
Rodent Models of Diabetic Neuropathy, Role of Calcium Homeostasis in Pain and KB-R7943 as a Potential Therapeutic



Abstract
1. Introduction
2. In Vivo Animal Models of DN: Drug- and Diet-Induced
2.1. Chemically-Induced DN Rodent Models
2.1.1. Streptozotocin (STZ)-Induced Diabetes
2.1.2. Alloxan-Induced Diabetes
2.2. Diet-Induced DN Rodent Models
2.3. Combined Chemically- and Diet-Induced DN Rodent Models
3. Calcium Dyshomeostasis in Diabetic Neuralgia
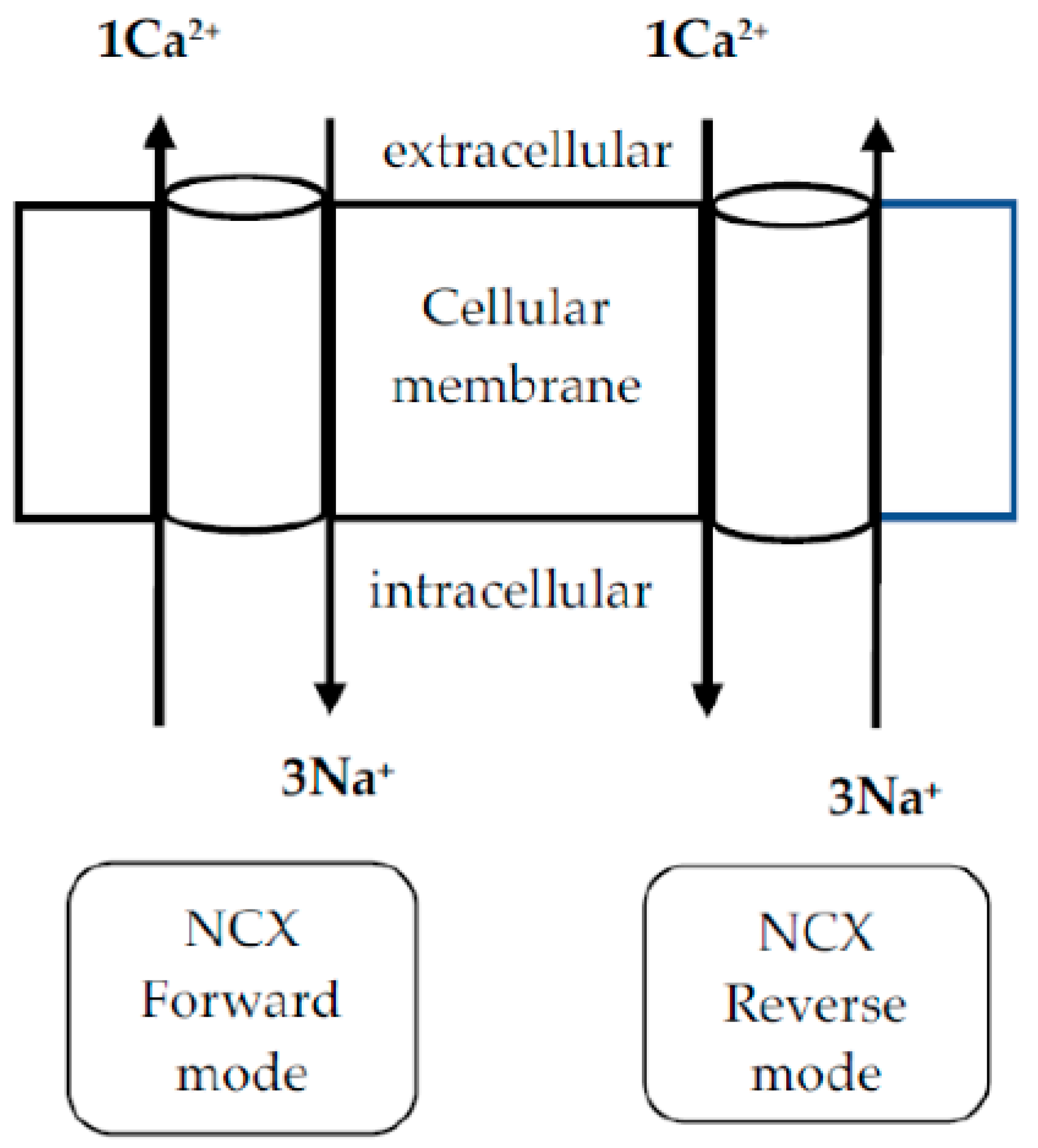
4. Na+/Ca2+ Exchanger (NCX) in PDN
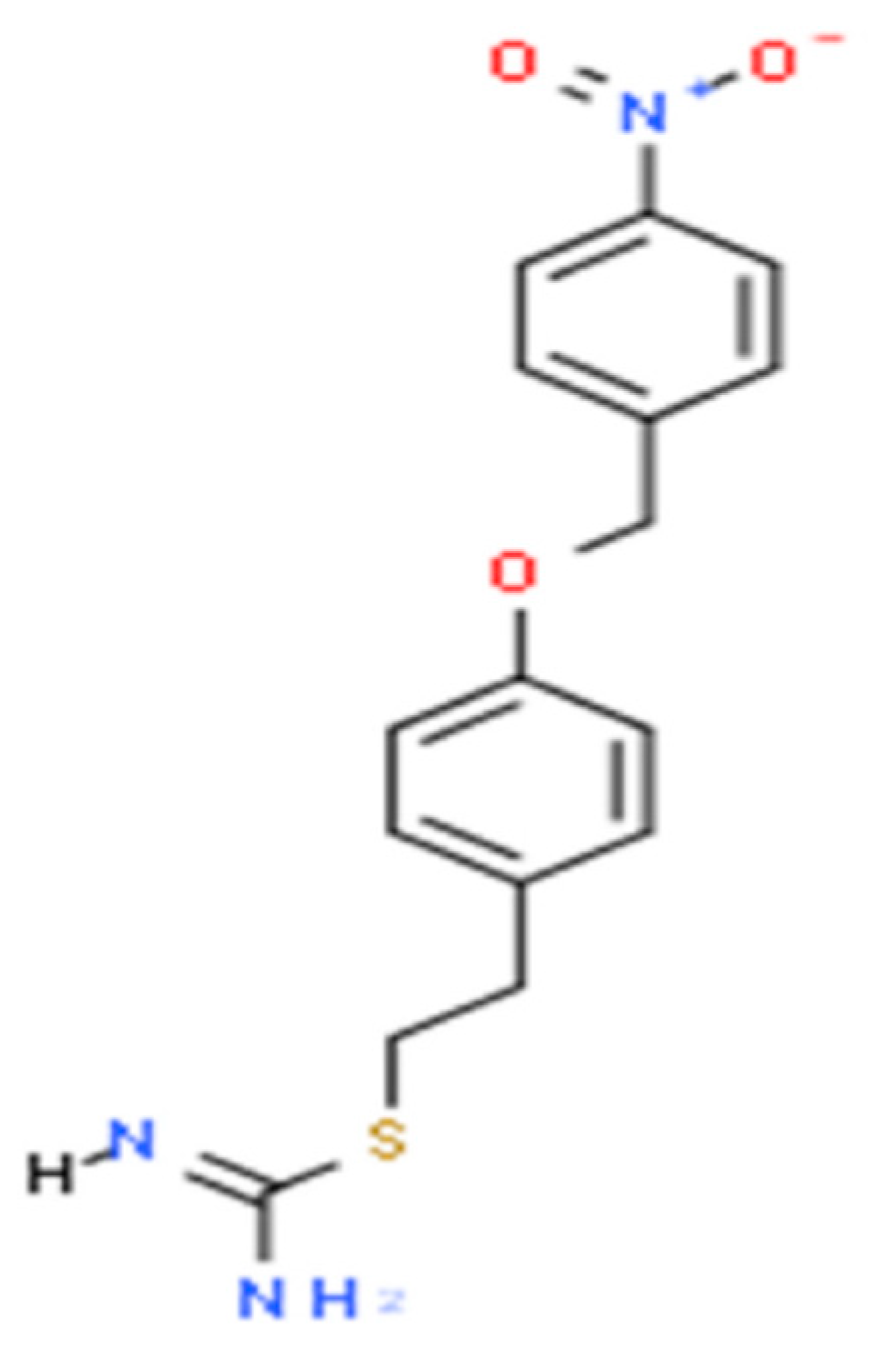
5. KB-R7943 in the Treatment of PDN
5.1. Other Therapeutic Targets and Drugs for PDN
5.2. Current Challenges and Future Directions
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| DN | diabetic neuropathy |
| PDN | painful diabetic neuropathy |
| NCX | Na+/Ca2+ exchanger |
| NCXrev | NCX reverse mode |
| STZ | streptozotocin |
| GLUT | glucose transporter |
| Ca2+ | calcium |
| NMDA | N-methyl-D-aspartate |
| AMPA | alpha-amino-3-hydroxy-5-methylisoxazole-4-propionicacid |
References
- Feldman, E. Diabetic Neuropathy. Nat. Rev. Dis. Primers 2019, 5, 42. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Rayner, A.W.; Gregg, E.W.; Sheffer, K.E.; Carrillo-Larco, R.M.; Bennett, J.E.; Shaw, J.E.; Paciorek, C.J.; Singleton, R.K.; Barradas Pires, A.; et al. Worldwide Trends in Diabetes Prevalence and Treatment from 1990 to 2022: A Pooled Analysis of 1108 Population-Representative Studies with 141 Million Participants. Lancet 2024, 404, 2077–2093. [Google Scholar] [CrossRef] [PubMed]
- Said, G. Diabetic Neuropathy—A Review. Nat. Rev. Neurol. 2007, 3, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Bansal, V.; Kalita, J.; Misra, U.K. Diabetic Neuropathy. Postgrad. Med. J. 2006, 82, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Pop-Busui, R.; Boulton, A.J.M.; Feldman, E.L.; Bril, V.; Freeman, R.; Malik, R.A.; Sosenko, J.M.; Ziegler, D. Diabetic Neuropathy: A Position Statement by the American Diabetes Association. Diabetes Care 2017, 40, 136–154. [Google Scholar] [CrossRef]
- Pan, Q.; Fei, S.; Zhang, L.; Chen, H.; Luo, J.; Wang, W.; Xiao, F.; Guo, L. How Does Diabetic Peripheral Neuropathy Impact Patients’ Burden of Illness and the Economy? A Retrospective Study in Beijing, China. Front. Public Health 2023, 11, 1164536. [Google Scholar] [CrossRef] [PubMed]
- Hicks, C.W.; Selvin, E. Epidemiology of Peripheral Neuropathy and Lower Extremity Disease in Diabetes. Curr. Diabetes Rep. 2019, 19, 86. [Google Scholar] [CrossRef]
- Preston, F.G.; Riley, D.R.; Azmi, S.; Alam, U. Painful Diabetic Peripheral Neuropathy: Practical Guidance and Challenges for Clinical Management. Diabetes Metab. Syndr. Obes. 2023, 16, 1595–1612. [Google Scholar] [CrossRef]
- Schreiber, A.K. Diabetic Neuropathic Pain: Physiopathology and Treatment. World J. Diabetes 2015, 6, 432. [Google Scholar] [CrossRef]
- O’Brien, P.D.; Hinder, L.M.; Sakowski, S.A.; Feldman, E.L. ER Stress in Diabetic Peripheral Neuropathy: A New Therapeutic Target. Antioxid. Redox Signal. 2014, 21, 621–633. [Google Scholar] [CrossRef] [PubMed]
