

Theoretical Study on the Metabolic Mechanism of Heptachlor in Human Cytochrome P450 Enzymes
Abstract

1. Introduction
2. Results and Discussion
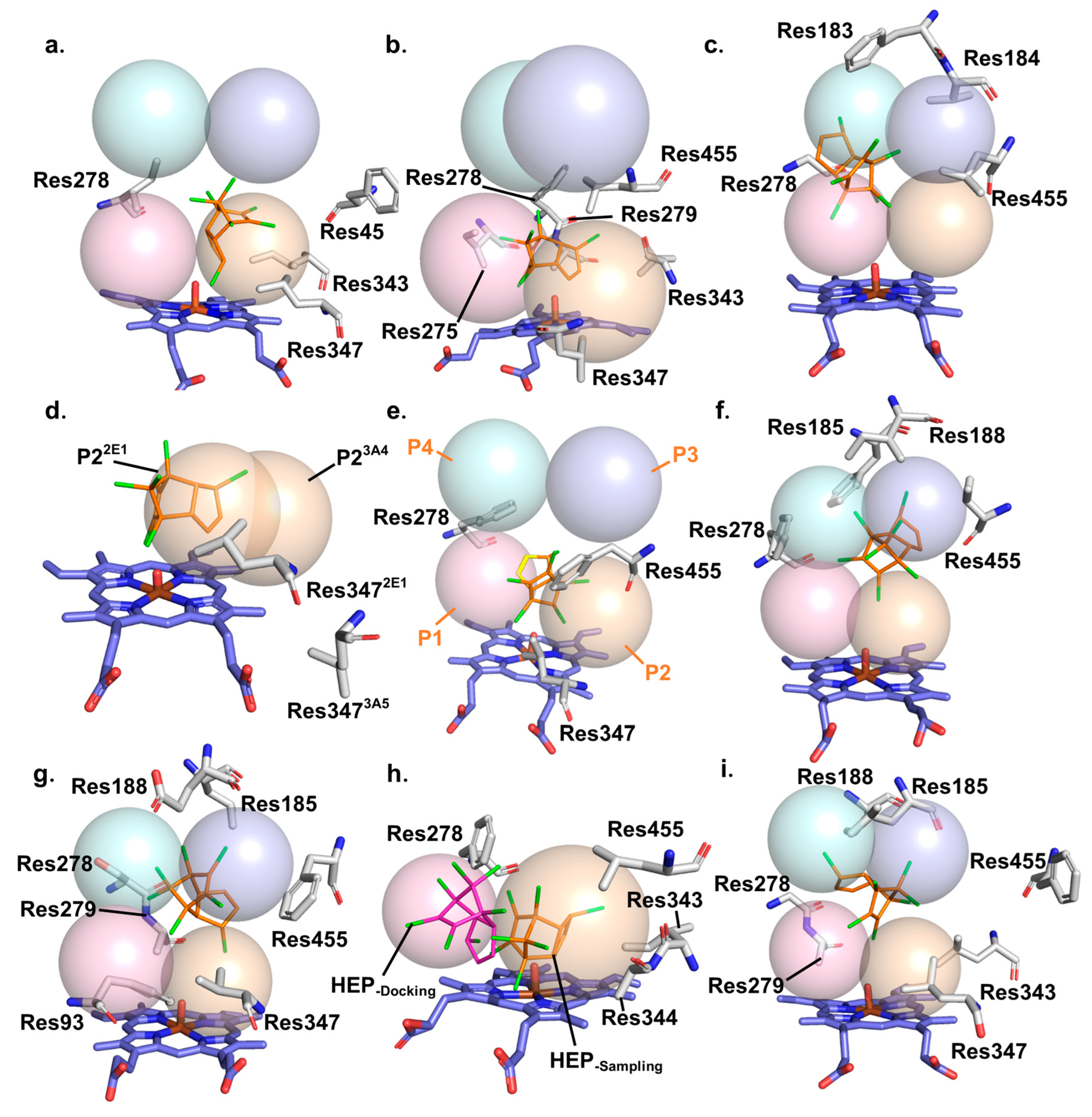
2.1. Different Binding Modes of HEP to Eight CYP Enzymes and Influencing Factors
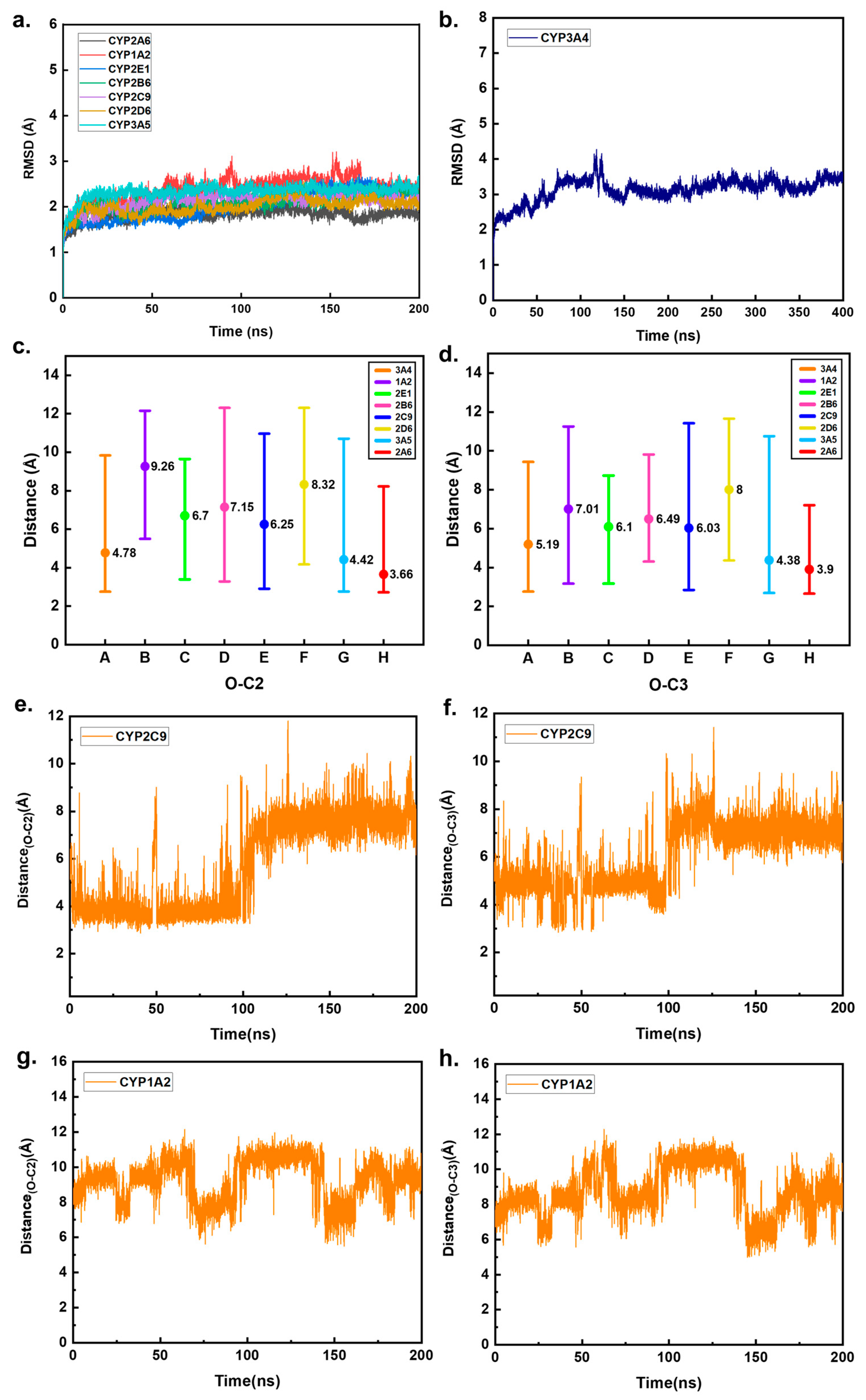
2.2. Preliminary Prediction of CYP Enzymes That May Metabolize HEP in Humans
2.3. Key Factors Affecting the Binding Site of HEP in CYP Enzymes
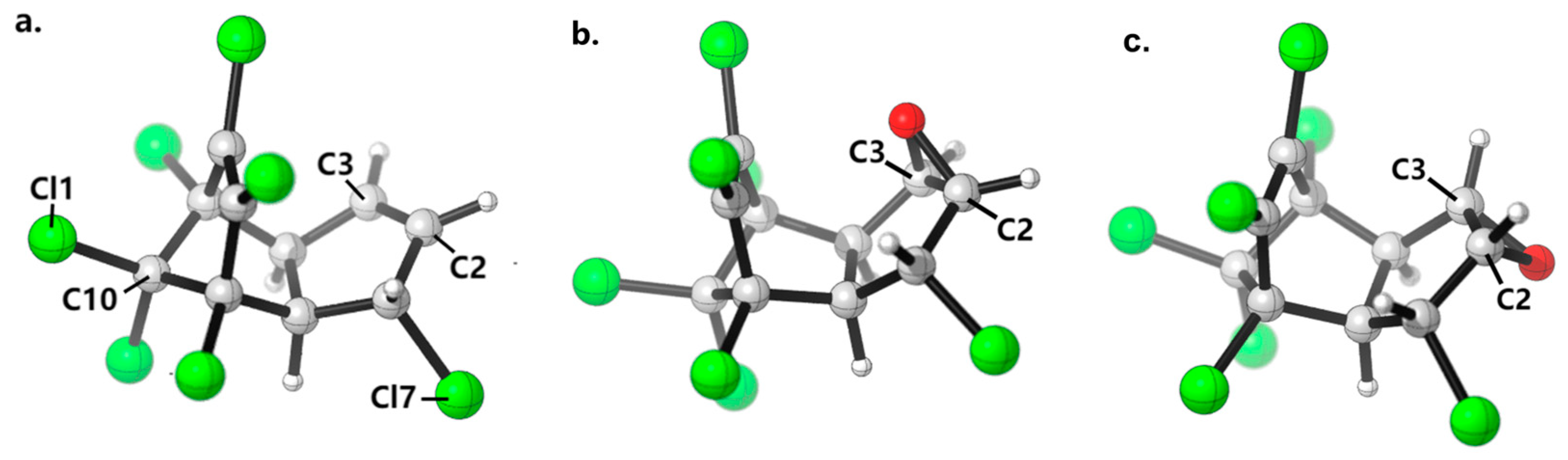
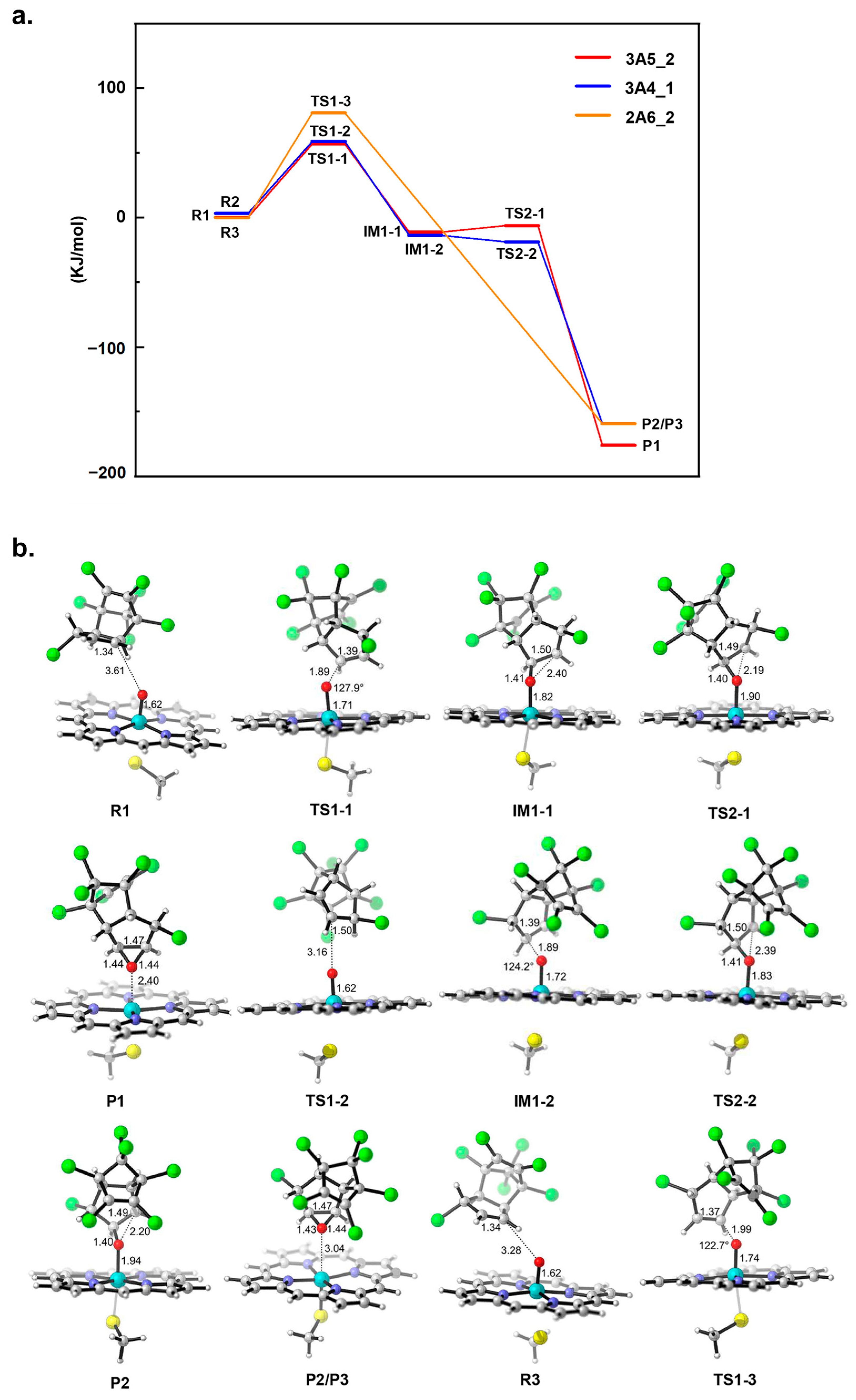
2.4. Potential Energy Surface Analysis of HEP Epoxidation Catalyzed by Different CYP Enzymes
3. Materials and Methods
3.1. Model Preparation
3.2. Molecular Docking and Molecular Dynamics Simulation
3.3. Binding Free-Energy Calculation
3.4. Quantum Mechanical Calculation
4. Contributions and Future Plans
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fendick, E.A.; Mather-Mihaich, E.; Houck, K.A.; St. Clair, M.B.; Faust, J.B.; Rockwell, C.H.; Owens, M. Ecological Toxicology and Human Health Effects of Heptachlor. In Reviews of Environmental Contamination and Toxicology; Ware, G.W., Ed.; Reviews of Environmental Contamination and Toxicology; Springer New York: New York, NY, USA, 1990; Volume 111, pp. 61–142. ISBN 978-1-4612-7972-3. [Google Scholar]
