholar] [CrossRef]
- Nakagawa, S.; Ip, J.Y.; Shioi, G.; Tripathi, V.; Zong, X.; Hirose, T.; Prasanth, K.V. Malat1 is not an essential component of nuclear speckles in mice. RNA 2012, 18, 1487–1499. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Chen, J.; Chen, N. Long noncoding RNA MALAT1 promotes hepatic steatosis and insulin resistance by increasing nuclear SREBP-1c protein stability. Sci. Rep. 2016, 6, 22640. [Google Scholar] [CrossRef]
- Chen, J.; Ke, S.; Zhong, L.; Wu, J.; Tseng, A.; Morpurgo, B.; Golovko, A.; Wang, G.; Cai, J.J.; Ma, X.; et al. Long noncoding RNA MALAT1 regulates generation of reactive oxygen species and the insulin responses in male mice. Biochem. Pharmacol. 2018, 152, 94–103. [Google Scholar] [CrossRef]
- Wang, J.; Yang, W.; Chen, Z.; Chen, J.; Meng, Y.; Feng, B.; Sun, L.; Dou, L.; Li, J.; Cui, Q.; et al. Long Noncoding RNA lncSHGL Recruits hnRNPA1 to Suppress Hepatic Gluconeogenesis and Lipogenesis. Diabetes 2018, 67, 581–593. [Google Scholar] [CrossRef]
- Gui, W.; Zhu, W.F.; Zhu, Y.; Tang, S.; Zheng, F.; Yin, X.; Lin, X.; Li, H. LncRNAH19 improves insulin resistance in skeletal muscle by regulating heterogeneous nuclear ribonucleoprotein A1. Cell Commun. Signal. 2020, 18, 173. [Google Scholar] [CrossRef]
- Gao, Y.; Wu, F.; Zhou, J.; Yan, L.; Jurczak, M.J.; Lee, H.Y.; Yang, L.; Mueller, M.; Zhou, X.B.; Dandolo, L.; et al. The H19/let-7 double-negative feedback loop contributes to glucose metabolism in muscle cells. Nucleic Acids Res. 2014, 42, 13799–13811. [Google Scholar] [CrossRef]
- Geng, T.; Liu, Y.; Xu, Y.; Jiang, Y.; Zhang, N.; Wang, Z.; Carmichael, G.G.; Taylor, H.S.; Li, D.; Huang, Y. H19 lncRNA Promotes Skeletal Muscle Insulin Sensitivity in Part by Targeting AMPK. Diabetes 2018, 67, 2183–2198. [Google Scholar] [CrossRef]
- Liao, W.; Xu, N.; Zhang, H.; Liao, W.; Wang, Y.; Wang, S.; Zhang, S.; Jiang, Y.; Xie, W.; Zhang, Y. Persistent high glucose induced EPB41L4A-AS1 inhibits glucose uptake via GCN5 mediating crotonylation and acetylation of histones and non-histones. Clin. Transl. Med. 2022, 12, e699. [Google Scholar] [CrossRef]
- Tang, X.; Qin, Q.; Xu, W.; Zhang, X. Long Non-Coding RNA TUG1 Attenuates Insulin Resistance in Mice with Gestational Diabetes Mellitus via Regulation of the MicroRNA-328-3p/SREBP-2/ERK Axis. Diabetes Metab. J. 2023, 47, 267–286. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.; Dhaouadi, I.; Gaziano, I.; Oliverio, M.; Klemm, P.; Awazawa, M.; Mitterer, G.; Fernandez-Rebollo, E.; Pradas-Juni, M.; Wagner, W.; et al. LincRNA H19 protects from dietary obesity by constraining expression of monoallelic genes in brown fat. Nat. Commun. 2018, 9, 3622. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.X.; Zheng, F.; Xie, K.L.; Xie, M.R.; Jiang, L.J.; Cai, Y. Exercise Reduces Insulin Resistance in Type 2 Diabetes Mellitus via Mediating the lncRNA MALAT1/MicroRNA-382-3p/Resistin Axis. Mol. Ther. Nucleic Acids 2019, 18, 34–44. [Google Scholar] [CrossRef]