- Sandireddy, R.; Yerra, V.G.; Areti, A.; Komirishetty, P.; Kumar, A. Neuroinflammation and Oxidative Stress in Diabetic Neuropathy: Futuristic Strategies Based on These Targets. Int. J. Endocrinol. 2014, 2014, 674987. [Google Scholar] [CrossRef] [PubMed]
- Fernyhough, P.; Calcutt, N.A. Abnormal Calcium Homeostasis in Peripheral Neuropathies. Cell Calcium 2010, 47, 130–139. [Google Scholar] [CrossRef]
- Bryda, E.C. The Mighty Mouse: The Impact of Rodents on Advances in Biomedical Research. Mo. Med. 2013, 110, 207–211. [Google Scholar] [PubMed]
- Islam, M.S. Animal Models of Diabetic Neuropathy: Progress Since 1960s. J. Diabetes Res. 2013, 2013, 149452. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, P.D.; Sakowski, S.A.; Feldman, E.L. Mouse Models of Diabetic Neuropathy. ILAR J. 2014, 54, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Saini, T.; Mazumder, P.M. Current Advancement in the Preclinical Models Used for the Assessment of Diabetic Neuropathy. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2024, 397, 2727–2745. [Google Scholar] [CrossRef]
- Shaikh, A.; Somani, R. Animal Models and Biomarkers of Neuropathy in Diabetic Rodents. Indian J. Pharmacol. 2010, 42, 129. [Google Scholar] [CrossRef]
- Pham, V.; Matsumura, S.; Katano, T.; Funatsu, N.; Ito, S. Diabetic Neuropathy Research: From Mouse Models to Targets for Treatment. Neural Regen. Res. 2019, 14, 1870. [Google Scholar] [CrossRef]
- Singh, R.; Gholipourmalekabadi, M.; Shafikhani, S.H. Animal Models for Type 1 and Type 2 Diabetes: Advantages and Limitations. Front. Endocrinol. 2024, 15, 1359685. [Google Scholar] [CrossRef] [PubMed]
- Mehta, B.K.; Nerkar, D.; Banerjee, S. Characterization of Peripheral Neuropathy in Rat Model of Type 2 Diabetes. Indian J. Pharm. Educ. Res. 2017, 51, 92–101. [Google Scholar] [CrossRef]
- Skovsø, S. Modeling Type 2 Diabetes in Rats Using High Fat Diet and Streptozotocin. J. Diabetes Investig. 2014, 5, 349–358. [Google Scholar] [CrossRef]
- Flier, J.S. Insulin Receptors and Insulin Resistance. Annu. Rev. Med. 1983, 34, 145–160. [Google Scholar] [CrossRef]
- Andreeva-Gateva, P.; Hristov, M.; Strokova-Stoilova, M.; Ivanova, N.; Sabit, Z.; Surcheva, S.; Beliakov, M.; Karakashev, G.; Sukhov, I.; Belinskaya, D.; et al. Therapeutic Potential of Orally Applied KB-R7943 in Streptozotocin-Induced Neuropathy in Rats. Heliyon 2024, 10, e27367. [Google Scholar] [CrossRef] [PubMed]
- King, A.J. The Use of Animal Models in Diabetes Research. Br. J. Pharmacol. 2012, 166, 877–894. [Google Scholar] [CrossRef] [PubMed]
- Biessels, G.J.; Bril, V.; Calcutt, N.A.; Cameron, N.E.; Cotter, M.A.; Dobrowsky, R.; Feldman, E.L.; Fernyhough, P.; Jakobsen, J.; Malik, R.A.; et al. Phenotyping Animal Models of Diabetic Neuropathy: A Consensus Statement of the Diabetic Neuropathy Study Group of the EASD (Neurodiab). J. Peripher. Nerv. Sys. 2014, 19, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Farooq, S.A.; Mannan, A.; Singh, T.G.; Najda, A.; Grażyna, Z.; Albadrani, G.M.; Sayed, A.A.; Abdel-Daim, M.M. Animal Models of Diabetic Microvascular Complications: Relevance to Clinical Features. Biomed. Pharmacother. 2022, 145, 112305. [Google Scholar] [CrossRef] [PubMed]
- Filho, O.A.R.; Fazan, V.P.S. Streptozotocin Induced Diabetes as a Model of Phrenic Nerve Neuropathy in Rats. J. Neurosci. Methods 2006, 151, 131–138. [Google Scholar] [CrossRef]