- Nomata, K.; Kang, K.-S.; Hayashi, T.; Matesic, D.; Lockwood, L.; Chang, C.C.; Trosko, J.E. Inhibition of Gap Junctional Intercellular Communication in Heptachlor- and Heptachlor Epoxide-Treated Normal Human Breast Epithelial Cells. Cell Biol. Toxicol. 1996, 12, 69–78. [Google Scholar] [CrossRef]
- Epstein, S.S. Carcinogenicity of Heptachlor and Chlordane. Sci. Total Environ. 1976, 6, 103–154. [Google Scholar] [CrossRef]
- Rincón-Rubio, A.; Mérida-Ortega, Á.; Ugalde-Resano, R.; Gamboa-Loira, B.; Rothenberg, S.J.; González, F.B.; Cebrián, M.E.; López-Carrillo, L. Carcinogenic, Non-Carcinogenic Risk, and Attributable Cases to Organochlorine Pesticide Exposure in Women from Northern Mexico. Environ. Monit. Assess. 2024, 196, 421. [Google Scholar] [CrossRef]
- Luderer, U.; Kesner, J.S.; Fuller, J.M.; Krieg, E.F.; Meadows, J.W.; Tramma, S.L.; Yang, H.; Baker, D. Effects of Gestational and Lactational Exposure to Heptachlor Epoxide on Age at Puberty and Reproductive Function in Men and Women. Environ. Res. 2013, 121, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Prado, G.; Bhalli, J.A.; Marcos, R. Genotoxicity of Heptachlor and Heptachlor Epoxide in Human TK6 Lymphoblastoid Cells. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2009, 673, 87–91. [Google Scholar] [CrossRef]
- Abbott, D. Environmental-Health Criteria.38. Heptachlor—Who. J. R. Soc. Health 1985, 105, 190. [Google Scholar]