- Giri, B.; Dey, S.; Das, T.; Sarkar, M.; Banerjee, J.; Dash, S.K. Chronic hyperglycemia mediated physiological alteration and metabolic distortion leads to organ dysfunction, infection, cancer progression and other pathophysiological consequences: An update on glucose toxicity. Biomed. Pharmacother. 2018, 107, 306–328. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Liu, Q.; He, J.; Li, Y. Immune responses in diabetic nephropathy: Pathogenic mechanisms and therapeutic target. Front. Immunol. 2022, 13, 958790. [Google Scholar] [CrossRef]
- Long, J.; Badal, S.S.; Ye, Z.; Wang, Y.; Ayanga, B.A.; Galvan, D.L.; Green, N.H.; Chang, B.H.; Overbeek, P.A.; Danesh, F.R. Long noncoding RNA Tug1 regulates mitochondrial bioenergetics in diabetic nephropathy. J. Clin. Investig. 2016, 126, 4205–4218. [Google Scholar] [CrossRef]
- Long, J.; Galvan, D.L.; Mise, K.; Kanwar, Y.S.; Li, L.; Poungavrin, N.; Overbeek, P.A.; Chang, B.H.; Danesh, F.R. Role for carbohydrate response element-binding protein (ChREBP) in high glucose-mediated repression of long noncoding RNA Tug1. J. Biol. Chem. 2020, 295, 15840–15852. [Google Scholar] [CrossRef]
- Li, L.; Long, J.; Mise, K.; Galvan, D.L.; Overbeek, P.A.; Tan, L.; Kumar, S.V.; Chan, W.K.; Lorenzi, P.L.; Chang, B.H.; et al. PGC1alpha is required for the renoprotective effect of lncRNA Tug1 in vivo and links Tug1 with urea cycle metabolites. Cell Rep. 2021, 36, 109510. [Google Scholar] [CrossRef]
- Hu, M.; Wang, R.; Li, X.; Fan, M.; Lin, J.; Zhen, J.; Chen, L.; Lv, Z. LncRNA MALAT1 is dysregulated in diabetic nephropathy and involved in high glucose-induced podocyte injury via its interplay with beta-catenin. J. Cell Mol. Med. 2017, 21, 2732–2747. [Google Scholar] [CrossRef]
- Liu, D.W.; Zhang, J.H.; Liu, F.X.; Wang, X.T.; Pan, S.K.; Jiang, D.K.; Zhao, Z.H.; Liu, Z.S. Silencing of long noncoding RNA PVT1 inhibits podocyte damage and apoptosis in diabetic nephropathy by upregulating FOXA1. Exp. Mol. Med. 2019, 51, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Zeng, J.; Xue, J.; Du, A.; Xu, Y. Knockdown of lncRNA PVT1 alleviates high glucose-induced proliferation and fibrosis in human mesangial cells by miR-23b-3p/WT1 axis. Diabetol. Metab. Syndr. 2020, 12, 33. [Google Scholar] [CrossRef] [PubMed]
- Yi, H.; Peng, R.; Zhang, L.Y.; Sun, Y.; Peng, H.M.; Liu, H.D.; Yu, L.J.; Li, A.L.; Zhang, Y.J.; Jiang, W.H.; et al. LincRNA-Gm4419 knockdown ameliorates NF-kappaB/NLRP3 inflammasome-mediated inflammation in diabetic nephropathy. Cell Death Dis. 2017, 8, e2583. [Google Scholar] [CrossRef]
- Yang, Y.L.; Xue, M.; Jia, Y.J.; Hu, F.; Zheng, Z.J.; Wang, L.; Si, Z.K.; Xue, Y.M. Long noncoding RNA NEAT1 is involved in the protective effect of Klotho on renal tubular epithelial cells in diabetic kidney disease through the ERK1/2 signaling pathway. Exp. Mol. Med. 2020, 52, 266–280. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Xu, Y.; Ge, X.; Xu, B.; Peng, W.; Jiang, X.; Shen, L.; Xia, L. Long noncoding RNA NEAT1 accelerates the proliferation and fibrosis in diabetic nephropathy through activating Akt/mTOR signaling pathway. J. Cell Physiol. 2019, 234, 11200–11207. [Google Scholar] [CrossRef]