- Raafat, K.; Aboul-Ela, M.; El-Lakany, A. Alloxan-Induced Diabetic Thermal Hyperalgesia, Prophylaxis and Phytotherapeutic Effects of Rheum ribes L. in Mouse Model. Arch. Pharm. Res. 2021, 44, 1–10. [Google Scholar] [CrossRef]
- Davidson, E.P.; Coppey, L.J.; Calcutt, N.A.; Oltman, C.L.; Yorek, M.A. Diet-Induced Obesity in Sprague-Dawley Rats Causes Microvascular and Neural Dysfunction. Diabetes Metab. Res. Rev. 2010, 26, 306–318. [Google Scholar] [CrossRef] [PubMed]
- Preguiça, I.; Alves, A.; Nunes, S.; Gomes, P.; Fernandes, R.; Viana, S.D.; Reis, F. Diet-Induced Rodent Models of Diabetic Peripheral Neuropathy, Retinopathy and Nephropathy. Nutrients 2020, 12, 250. [Google Scholar] [CrossRef] [PubMed]
- Guilford, B.L.; Ryals, J.M.; Wright, D.E. Phenotypic Changes in Diabetic Neuropathy Induced by a High-Fat Diet in Diabetic C57BL/6 Mice. Exp. Diabetes Res. 2011, 2011, 848307. [Google Scholar] [CrossRef] [PubMed]
- Obrosova, I.G.; Ilnytska, O.; Lyzogubov, V.V.; Pavlov, I.A.; Mashtalir, N.; Nadler, J.L.; Drel, V.R. High-Fat Diet–Induced Neuropathy of Pre-Diabetes and Obesity. Diabetes 2007, 56, 2598–2608. [Google Scholar] [CrossRef] [PubMed]
- Winzell, M.S.; Ahrén, B. The High-Fat Diet-Fed Mouse: A Model for Studying Mechanisms and Treatment of Impaired Glucose Tolerance and Type 2 Diabetes. Diabetes 2004, 53 (Suppl. 3), S215–S219. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.J.; Kendig, M.D.; Letton, M.E.; Morris, M.J.; Arnold, R. Peripheral Neuropathy Phenotyping in Rat Models of Type 2 Diabetes Mellitus: Evaluating Uptake of the Neurodiab Guidelines and Identifying Future Directions. Diabetes Metab. J. 2022, 46, 198–221. [Google Scholar] [CrossRef] [PubMed]
- Coppey, L.J.; Shevalye, H.; Obrosov, A.; Davidson, E.P.; Yorek, M.A. Determination of Peripheral Neuropathy in High-Fat Diet Fed Low-Dose Streptozotocin-Treated Female C57Bl/6J Mice and Sprague-Dawley Rats. J. Diabetes Investig. 2018, 9, 1033–1040. [Google Scholar] [CrossRef]
- Szkudelski, T. The Mechanism of Alloxan and Streptozotocin Action in B Cells of the Rat Pancreas. Physiol. Res. 2001, 50, 537–546. [Google Scholar] [CrossRef]
- Kostyuk, E.; Svichar, N.; Shishkin, V.; Kostyuk, P. Role of Mitochondrial Dysfunction in Calcium Signalling Alterations in Dorsal Root Ganglion Neurons of Mice with Experimentally-Induced Diabetes. Neuroscience 1999, 90, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Lee-Kubli, C.A.; Mixcoatl-Zecuatl, T.; Jolivalt, C.G.; Calcutt, N.A. Animal Models of Diabetes-Induced Neuropathic Pain. In Behavioral Neurobiology of Chronic Pain; Taylor, B.K., Finn, D.P., Eds.; Current Topics in Behavioral Neurosciences; Springer: Berlin/Heidelberg, Germany, 2014; Volume 20, pp. 147–170. ISBN 978-3-662-45093-2. [Google Scholar]
- Ghasemi, A.; Jeddi, S. Streptozotocin as a Tool for Induction of Rat Models of Diabetes: A Practical Guide. EXCLI J. 2023, 22, 274–294. [Google Scholar] [CrossRef] [PubMed]
- Jaffey, P.B.; Gelman, B.B. Increased Vulnerability to Demyelination in Streptozotocin Diabetic Rats. J. Comp. Neurol. 1996, 373, 55–61. [Google Scholar] [CrossRef]