- Roque, I.; Lourenço, R.; Marques, A.; Martínez-López, E.; Espín, S.; Gómez-Ramirez, P.; García-Fernández, A.J.; Roulin, A.; Rabaça, J.E. A First Record of Organochlorine Pesticides in Barn Owls (Tyto alba) from Portugal: Assessing Trends from Variation in Feather and Liver Concentrations. Bull. Environ. Contam. Toxicol. 2022, 109, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Chen, X.; Fan, L.; Hu, G.; Qiu, L.; Song, C.; Xie, Y.; Giesy, J.P.; Wang, C.; Meng, S. Environment Consistently Impact on Aquaculture: The Predominant Source of Residual Pollutants in Cultured Chinese Mitten Crab (Eriocheir sinensis) across China. Heliyon 2024, 10, e32418. [Google Scholar] [CrossRef] [PubMed]
- Sahyoun, W.; Net, S.; López-Maldonado, E.A.; Baroudi, M.; Ouddane, B. Occurrence and Health Risk Estimate of Organochlorine Pesticides in Fruits and Vegetables Matrices. Environ. Sci. Pollut. Res. 2024, 31, 1–16. [Google Scholar] [CrossRef]
- Wang, N.; Cui, Z.; Wang, Y.; Zhang, J. Characteristics and Residual Health Risk of Organochlorine Pesticides in Fresh Vegetables in the Suburb of Changchun, Northeast China. Int. J. Environ. Res. Public Health 2022, 19, 12547. [Google Scholar] [CrossRef] [PubMed]