- Wang, X.; Xu, Y.; Zhu, Y.C.; Wang, Y.K.; Li, J.; Li, X.Y.; Ji, T.; Bai, S.J. LncRNA NEAT1 promotes extracellular matrix accumulation and epithelial-to-mesenchymal transition by targeting miR-27b-3p and ZEB1 in diabetic nephropathy. J. Cell Physiol. 2019, 234, 12926–12933. [Google Scholar] [CrossRef]
- Liao, L.; Chen, J.; Zhang, C.; Guo, Y.; Liu, W.; Liu, W.; Duan, L.; Liu, Z.; Hu, J.; Lu, J. LncRNA NEAT1 Promotes High Glucose-Induced Mesangial Cell Hypertrophy by Targeting miR-222-3p/CDKN1B Axis. Front. Mol. Biosci. 2020, 7, 627827. [Google Scholar] [CrossRef]
- Wang, J.; Zhao, S.M. LncRNA-antisense non-coding RNA in the INK4 locus promotes pyroptosis via miR-497/thioredoxin-interacting protein axis in diabetic nephropathy. Life Sci. 2021, 264, 118728. [Google Scholar] [CrossRef]
- Thomas, A.A.; Feng, B.; Chakrabarti, S. ANRIL regulates production of extracellular matrix proteins and vasoactive factors in diabetic complications. Am. J. Physiol. Endocrinol. Metab. 2018, 314, E191–E200. [Google Scholar] [CrossRef]
- Du, L.; Lu, Y.; Wang, J.; Zheng, Y.; Li, H.; Liu, Y.; Wu, X.; Zhou, J.; Wang, L.; He, L.; et al. LncRNA KIFAP3-5:1 inhibits epithelial-mesenchymal transition of renal tubular cell through PRRX1 in diabetic nephropathy. Cell Biol. Toxicol. 2024, 40, 47. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Jiang, T.; Liang, X.; Shu, S.; Xiang, X.; Zhang, W.; Guo, T.; Xie, W.; Deng, W.; Tang, X. lncRNA MALAT1 mediated high glucose-induced HK-2 cell epithelial-to-mesenchymal transition and injury. J. Physiol. Biochem. 2019, 75, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Song, D.; Wang, Y.; Tang, L. lncRNA MALAT1 Promotes Diabetic Nephropathy Progression via miR-15b-5p/TLR4 Signaling Axis. J. Immunol. Res. 2022, 2022, 8098001. [Google Scholar] [CrossRef]
- Song, P.; Chen, Y.; Liu, Z.; Liu, H.; Xiao, L.; Sun, L.; Wei, J.; He, L. LncRNA MALAT1 Aggravates Renal Tubular Injury via Activating LIN28A and the Nox4/AMPK/mTOR Signaling Axis in Diabetic Nephropathy. Front. Endocrinol. 2022, 13, 895360. [Google Scholar] [CrossRef]
- Cao, X.; Xue, L.D.; Di, Y.; Li, T.; Tian, Y.J.; Song, Y. MSC-derived exosomal lncRNA SNHG7 suppresses endothelial-mesenchymal transition and tube formation in diabetic retinopathy via miR-34a-5p/XBP1 axis. Life Sci. 2021, 272, 119232. [Google Scholar] [CrossRef]
- Mohammad, G.; Kumar, J.; Kowluru, R.A. Mitochondrial Genome-Encoded Long Noncoding RNA Cytochrome B and Mitochondrial Dysfunction in Diabetic Retinopathy. Antioxid. Redox Signal. 2023, 39, 817–828. [Google Scholar] [CrossRef] [PubMed]
- Kumar, J.; Mohammad, G.; Alka, K.; Kowluru, R.A. Mitochondrial Genome-Encoded Long Noncoding RNA and Mitochondrial Stability in Diabetic Retinopathy. Diabetes 2023, 72, 520–531. [Google Scholar] [CrossRef]
- Kumar, J.; Malaviya, P.; Kowluru, R.A. Mitochondrial Genome-Encoded Long Noncoding RNA Cytochrome B (LncCytB) and Mitochondrial Ribonucleases in Diabetic Retinopathy. Biomedicines 2024, 12, 1637. [Google Scholar] [CrossRef]
