
KinasePred: A Computational Tool for Small-Molecule Kinase Target Prediction
 , , , , ,
, , , , ,  ,
, 

Abstract
1. Introduction
2. Results and Discussion
2.1. Generation and Evaluation of ML Operational Models
2.2. Explainability of the Best Model
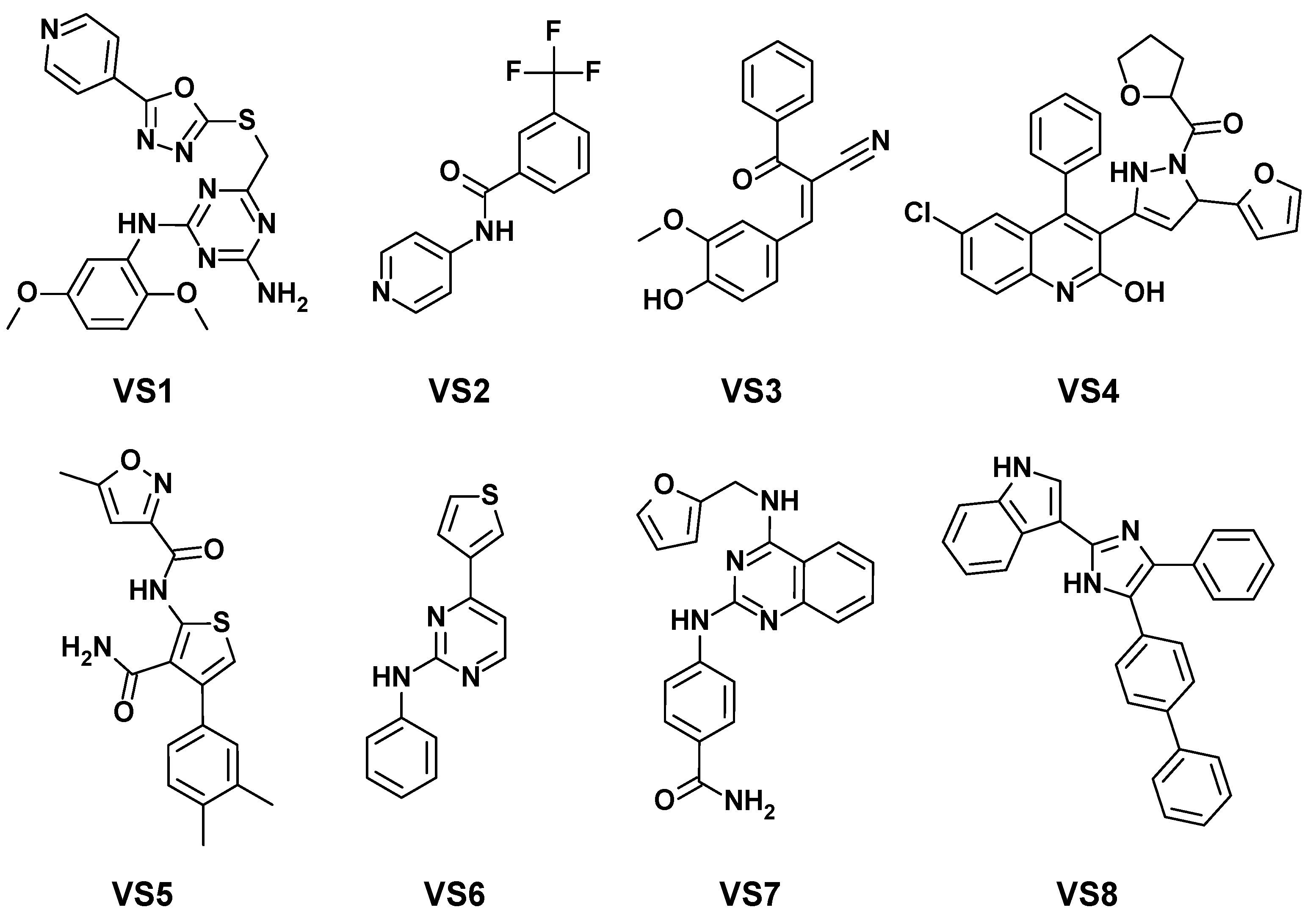
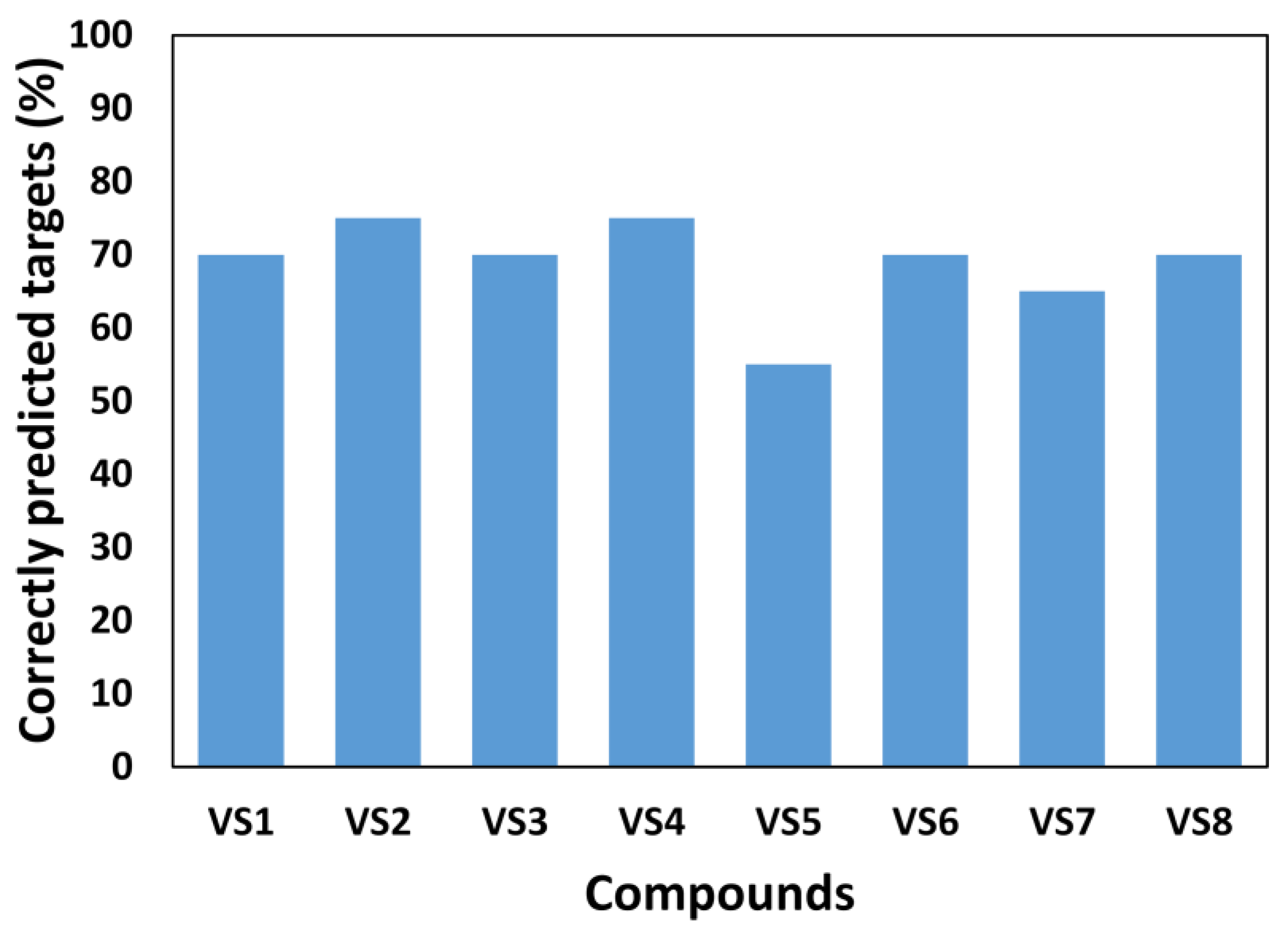
2.3. Virtual Screening and Experimental Evaluation
2.4. Generation and Evaluation of Target-Specific Models
2.5. Experimental Validation
3. Materials and Methods
3.1. Modeling Datasets
3.2. Representation of Molecules
3.3. Machine Learning Algorithms
3.4. Performance Metrics
- Precision
- Recall
- Negative Predictive Value (NPV)
- Specificity
- Balanced Accuracy (BA)
- Matthews Correlation Coefficient (MCC)
3.5. Generation and Evaluation of Models
3.6. Virtual Screening Dataset
3.7. Feature Contribution
3.8. Panel Inhibition Assay
3.9. Target-Specific Models
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ML | machine learning |
| VS | virtual screening |
| CNN | convolutional neural networks |
| XAI | explainable artificial intelligence |
| SHAP | SHapley Additive exPlanations |
| LIME | Local Interpretable Model-agnostic Explanations |
| RF | Random Forest |
| GNB | Gaussian Naïve Bayes |
| MLP | Multi-Layer Perceptron |
| FPs | Fingerprints |
| CV | cross-validation |
| MCC | Matthews Correlation Coefficient |
| BA | Balanced Accuracy |
| NPV | Negative Predictive Value |
| ROCK2 | Rho-Associated Coiled-Coil Containing Protein Kinase 2 |
| BTK | Bruton’s tyrosine kinase |
| GSK-3β | Glycogen Synthase Kinase 3 Beta |
| TP | true positive |
| FP | false positive |
| TN | true negative |
| FN | false negative |
References
- Graves, J.D.; Campbell, J.S.; Krebs, E.G. Protein Serine/Threonine Kinases of the MAPK Cascade. Ann. New York Acad. Sci. 1995, 766, 320–343. [Google Scholar] [CrossRef]