- Eftekharpour, E.; Fernyhough, P. Oxidative Stress and Mitochondrial Dysfunction Associated with Peripheral Neuropathy in Type 1 Diabetes. Antioxid. Redox Signal. 2022, 37, 578–596. [Google Scholar] [CrossRef]
- Vargas-Soria, M.; García-Alloza, M.; Corraliza-Gómez, M. Effects of Diabetes on Microglial Physiology: A Systematic Review of in Vitro, Preclinical and Clinical Studies. J. Neuroinflammation 2023, 20, 57. [Google Scholar] [CrossRef] [PubMed]
- Ighodaro, O.M.; Adeosun, A.M.; Akinloye, O.A. Alloxan-Induced Diabetes, a Common Model for Evaluating the Glycemic-Control Potential of Therapeutic Compounds and Plants Extracts in Experimental Studies. Medicina 2017, 53, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Boquist, L. A New Hypothesis for Alloxan Diabetes. Acta Pathol. Microbiol. Scand. Sect. A Pathol. 1980, 88A, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Bansal, R.; Ahmad, N.; Kidwai, J.R. Alloxan-Glucose Interaction: Effect on Incorporation of14C-Leucine into Pancreatic Islets of Rat. Acta Diabetol. Lat. 1980, 17, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Wattiez, A.-S.; Barrière, D.A. Rodent Models of Painful Diabetic Neuropathy: What Can We Learn from Them? J. Diabetes Metab. 2012. [Google Scholar] [CrossRef]
- Powell, H.; Knox, D.; Lee, S.; Charters, A.C.; Orloff, M.; Garrett, R.; Lampert, P. Alloxan Diabetic Neuropathy: Electron Microscopic Studies. Neurology 1977, 27, 60. [Google Scholar] [CrossRef]
- Belosludtsev, K.N.; Belosludtseva, N.V.; Dubinin, M.V. Diabetes Mellitus, Mitochondrial Dysfunction and Ca2+-Dependent Permeability Transition Pore. Int. J. Mol. Sci. 2020, 21, 6559. [Google Scholar] [CrossRef]
- Abd El-Emam, M.M.; Behairy, A.; Mostafa, M.; Khamis, T.; Osman, N.M.S.; Alsemeh, A.E.; Mansour, M.F. Chrysin-Loaded PEGylated Liposomes Protect against Alloxan-Induced Diabetic Neuropathy in Rats: The Interplay between Endoplasmic Reticulum Stress and Autophagy. Biol. Res. 2024, 57, 45. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Ding, Y.; Tanaka, Y.; Zhang, W. Risk Factors Contributing to Type 2 Diabetes and Recent Advances in the Treatment and Prevention. Int. J. Med. Sci. 2014, 11, 1185–1200. [Google Scholar] [CrossRef]
- Klein, S.; Gastaldelli, A.; Yki-Järvinen, H.; Scherer, P.E. Why Does Obesity Cause Diabetes? Cell Metab. 2022, 34, 11–20. [Google Scholar] [CrossRef]
- Amelia, R.; Wahyuni, A.S.; Yunanda, Y. Diabetic Neuropathy among Type 2 Diabetes Mellitus Patients at Amplas Primary Health Care in Medan City. Open Access Maced. J. Med. Sci. 2019, 7, 3400–3403. [Google Scholar] [CrossRef] [PubMed]
- Speakman, J.R. Use of High-Fat Diets to Study Rodent Obesity as a Model of Human Obesity. Int. J. Obes. 2019, 43, 1491–1492. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Lv, X.-Y.; Li, J.; Xu, Z.-G.; Chen, L. The Characterization of High-Fat Diet and Multiple Low-Dose Streptozotocin Induced Type 2 Diabetes Rat Model. J. Diabetes Res. 2008, 2008, 704045. [Google Scholar] [CrossRef] [PubMed]