- Sehgal, P.; Mathew, S.; Sivadas, A.; Ray, A.; Tanwar, J.; Vishwakarma, S.; Ranjan, G.; Shamsudheen, K.V.; Bhoyar, R.C.; Pateria, A.; et al. LncRNA VEAL2 regulates PRKCB2 to modulate endothelial permeability in diabetic retinopathy. EMBO J. 2021, 40, e107134. [Google Scholar] [CrossRef]
- Yan, B.; Tao, Z.F.; Li, X.M.; Zhang, H.; Yao, J.; Jiang, Q. Aberrant expression of long noncoding RNAs in early diabetic retinopathy. Invest. Ophthalmol. Vis. Sci. 2014, 55, 941–951. [Google Scholar] [CrossRef]
- Liu, J.Y.; Yao, J.; Li, X.M.; Song, Y.C.; Wang, X.Q.; Li, Y.J.; Yan, B.; Jiang, Q. Pathogenic role of lncRNA-MALAT1 in endothelial cell dysfunction in diabetes mellitus. Cell Death Dis. 2014, 5, e1506. [Google Scholar] [CrossRef] [PubMed]
- Zeng, R.; Zhang, R.; Song, X.; Ni, L.; Lai, Z.; Liu, C.; Ye, W. The long non-coding RNA MALAT1 activates Nrf2 signaling to protect human umbilical vein endothelial cells from hydrogen peroxide. Biochem. Biophys. Res. Commun. 2018, 495, 2532–2538. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Dan, Y.; Gao, X.; Huang, L.; Lv, H.; Chen, J. DNMT1-mediated lncRNA MEG3 methylation accelerates endothelial-mesenchymal transition in diabetic retinopathy through the PI3K/Akt/mTOR signaling pathway. Am. J. Physiol. Endocrinol. Metab. 2021, 320, E598–E608. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Sun, W.; Liu, L.; Xiao, J.; Xiong, J.; Hu, Y.; Zhou, Q.; Yin, X. Muller Cells Harboring Exosomal lncRNA OGRU Modulate Microglia Polarization in Diabetic Retinopathy by Serving as miRNA Sponges. Diabetes 2024, 73, 1919–1934. [Google Scholar] [CrossRef]
- Yang, J.; Zhao, S.; Tian, F. SP1-mediated lncRNA PVT1 modulates the proliferation and apoptosis of lens epithelial cells in diabetic cataract via miR-214-3p/MMP2 axis. J. Cell Mol. Med. 2020, 24, 554–561. [Google Scholar] [CrossRef]
- Beydag-Tasoz, B.S.; Yennek, S.; Grapin-Botton, A. Towards a better understanding of diabetes mellitus using organoid models. Nat. Rev. Endocrinol. 2023, 19, 232–248. [Google Scholar] [CrossRef]
- Chen, L.L.; Kim, V.N. Small and long non-coding RNAs: Past, present, and future. Cell 2024, 187, 6451–6485. [Google Scholar] [CrossRef]
- Johnsson, P.; Lipovich, L.; Grander, D.; Morris, K.V. Evolutionary conservation of long non-coding RNAs; sequence, structure, function. Biochim. Biophys. Acta 2014, 1840, 1063–1071. [Google Scholar] [CrossRef]
- Nemeth, K.; Bayraktar, R.; Ferracin, M.; Calin, G.A. Non-coding RNAs in disease: From mechanisms to therapeutics. Nat. Rev. Genet. 2024, 25, 211–232. [Google Scholar] [CrossRef] [PubMed]
- Coan, M.; Haefliger, S.; Ounzain, S.; Johnson, R. Targeting and engineering long non-coding RNAs for cancer therapy. Nat. Rev. Genet. 2024, 25, 578–595. [Google Scholar] [CrossRef]
- Hoy, S.M. Patisiran: First Global Approval. Drugs 2018, 78, 1625–1631. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. FDA approves landmark RNAi drug. Nat. Rev. Drug Discov. 2018, 17, 613. [Google Scholar] [CrossRef] [PubMed]