- Ahsan, R.; Khan, M.M.; Mishra, A.; Noor, G.; Ahmad, U. Protein Kinases and Their Inhibitors Implications in Modulating Disease Progression. Protein J. 2023, 42, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Silnitsky, S.; Rubin, S.J.S.; Zerihun, M.; Qvit, N. An Update on Protein Kinases as Therapeutic Targets-Part I: Protein Kinase C Activation and Its Role in Cancer and Cardiovascular Diseases. Int. J. Mol. Sci. 2023, 24, 17600. [Google Scholar]
- Kannaiyan, R.; Mahadevan, D. A Comprehensive Review of Protein Kinase Inhibitors for Cancer Therapy. Expert. Rev. Anticancer. Ther. 2018, 18, 1249–1270. [Google Scholar] [CrossRef]
- Song, J.; Wang, H.; Wang, J.; Leier, A.; Marquez-Lago, T.; Yang, B.; Zhang, Z.; Akutsu, T.; Webb, G.I.; Daly, R.J. PhosphoPredict: A Bioinformatics Tool for Prediction of Human Kinase-Specific Phosphorylation Substrates and Sites by Integrating Heterogeneous Feature Selection. Sci. Rep. 2017, 7, 6862. [Google Scholar] [CrossRef]
- Galati, S.; Di Stefano, M.; Martinelli, E.; Poli, G.; Tuccinardi, T. Recent Advances in In Silico Target Fishing. Molecules 2021, 26, 5124. [Google Scholar] [CrossRef] [PubMed]
- Gehringer, M.; Muth, F.; Koch, P.; Laufer, S.A. C-Jun N-Terminal Kinase Inhibitors: A Patent Review (2010—2014). Expert. Opin. Ther. Pat. 2015, 25, 849–872. [Google Scholar] [CrossRef] [PubMed]
- Olsen, J.V.; Mann, M. Status of Large-Scale Analysis of Post-Translational Modifications by Mass Spectrometry. Mol. Cell Proteomics 2013, 12, 3444–3452. [Google Scholar] [CrossRef]
- Galati, S.; Di Stefano, M.; Bertini, S.; Granchi, C.; Giordano, A.; Gado, F.; Macchia, M.; Tuccinardi, T.; Poli, G. Identification of New GSK3β Inhibitors through a Consensus Machine Learning-Based Virtual Screening. Int. J. Mol. Sci. 2023, 24, 17233. [Google Scholar] [CrossRef] [PubMed]
- Di Stefano, M.; Galati, S.; Ortore, G.; Caligiuri, I.; Rizzolio, F.; Ceni, C.; Bertini, S.; Bononi, G.; Granchi, C.; Macchia, M.; et al. Machine Learning-Based Virtual Screening for the Identification of Cdk5 Inhibitors. Int. J. Mol. Sci. 2022, 23, 10653. [Google Scholar] [CrossRef] [PubMed]
- Cockroft, N.T.; Cheng, X.; Fuchs, J.R. STarFish: A Stacked Ensemble Target Fishing Approach and Its Application to Natural Products. J. Chem. Inf. Model. 2019, 59, 4906–4920. [Google Scholar] [CrossRef] [PubMed]