- Kumah, E.K.; Arah, I.K.; Anku, E.K.; Akuaku, J.; Aidoo, M.K. Determination of Levels of Organochlorine Pesticide Residues in Some Common Grown and Consumed Vegetables Purchased from Ho Municipal Markets, Ghana. Cogent Food Agric. 2023, 9, 2191810. [Google Scholar] [CrossRef]
- Tashiro, S.; Matsumura, F. Metabolism Oftrans-Nonachlor and Related Chlordane Components in Rat and Man. Arch. Environ. Contam. Toxicol. 1978, 7, 113–127. [Google Scholar] [CrossRef]
- Radomski, J.L.; Davidow, B. The Metabolite of Heptachlor, Its Estimation Storage, and Toxicity. J. Pharmacol. Exp. Ther. 1953, 107, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, P.; Liang, Y.; Zhan, J.; Liu, D.; Zhou, Z. Enantioselective Characteristics, Bioaccumulation and Toxicological Effects of Chlordane-Related Compounds in Laying Hens. Chemosphere 2022, 300, 134486. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, F.; Nelson, J.O. Indentification of the Major Metabolic Product of Heptachlor Epoxide in Rat Feces. Bull. Environ. Contam. Toxicol. 1970, 5, 489–492. [Google Scholar] [CrossRef]
- Butler Walker, J.; Seddon, L.; McMullen, E.; Houseman, J.; Tofflemire, K.; Corriveau, A.; Weber, J.-P.; Mills, C.; Smith, S.; Van Oostdam, J. Organochlorine Levels in Maternal and Umbilical Cord Blood Plasma in Arctic Canada. Sci. Total Environ. 2003, 302, 27–52. [Google Scholar] [CrossRef]