- Dey, S.K.; Jaffrey, S.R. RIBOTACs: Small Molecules Target RNA for Degradation. Cell Chem. Biol. 2019, 26, 1047–1049. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Cui, J.; Zhu, J.; Kim, B.; Kuo, M.L.; Potts, P.R. RNATACs: Multispecific small molecules targeting RNA by induced proximity. Cell Chem. Biol. 2024, 31, 1101–1117. [Google Scholar] [CrossRef]
- Gao, J.; Wang, W.; Wang, F.; Guo, C. LncRNA-NR_033515 promotes proliferation, fibrogenesis and epithelial-to-mesenchymal transition by targeting miR-743b-5p in diabetic nephropathy. Biomed. Pharmacother. 2018, 106, 543–552. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| LncRNAs | Mechanism in ER Stress or OS | Impact on Diabetes | Reference |
|---|---|---|---|
| ANRIL | ANRIL knockdown decreased the level of MDA. | Accelerates podocytes inflammation in HG condition | [34] |
| CTBP1-AS2 | CTBP1-AS2 overexpression reduced ROS and MDA levels, and increased SOD activity. | Protects HGMCs from HG-induced accumulation of extracellular matrix, and inflammation | [35] |
| FLG-AS1 | FLG-AS1 overexpression inhibited ROS, MDA levels. | Protects retinal pigment epithelial cells from HG-induced inflammation, and apoptosis | [36] |
| GAS5 | GAS5 overexpression reduced ATF4, CHOP, as well as p-PERK/PERK and p-eIF2α/eIF2α levels. | Protects retinal epithelial cells from HG-induced apoptosis and inflammation | [37] |
| H19 | H19 overexpression enhanced the expression of XBP1s. | Protects retinal pigment epithelial cells from HG-induced inflammation | [38] |
| LINC01619 | LINC01619 knockdown upregulated CHOP and GRP78 levels. | Protects podocytes from HG-induced injury | [39] |
| MGC | CHOP upregulated lnc RNA MGC expression in glomeruli. | Accelerate mesangial cell hypertrophy and the accumulation of extracellular matrix in HG condition | [40] |
| SNHG14 | SNHG14 knockdown inhibited ROS levels. | Accelerates renal epithelial cells apoptosis in HG condition | [41] |
| Tug1 | TUG1 overexpression reduced GRP78, CHOP, p—PERK, and p-eIF2α levels. | Protects renal tubular epithelial cells from HG-induced apoptosis | [42] |
| ZFAS1 | ZFAS1 knockdown inhibited ROS, MDA levels and increased SOD activity. | Accelerates mesangial cells fibrosis, and inflammation in HG condition | [43] |
| LncRNAs | Diabetic Complications | Expression Levels in Diabetes | Association to Stresses and Mechanism in Diabetes | Reference |
|---|---|---|---|---|
| ANRIL | DN and DM | Up | Silencing ANRIL through MME alleviates inflammation, OS, and apoptosis in podocytes while also reducing myocardial injury in diabetes by inhibiting myocardial OS. | [34,44] |
| CTBP1-AS2 | DN | Down | CTBP1-AS2 alleviated HG-induced OS, ECM accumulation, and inflammation in HGMCs via miR-155-5p/FOXO1 axis. | [35] |
| FLG-AS1 | DR | Down | Overexpression of FLG-AS1 reduced inflammation, OS, and apoptosis of HG-treated human retinal pigment epithelial cells via the miR-380-3p/SOCS6 axis. | [36] |