- Rifaioglu, A.S.; Atas, H.; Martin, M.J.; Cetin-Atalay, R.; Atalay, V.; Doǧan, T. Recent Applications of Deep Learning and Machine Intelligence on in Silico Drug Discovery: Methods, Tools and Databases. Brief. Bioinform. 2019, 20, 1878–1912. [Google Scholar] [CrossRef] [PubMed]
- Cichonska, A.; Ravikumar, B.; Parri, E.; Timonen, S.; Pahikkala, T.; Airola, A.; Wennerberg, K.; Rousu, J.; Aittokallio, T. Computational-Experimental Approach to Drug-Target Interaction Mapping: A Case Study on Kinase Inhibitors. PLoS Comput. Biol. 2017, 13, e1005678. [Google Scholar] [CrossRef] [PubMed]
- Gagic, Z.; Ruzic, D.; Djokovic, N.; Djikic, T.; Nikolic, K. In Silico Methods for Design of Kinase Inhibitors as Anticancer Drugs. Front. Chem. 2020, 7, 873. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, S.M.; Erion, G.; Chen, H.; DeGrave, A.; Prutkin, J.M.; Nair, B.; Katz, R.; Himmelfarb, J.; Bansal, N.; Lee, S.-I. From Local Explanations to Global Understanding with Explainable AI for Trees. Nat. Mach. Intell. 2020, 2, 56–67. [Google Scholar] [CrossRef]
- Di Stefano, M.; Galati, S.; Piazza, L.; Granchi, C.; Mancini, S.; Fratini, F.; Macchia, M.; Poli, G.; Tuccinardi, T. VenomPred 2.0: A Novel In Silico Platform for an Extended and Human Interpretable Toxicological Profiling of Small Molecules. J. Chem. Inf. Model. 2023, 64, 2275–2289. [Google Scholar] [CrossRef]
- Zdrazil, B.; Felix, E.; Hunter, F.; Manners, E.J.; Blackshaw, J.; Corbett, S.; de Veij, M.; Ioannidis, H.; Lopez, D.M.; Mosquera, J.F.; et al. The ChEMBL Database in 2023: A Drug Discovery Platform Spanning Multiple Bioactivity Data Types and Time Periods. Nucleic Acids Res 2024, 52, D1180–D1192. [Google Scholar] [CrossRef]
- Pala, D.; Clark, D.; Edwards, C.; Pasqua, E.; Tigli, L.; Pioselli, B.; Malysa, P.; Facchinetti, F.; Rancati, F.; Accetta, A. Design and Synthesis of Novel 8-(Azaindolyl)-Benzoazepinones as Potent and Selective ROCK Inhibitors. RSC Med. Chem. 2024, 15, 3862–3879. [Google Scholar] [CrossRef]