- Cavaliere, G.; Trinchese, G.; Penna, E.; Cimmino, F.; Pirozzi, C.; Lama, A.; Annunziata, C.; Catapano, A.; Mattace Raso, G.; Meli, R.; et al. High-Fat Diet Induces Neuroinflammation and Mitochondrial Impairment in Mice Cerebral Cortex and Synaptic Fraction. Front. Cell. Neurosci. 2019, 13, 509. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Bai, Y.; Song, F. High-Fat Diet and Neuroinflammation: The Role of Mitochondria. Pharmacol. Res. 2025, 212, 107615. [Google Scholar] [CrossRef] [PubMed]
- Marques Miranda, C.; De Lima Campos, M.; Leite-Almeida, H. Diet, Body Weight and Pain Susceptibility—A Systematic Review of Preclinical Studies. Neurobiol. Pain 2021, 10, 100066. [Google Scholar] [CrossRef]
- Burgoyne, R.D.; Haynes, L.P. Understanding the Physiological Roles of the Neuronal Calcium Sensor Proteins. Mol. Brain 2012, 5, 2. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.-J.; Sayers, N.M.; Fernyhough, P.; Verkhratsky, A. Diabetes-Induced Alterations in Calcium Homeostasis in Sensory Neurones of Streptozotocin-Diabetic Rats Are Restricted to Lumbar Ganglia and Are Prevented by Neurotrophin-3. Diabetologia 2002, 45, 560–570. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.; Gavin, J.R.; Sowers, J.R. Diabetes Mellitus: A Disease of Abnormal Cellular Calcium Metabolism? Am. J. Med. 1994, 96, 260–273. [Google Scholar] [CrossRef]
- Lowery, J.M.; Eichberg, J.; Saubermann, A.J.; LoPachin, R.M. Distribution of Elements and Water in Peripheral Nerve of Streptozocin-Induced Diabetic Rats. Diabetes 1990, 39, 1498–1503. [Google Scholar] [CrossRef] [PubMed]
- Obradovic, A.L.; Hwang, S.M.; Scarpa, J.; Hong, S.J.; Todorovic, S.M.; Jevtovic-Todorovic, V. CaV3.2 T-Type Calcium Channels in Peripheral Sensory Neurons Are Important for Mibefradil-Induced Reversal of Hyperalgesia and Allodynia in Rats with Painful Diabetic Neuropathy. PLoS ONE 2014, 9, e91467. [Google Scholar] [CrossRef] [PubMed]
- Amran, M.S.; Homma, N.; Hashimoto, K. Pharmacology of KB-R7943: A Na+–Ca2+ Exchange Inhibitor. Cardiovasc. Drug Rev. 2003, 21, 255–276. [Google Scholar] [CrossRef]
- Lu, J.; Yang, L.; Xu, Y.; Ai, L.; Chen, J.; Xiong, F.; Hu, L.; Chen, H.; Liu, J.; Yan, X.; et al. The Modulatory Effect of Motor Cortex Astrocytes on Diabetic Neuropathic Pain. J. Neurosci. 2021, 41, 5287–5302. [Google Scholar] [CrossRef]
- Annunziato, L.; Pignataro, G.; Di Renzo, G.F. Pharmacology of Brain Na+/Ca2+ Exchanger: From Molecular Biology to Therapeutic Perspectives. Pharmacol. Rev. 2004, 56, 633–654. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, T.; Shigekawa, M. Differential Inhibition of Na+/Ca2+ Exchanger Isoforms by Divalent Cations and Isothiourea Derivative. Am. J. Physiol.-Cell Physiol. 1998, 275, C423–C430. [Google Scholar] [CrossRef]
- Herchuelz, A.; Nguidjoe, E.; Jiang, L.; Pachera, N. Na+/Ca2+ Exchange and the Plasma Membrane Ca2+-ATPase in β-Cell Function and Diabetes. In Sodium Calcium Exchange: A Growing Spectrum of Pathophysiological Implications; Annunziato, L., Ed.; Advances in Experimental Medicine and Biology; Springer: Boston, MA, USA, 2013; Volume 961, pp. 385–394. ISBN 978-1-4614-4755-9. [Google Scholar]
- Li, Y.; Woo, V.; Bose, R. Platelet Hyperactivity and Abnormal Ca2+ Homeostasis in Diabetes Mellitus. Am. J. Physiol.-Heart Circ. Physiol. 2001, 280, H1480–H1489. [Google Scholar] [CrossRef]