| GAS5 | DN/DR | Down | GAS5 overexpression inhibited inflammation, OS, and pyroptosis in renal tubular cells by downregulating miR-452-5p expression, while also reducing ER stress-related apoptosis and inflammation in retinal pigment epithelium cells via SERCA2b. | [37,45] |
| H19 | DR | Down | H19 overexpression inhibited inflammatory processes via XBP1s/miR-93 in human retinal pigment epithelial cells. | [38] |
| HOTAIR | DCM | Down | HOTAIR overexpression decreased OS and inflammation, and attenuated myocyte death via miR-34a/SIRT1 signaling. | [46] |
| LINC01619 | DN | Down | LINC01619 downregulation triggered ER stress and podocyte injury via miR-27a/FOXO1. | [39] |
| MALAT1 | DC | Up | MALAT1 overexpressed promoted apoptosis and OS via the p38MAPK pathway in human lens epithelial cells. | [47] |
| MGC | DN and DM | Up | The ER stress-related transcription factor CHOP upregulated MGC in glomeruli, while lowering MGC reduced cell death in pancreatic islets. | [40,48] |
| SNHG14 | DN | Up | Silencing SNHG14 inhibited apoptosis, OS, and inflammation through the miR-483-5p/HDAC4 pathway to mitigate renal tubular damage. | [41] |
| TUG1 | DN | Down | Overexpressed TUG1 could prevent HG-induced apoptosis and alleviate ER Stress in renal epithelial cells via miR-29c-3p/SIRT1 and PU.1/RTN1 pathway. | [42,49] |
| ZFAS1 | DN | Up | Silencing ZFAS1 had a protective effect on HG-induced proliferation, OS, fibrosis, and inflammation in HGMCs. | [43] |
| LncRNAs | Model of DM | Expression Levels in DM | Functions in DM | Target | Reference |
|---|---|---|---|---|---|
| ADIPINT | T2DM | Upregulated in abdominal subcutaneous adipose tissue with obese women | Enhances the synthesis of triglycerides and increases the size of the lipid droplet/fat cell | Pyruvate carboxylase | [67] |
| ASMER-1/2 | T2DM | Upregulated in adipocyte tissue with obese women | Enhances lipolysis, adiponectin release, and triglyceride accumulation | PPARG and INSR | [66] |
| betaFaar | T2DM | Downregulated in the islets of the obese mice | Enhances β cell apoptosis to decrease insulin transcription and secretion | TRAF3IP2/NF-κB (β cell apoptosis), miR-138-5p (insulin transcription) | [59] |
| EPB41L4A-AS1 | T2DM | Upregulated in the liver of patients with T2DM, muscle cells of patients with IR, and T2DM cell models | Inhibits glucose uptake and mitochondrial respiration in liver cells | GLUT2/4/TXNIP | [82] |
| Gomafu | T2DM | Upregulated in the livers of obese mice | Enhances hepatic gluconeogenesis and decreases insulin sensitivity | miR-139/FOXO1 | [21] |
| H19 | T2DM | Downregulated in skeletal muscle of humans with T2DM, HFD mice and db/db mice | Impairs systemic glucose metabolism, decreases expression of insulin receptor and lipoprotein lipase, enhances lipid deposition in skeletal muscle, impairs adipogenesis, oxidative metabolism, and mitochondrial respiration in brown adipocytes | hnRNPA1/PGC1a, miRNA let-7, DUSP27/AMPK, PEG-inactivating H19-MBD1 complexes | [79,80,81,84] |