- Montoya, S.; Bourcier, J.; Noviski, M.; Lu, H.; Thompson, M.C.; Chirino, A.; Jahn, J.; Sondhi, A.K.; Gajewski, S.; Tan, Y.S.; et al. Kinase-Impaired BTK Mutations Are Susceptible to Clinical-Stage BTK and IKZF1/3 Degrader NX-2127. Science 2024, 383, eadi5798. [Google Scholar] [CrossRef] [PubMed]
- Góral, I.; Wichur, T.; Sługocka, E.; Grygier, P.; Głuch-Lutwin, M.; Mordyl, B.; Honkisz-Orzechowska, E.; Szałaj, N.; Godyń, J.; Panek, D.; et al. Exploring Novel GSK-3β Inhibitors for Anti-Neuroinflammatory and Neuroprotective Effects: Synthesis, Crystallography, Computational Analysis, and Biological Evaluation. ACS Chem. Neurosci. 2024, 15, 3181–3201. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the Sensitivity of Progressive Multiple Sequence Alignment through Sequence Weighting, Position-Specific Gap Penalties and Weight Matrix Choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed]
- Gilson, P.R.; Tan, C.; Jarman, K.E.; Lowes, K.N.; Curtis, J.M.; Nguyen, W.; Di Rago, A.E.; Bullen, H.E.; Prinz, B.; Duffy, S.; et al. Optimization of 2-Anilino 4-Amino Substituted Quinazolines into Potent Antimalarial Agents with Oral in Vivo Activity. J. Med. Chem. 2017, 60, 1171–1188. [Google Scholar] [CrossRef]
- Rui, Q.; Ni, H.; Li, D.; Gao, R.; Chen, G. The Role of LRRK2 in Neurodegeneration of Parkinson Disease. Curr. Neuropharmacol. 2018, 16, 1348–1357. [Google Scholar] [CrossRef]
- Wen, J.; Wang, S.; Guo, R.; Liu, D. CSF1R Inhibitors Are Emerging Immunotherapeutic Drugs for Cancer Treatment. Eur. J. Med. Chem. 2023, 245, 114884. [Google Scholar] [CrossRef]
- Cocco, E.; Scaltriti, M.; Drilon, A. NTRK Fusion-Positive Cancers and TRK Inhibitor Therapy. Nat. Rev. Clin. Oncol. 2018, 15, 731–747. [Google Scholar] [CrossRef]
- Paul, R.; Hallett, W.A.; Hanifin, J.W.; Reich, M.F.; Johnson, B.D.; Lenhard, R.H.; Dusza, J.P.; Kerwar, S.S.; Lin, Y.I.; Pickett, W.C.; et al. Preparation of Substituted N-Phenyl-4-Aryl-2-Pyrimidinamines as Mediator Release Inhibitors. J. Med. Chem. 1993, 36, 2716–2725. [Google Scholar] [CrossRef]