- Liu, D.; Cui, K.-Z.; Sun, Y.-M.; Liu, J.-W.; Li, Y.-B.; Su, Y. Protective Effects of the Sodium/Calcium Exchanger Inhibitor on Endothelial Dysfunction Induced by High Glucose. Exp. Clin. Endocrinol. Diabetes 2014, 123, 7–10. [Google Scholar] [CrossRef]
- Liamis, G.; Liberopoulos, E.; Barkas, F.; Elisaf, M. Diabetes Mellitus and Electrolyte Disorders. World J. Clin. Cases 2014, 2, 488–496. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, C.; Huang, L.; Shen, X.; Zhao, F.; Wu, C.; Yan, S. Relationship between Hyponatremia and Peripheral Neuropathy in Patients with Diabetes. J. Diabetes Res. 2021, 2021, 9012887. [Google Scholar] [CrossRef]
- Matchkov, V.V.; Gustafsson, H.; Rahman, A.; Briggs Boedtkjer, D.M.; Gorintin, S.; Hansen, A.K.; Bouzinova, E.V.; Praetorius, H.A.; Aalkjaer, C.; Nilsson, H. Interaction Between Na+/K+-Pump and Na+/Ca2+-Exchanger Modulates Intercellular Communication. Circ. Res. 2007, 100, 1026–1035. [Google Scholar] [CrossRef]
- Snutch, T.P. Targeting Chronic and Neuropathic Pain: The N-Type Calcium Channel Comes of Age. Neurotherapeutics 2005, 2, 662–670. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.D.; Calcutt, N.A.; Higuera, E.S.; Valder, C.R.; Song, Y.-H.; Svensson, C.I.; Myers, R.R. Injury Type-Specific Calcium Channel α2 δ-1 Subunit Up-Regulation in Rat Neuropathic Pain Models Correlates with Antiallodynic Effects of Gabapentin. J. Pharmacol. Exp. Ther. 2002, 303, 1199–1205. [Google Scholar] [CrossRef] [PubMed]
- Dray, A. Neuropathic Pain: Emerging Treatments. Br. J. Anaesth. 2008, 101, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Song, X.-J. Diabetic Neuropathic Pain: Directions for Exploring Treatments. Biomedicines 2024, 12, 589. [Google Scholar] [CrossRef] [PubMed]
- Bril, V.; Breiner, A.; Perkins, B.A.; Zochodne, D. Neuropathy. Can. J. Diabetes 2018, 42, S217–S221. [Google Scholar] [CrossRef] [PubMed]
- Price, R.; Smith, D.; Franklin, G.; Gronseth, G.; Pignone, M.; David, W.S.; Armon, C.; Perkins, B.A.; Bril, V.; Rae-Grant, A.; et al. Oral and Topical Treatment of Painful Diabetic Polyneuropathy: Practice Guideline Update Summary: Report of the AAN Guideline Subcommittee. Neurology 2022, 98, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Finnerup, N.B.; Attal, N.; Haroutounian, S.; McNicol, E.; Baron, R.; Dworkin, R.H.; Gilron, I.; Haanpää, M.; Hansson, P.; Jensen, T.S.; et al. Pharmacotherapy for Neuropathic Pain in Adults: A Systematic Review and Meta-Analysis. Lancet Neurol. 2015, 14, 162–173. [Google Scholar] [CrossRef]
- Brustovetsky, T.; Brittain, M.K.; Sheets, P.L.; Cummins, T.R.; Pinelis, V.; Brustovetsky, N. KB-R7943, an Inhibitor of the Reverse Na+ /Ca2+ Exchanger, Blocks N-methyl-D-aspartate Receptor and Inhibits Mitochondrial Complex I. Br. J. Pharmacol. 2011, 162, 255–270. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Wang, Y.; Chen, X.; Chen, H.; Kintner, D.B.; Shull, G.E.; Philipson, K.D.; Sun, D. Increased Tolerance to Ischemic Neuronal Damage by Knockdown of Na+–Ca2+ Exchanger Isoform 1. Ann. N. Y. Acad. Sci. 2007, 1099, 292–305. [Google Scholar] [CrossRef]
- Hernandez-Ojeda, M.; Ureña-Guerrero, M.E.; Gutierrez-Barajas, P.E.; Cardenas-Castillo, J.A.; Camins, A.; Beas-Zarate, C. KB-R7943 Reduces 4-Aminopyridine-Induced Epileptiform Activity in Adult Rats after Neuronal Damage Induced by Neonatal Monosodium Glutamate Treatment. J. Biomed. Sci. 2017, 24, 27. [Google Scholar] [CrossRef] [PubMed]