| MALAT1 | T2DM | Upregulated in patients with T2DM, in hepatocytes exposed to palmitate, livers of ob/ob mice, and palmitate-treated primary hepatocytes | Inhibits glucose uptake and insulin signaling response, enhances lipid accumulation in hepatocytes, impairs insulin secretion, and decreases pancreatic islet cellularity | SREBP-1c, Nrf2, miR-382-3p/resistin, | [76,77,85] |
| MEG3 | T2DM | Upregulated in the livers of HFD and ob/ob mice, palmitate-treated primary hepatocytes, in adipocytes treated with TNF-α and HFD mice | Impairs glucose and insulin tolerance, enhances hepatic lipid accumulation, Enhances adipocyte inflammation injury | miR-185-5p/Egr2, FOXO1, miR-214/ATF4, IGF2BP2/TLR4/NF-κB | [68,70,71,72] |
| MIR503HG | T1DM | Not detected | SC-β cell differentiation and insulin production | CDH1, HES1 | [61] |
| SHGL | T2DM | Downregulated in obese mouse livers | Inhibits hepatic gluconeogenesis and lipogenesis | hnRNPA1/CaM/Akt | [78] |
| TUG1 | GDM | Downregulated in islet tissues of mice with HFD-induced GDM | Enhances β cell apoptosis | miR-328-3p/SREBP-2/ERK | [83] |
| TUNAR | T2DM | Downregulated in β cells of T2DM patients | Inhibits β cell proliferation | Wnt/DKK3 | [60] |
| LncRNAs | Expression Levels in DR | Functions in DR | Target | Reference |
|---|---|---|---|---|
| CytB | Downregulated in the HG-induced retinal microvessels | Increases ROS production and drives mitochondrial dysfunction | cytochrome B complex III | [108] |
| MALAT1 | Upregulated in the retinas of diabetic patients and mouse models, as well as in retinal endothelial cells under HG | Accelerates retinal vessel impairment and inflammation | p38 mitogen-activated protein kinase (MAPK) | [111] |
| MEG3 | Downregulated in the retinas of DR rat and HG-treated microvascular endothelial cells obtained from retina | Inhibits EndMT | PI3K/Akt/mTOR | [113] |
| OGRU | Upregulated in the retinas of diabetic patients and mouse models, as well as in HG-treated Müller cells | Accelerates inflammation | miR-221/USP14 | [22] |
| SNHG7 | Downregulated in the HG-treated HRMECs cells | Suppresses EndMT and tube formation in HRMECs | miR-34a-5p/XBP1 | [105] |
| VEAL2 | Downregulated in the choroid tissue of DR patients | Maintains endothelial permeability | PRKCB2 | [109] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, L.; Wu, Y.-Q.; Yang, J.-E. Stress-Related LncRNAs and Their Roles in Diabetes and Diabetic Complications. Int. J. Mol. Sci. 2025, 26, 2194. https://doi.org/10.3390/ijms26052194
Li L, Wu Y-Q, Yang J-E. Stress-Related LncRNAs and Their Roles in Diabetes and Diabetic Complications. International Journal of Molecular Sciences. 2025; 26(5):2194. https://doi.org/10.3390/ijms26052194
Chicago/Turabian StyleLi, Lian, Yu-Qi Wu, and Jin-E Yang. 2025. "Stress-Related LncRNAs and Their Roles in Diabetes and Diabetic Complications" International Journal of Molecular Sciences 26, no. 5: 2194. https://doi.org/10.3390/ijms26052194
APA StyleLi, L., Wu, Y.-Q., & Yang, J.-E. (2025). Stress-Related LncRNAs and Their Roles in Diabetes and Diabetic Complications. International Journal of Molecular Sciences, 26(5), 2194. https://doi.org/10.3390/ijms26052194