- Joshi, S. New Insights into SYK Targeting in Solid Tumors. Trends Pharmacol. Sci. 2024, 45, 904–918. [Google Scholar] [CrossRef]
- Fan, Z.; Duan, J.; Wang, L.; Xiao, S.; Li, L.; Yan, X.; Yao, W.; Wu, L.; Zhang, S.; Zhang, Y.; et al. PTK2 Promotes Cancer Stem Cell Traits in Hepatocellular Carcinoma by Activating Wnt/β-Catenin Signaling. Cancer Lett. 2019, 450, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Wicht, K.J.; Combrinck, J.M.; Smith, P.J.; Hunter, R.; Egan, T.J. Identification and SAR Evaluation of Hemozoin-Inhibiting Benzamides Active against Plasmodium Falciparum. J. Med. Chem. 2016, 59, 6512–6530. [Google Scholar] [CrossRef] [PubMed]
- Guiguemde, W.A.; Shelat, A.A.; Bouck, D.; Duffy, S.; Crowther, G.J.; Davis, P.H.; Smithson, D.C.; Connelly, M.; Clark, J.; Zhu, F.; et al. Chemical Genetics of Plasmodium Falciparum. Nature 2010, 465, 311–315. [Google Scholar] [CrossRef]
- Dechering, K.J.; Timmerman, M.; Rensen, K.; Koolen, K.M.J.; Honarnejad, S.; Vos, M.W.; Huijs, T.; Henderson, R.W.M.; Chenu, E.; Laleu, B.; et al. Replenishing the Malaria Drug Discovery Pipeline: Screening and Hit Evaluation of the MMV Hit Generation Library 1 (HGL1) against Asexual Blood Stage Plasmodium Falciparum, Using a Nano Luciferase Reporter Read-Out. SLAS Discov. 2022, 27, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Sterling, T.; Irwin, J.J. ZINC 15--Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Galati, S.; Di Stefano, M.; Macchia, M.; Poli, G.; Tuccinardi, T. MolBook UNIPI─Create, Manage, Analyze, and Share Your Chemical Data for Free. J. Chem. Inf. Model. 2023, 63, 3977–3982. [Google Scholar] [CrossRef] [PubMed]
- QUACPAC 2.2.2.0: OpenEye, Cadence Molecular Sciences, Santa Fe, NM. Available online: http://www.eyesopen.com (accessed on 20 January 2025).
- Landrum, G. RDKit: Open-Source Cheminformatics. Available online: https://www.Rdkit.Org (accessed on 1 February 2023).