- Schröder, U.H.; Breder, J.; Sabelhaus, C.F.; Reymann, K.G. The Novel Na+/Ca2+ Exchange Inhibitor KB-R7943 Protects CA1 Neurons in Rat Hippocampal Slices against Hypoxic/Hypoglycemic Injury. Neuropharmacology 1999, 38, 319–321. [Google Scholar] [CrossRef]
- Li, S.; Jiang, Q.; Stys, P.K. Important Role of Reverse Na+-Ca2+ Exchange in Spinal Cord White Matter Injury at Physiological Temperature. J. Neurophysiol. 2000, 84, 1116–1119. [Google Scholar] [CrossRef] [PubMed]
- Hoyt, K.R.; Arden, S.R.; Aizenman, E.; Reynolds, I.J. Reverse Na+/Ca2+ Exchange Contributes to Glutamate-Induced Intracellular Ca2+ Concentration Increases in Cultured Rat Forebrain Neurons. Mol. Pharmacol. 1998, 53, 742–749. [Google Scholar] [CrossRef] [PubMed]
- Newton, J.; Akinfiresoye, L.R.; N’Gouemo, P. Inhibition of the Sodium Calcium Exchanger Suppresses Alcohol Withdrawal-Induced Seizure Susceptibility. Brain Sci. 2021, 11, 279. [Google Scholar] [CrossRef] [PubMed]
- Bobylev, I.; Maru, H.; Joshi, A.R.; Lehmann, H.C. Toxicity to Sensory Neurons and Schwann Cells in Experimental Linezolid-Induced Peripheral Neuropathy. J. Antimicrob. Chemother. 2016, 71, 685–691. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Method | Model | Features of DN | Advantages | Disadvantages | ||
|---|---|---|---|---|---|---|
| Chemically-induced (type 1 diabetes) | STZ (single high doses or multiple low dose) | Nerve conduction velocity | Behavior | Nerve structure | ||
| Rats (single high doses of 35–60 mg/kg/day) [14,17,26,27] | Decreased sensory and motor conduction velocity | Allodynia; hyperalgesia to hypoalgesia | Axonal degeneration; decreased intraepidermal nerve fiber density; reduced corneal nerve fiber length; Schwann cell proliferation; reduced right and left fascicles of phrenic nerve; molecular changes of the peripheral nerves | Low cost; fast induction of diabetes; known duration of diabetes and DN development | High mortality rate; neuropathy phenotype variability; cell/organ toxicity; lack of severe neuropathy changes resembling human pathology; still to elucidate DN pathogenic aspects | |
| Mice (single high doses of 100–200 mg/kg/day or multiple low doses of 40–50 mg/kg/day) [15] | Slow sensory and motor conduction velocity | Allodynia; hypoalgesia | Decreased intraepidermal nerve fiber density; decreased size of dorsal root ganglia neuron; molecular changes of the peripheral nerves | Low cost; fast induction of diabetes; known duration of diabetes and DN development | Toxicity and late development of neuropathy in the single high-doses model; lack of severe neuropathy changes resembling human pathology; lack of neuropathy feature of some strains in the multiple low dose | |
| Alloxan | ||||||
| Rats and mice (single doses of 40–200 mg/kg for rats and 50–200 mg/kg for mice) [14,17,28] | Slow nerve conduction velocity | Allodynia; hyperalgesia; autonomic dysfunction | - | Low cost and affordability; fast induction of diabetes | High mortality rate; narrow diabetic dose; lack of late neuropathy changes; DN insufficient data | |