- Mariottini, M.; Guerranti, C.; Aurigi, S.; Corsi, I.; Focardi, S. Pesticides and Polychlorinated Biphenyl Residues in Human Adipose Tissue. Bull. Environ. Contam. Toxicol. 2002, 68, 72–78. [Google Scholar] [CrossRef]
- Jonsson, V.; Liu, G.J.K.; Armbruster, J.; Kettelhut, L.L.; Drucker, B. Chlorohydrocarbon Pesticide Residues in Human Milk in Greater St. Louis, Missouri 1977. Am. J. Clin. Nutr. 1977, 30, 1106–1109. [Google Scholar] [CrossRef]
- Adeshina, F.; Todd, E.L. Organochlorine Compounds in Human Adipose Tissue from North Texas. J. Toxicol. Environ. Health 1990, 29, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Nair, P.C.; McKinnon, R.A.; Miners, J.O. Cytochrome P450 Structure–Function: Insights from Molecular Dynamics Simulations. Drug Metab. Rev. 2016, 48, 434–452. [Google Scholar] [CrossRef]
- Davydov, R.; Perera, R.; Jin, S.; Yang, T.-C.; Bryson, T.A.; Sono, M.; Dawson, J.H.; Hoffman, B.M. Substrate Modulation of the Properties and Reactivity of the Oxy-Ferrous and Hydroperoxo-Ferric Intermediates of Cytochrome P450cam As Shown by Cryoreduction-EPR/ENDOR Spectroscopy. J. Am. Chem. Soc. 2005, 127, 1403–1413. [Google Scholar] [CrossRef]
- Song, W.J.; Ryu, Y.O.; Song, R.; Nam, W. Oxoiron(IV) Porphyrin π-Cation Radical Complexes with a Chameleon Behavior in Cytochrome P450 Model Reactions. J. Biol. Inorg. Chem. 2005, 10, 294–304. [Google Scholar] [CrossRef]