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Müller, A.; Nothman, J.; Louppe, G.; et al. Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Rodríguez-Pérez, R.; Bajorath, J. Interpretation of Machine Learning Models Using Shapley Values: Application to Compound Potency and Multi-Target Activity Predictions. J. Comput. Aided Mol. Des. 2020, 34, 1013–1026. [Google Scholar] [CrossRef] [PubMed]
- Galati, S.; Di Stefano, M.; Martinelli, E.; Macchia, M.; Martinelli, A.; Poli, G.; Tuccinardi, T. VenomPred: A Machine Learning Based Platform for Molecular Toxicity Predictions. Int. J. Mol. Sci. 2022, 23, 2105. [Google Scholar] [CrossRef]
- Al-Lawati, A.; Lucas, J.; Zhang, Z.; Mitra, P.; Wang, S. Graph-Based Molecular In-Context Learning Grounded on Morgan Fingerprints. arXiv 2025, arXiv:2502.05414. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Model | MCC | Recall | Specificity | BA | NPV | Precision |
|---|---|---|---|---|---|---|
| MLP–Morgan | 0.96 | 0.99 | 0.98 | 0.98 | 0.99 | 0.99 |
| MLP–PubChem | 0.94 | 0.96 | 0.96 | 0.96 | 0.96 | 0.96 |
| RF–Morgan | 0.93 | 0.96 | 0.96 | 0.96 | 0.96 | 0.96 |
| MLP–RDKit | 0.93 | 0.97 | 0.94 | 0.95 | 0.96 | 0.97 |
| RF–RDKit | 0.90 | 0.92 | 0.93 | 0.93 | 0.93 | 0.93 |
| RF–PubChem | 0.90 | 0.91 | 0.91 | 0.91 | 0.91 | 0.91 |
| GNB–Morgan | 0.78 | 0.90 | 0.87 | 0.89 | 0.90 | 0.88 |
| GNB–RDKit | 0.59 | 0.83 | 0.75 | 0.79 | 0.82 | 0.77 |
| GNB–PubChem | 0.53 | 0.95 | 0.54 | 0.74 | 0.91 | 0.67 |
| Model | MCC | Recall | Specificity | BA | NPV | Precision |
|---|---|---|---|---|---|---|
| MLP–Morgan | 0.88 | 0.90 | 0.98 | 0.94 | 0.90 | 0.97 |
| Kinase Target | VS1 | VS2 | VS3 | VS4 | VS5 | VS6 | VS7 | VS8 |
|---|---|---|---|---|---|---|---|---|
| GSK3B | - | - | - | - | - | - | - | - |
| ALK | - | - | - | - | - | - | - | - |
| RPS6KB1 | - | - | - | - | - | - | - | - |
| JAK3 | - | - | - | - | - | - | - | - |
| PIM2 | - | - | - | - | - | - | - | - |
| mTOR | - | - | - | - | - | - | - | - |
| MAPK8 | - | - | - | - | - | 63 | - | - |
| BRAF | - | - | - | 56 | - | - | - | - |
| IRAK4 | - | - | - | - | - | - | 80 | - |
| CHEK1 | - | - | - | - | - | - | - | - |