| Diet-induced (prediabetes/metabolic syndrome) | High-energy diet (high-fat, high-sugar, high-fat-high-sugar diets) | |||||
| Rats [17,29,30] (fat 25–60% and carbohydrates 10–80% of food content) | Decreased sensory conduction velocity | Allodynia; hypoalgesia | Decreased intraepidermal nerve fiber density and corneal nerve fiber length | Suitable to study insulin resistant and prediabetes related neuropathy | Extended time to DN development compared to drug-induced DN; lack of frank hyperglycemia | |
| Mice [26,30,31,32,33] (fat 25–60% and carbohydrates 10–80% of food content) | Decreased sensory and motor conduction velocity | Allodynia; early hyperalgesia, late hypoalgesia | Decreased intraepidermal nerve fiber density | Suitable to study insulin resistant and prediabetes related neuropathy | Extended time to DN development compared to drug-induced DN; lack of frank hyperglycemia | |
| Drug and diet combined (type 2 diabetes) | STR (low doses to moderate) + high-energy-diet | |||||
| Rats [29,30,34,35] | Slow sensory and motor conduction velocity | Hyper- or hypoalgesia; allodynia | Decreased intraepidermal nerve fiber density; reduction in corneal nerve fiber length; axonal swelling and degeneration; lymphocyte infiltration; Schwann cell damage and demyelination | Shorter time to DN development compared to matched mice; less toxic; gradual development of type 2 diabetes and neuropathy changes | Extended time compared to drug-induced DN development | |
| Mice [30,35] | Slow sensory and motor conduction velocity | Hypoalgesia | Decreased intraepidermal nerve fiber density | Less toxic; gradual development of type 2 diabetes and neuropathy changes | Extended time compared to drug-induced DN development | |
| Feature | NCX1 | NCX2 | NCX3 |
|---|---|---|---|
| Tissue Distribution | Heart, skeletal muscle, brain | Brain, retina, kidney | Skeletal muscle, brain |
| Main Function | Ca2+ extrusion in muscle, heart | Ca2+ regulation in neurons, glial cells | Ca2+ regulation in skeletal muscle |
| Pathophysiological Role | Heart failure, arrhythmias | Neurodegenerative diseases, synaptic dysfunction | Muscular disorders, neurodegeneration |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ivanova, N.; Hristov, M.; Gateva, P. Rodent Models of Diabetic Neuropathy, Role of Calcium Homeostasis in Pain and KB-R7943 as a Potential Therapeutic. Int. J. Mol. Sci. 2025, 26, 2094. https://doi.org/10.3390/ijms26052094
Ivanova N, Hristov M, Gateva P. Rodent Models of Diabetic Neuropathy, Role of Calcium Homeostasis in Pain and KB-R7943 as a Potential Therapeutic. International Journal of Molecular Sciences. 2025; 26(5):2094. https://doi.org/10.3390/ijms26052094
Chicago/Turabian StyleIvanova, Natasha, Milen Hristov, and Pavlina Gateva. 2025. "Rodent Models of Diabetic Neuropathy, Role of Calcium Homeostasis in Pain and KB-R7943 as a Potential Therapeutic" International Journal of Molecular Sciences 26, no. 5: 2094. https://doi.org/10.3390/ijms26052094
APA StyleIvanova, N., Hristov, M., & Gateva, P. (2025). Rodent Models of Diabetic Neuropathy, Role of Calcium Homeostasis in Pain and KB-R7943 as a Potential Therapeutic. International Journal of Molecular Sciences, 26(5), 2094. https://doi.org/10.3390/ijms26052094