| IGF1R | - | - | - | - | - | - | - | - |
| MET | - | - | - | - | - | - | - | - |
| LCK | - | - | - | - | - | - | 53 | - |
| ERBB2 | - | - | - | - | - | - | - | - |
| CSF1R | - | - | - | 90 | 54 | 89 | 89 | - |
| PTK2 | - | - | - | - | - | - | 66 | - |
| NTRK1 | - | - | - | - | - | - | 90 | - |
| SYK | - | - | - | - | - | 57 | 90 | - |
| LRKK2 | - | 50 | 54 | - | - | 60 | 100 | - |
| ROCK2 | - | 74 | - | - | - | - | - | - |
| Models Type | Models Number | BA | MCC | Precision | Recall | NPV | Specificity |
|---|---|---|---|---|---|---|---|
| RF | 230 | 0.83 ± 0.08 | 0.49 ± 0.23 | 0.89 ± 0.15 | 0.89 ± 0.08 | 0.55 ± 0.31 | 0.66 ± 0.26 |
| RF | 130 | 0.81 ± 0.07 | 0.59 ± 0.15 | 0.80 ± 0.16 | 0.85 ± 0.08 | 0.74 ± 0.19 | 0.78 ± 0.08 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Stefano, M.; Piazza, L.; Poles, C.; Galati, S.; Granchi, C.; Giordano, A.; Campisi, L.; Macchia, M.; Poli, G.; Tuccinardi, T. KinasePred: A Computational Tool for Small-Molecule Kinase Target Prediction. Int. J. Mol. Sci. 2025, 26, 2157. https://doi.org/10.3390/ijms26052157
Di Stefano M, Piazza L, Poles C, Galati S, Granchi C, Giordano A, Campisi L, Macchia M, Poli G, Tuccinardi T. KinasePred: A Computational Tool for Small-Molecule Kinase Target Prediction. International Journal of Molecular Sciences. 2025; 26(5):2157. https://doi.org/10.3390/ijms26052157
Chicago/Turabian StyleDi Stefano, Miriana, Lisa Piazza, Clarissa Poles, Salvatore Galati, Carlotta Granchi, Antonio Giordano, Luca Campisi, Marco Macchia, Giulio Poli, and Tiziano Tuccinardi. 2025. "KinasePred: A Computational Tool for Small-Molecule Kinase Target Prediction" International Journal of Molecular Sciences 26, no. 5: 2157. https://doi.org/10.3390/ijms26052157
APA StyleDi Stefano, M., Piazza, L., Poles, C., Galati, S., Granchi, C., Giordano, A., Campisi, L., Macchia, M., Poli, G., & Tuccinardi, T. (2025). KinasePred: A Computational Tool for Small-Molecule Kinase Target Prediction. International Journal of Molecular Sciences, 26(5), 2157. https://doi.org/10.3390/ijms26052157