- Abdo, W.; Hirata, A.; Sakai, H.; El-Sawak, A.; Nikami, H.; Yanai, T. Combined Effects of Organochlorine Pesticides Heptachlor and Hexachlorobenzene on the Promotion Stage of Hepatocarcinogenesis in Rats. Food Chem. Toxicol. 2013, 55, 578–585. [Google Scholar] [CrossRef]
- Zemoria, E.; Rosemond, G.; Todd, D.; Williams, M.; Paikoff, S.; Rhoades, J.; Llados, F.; Bosch, S. Toxicological Profile for Heptachlor and Heptachlor Epoxide; Agency for Toxic Substances and Disease Registry (ATSDR) Toxicological Profiles; Agency for Toxic Substances and Disease Registry: Atlanta, GA, USA, 2007.
- Zhao, L.; Sun, N.; Tian, L.; Zhao, S.; Sun, B.; Sun, Y.; Zhao, D. Strategies for the Development of Highly Selective Cytochrome P450 Inhibitors: Several CYP Targets in Current Research. Bioorganic Med. Chem. Lett. 2019, 29, 2016–2024. [Google Scholar] [CrossRef]
- Lonsdale, R.; Harvey, J.N.; Mulholland, A.J. Compound I Reactivity Defines Alkene Oxidation Selectivity in Cytochrome P450cam. J. Phys. Chem. B 2010, 114, 1156–1162. [Google Scholar] [CrossRef]
- Oláh, J.; Mulholland, A.J.; Harvey, J.N. Understanding the Determinants of Selectivity in Drug Metabolism through Modeling of Dextromethorphan Oxidation by Cytochrome P450. Proc. Natl. Acad. Sci. USA 2011, 108, 6050–6055. [Google Scholar] [CrossRef] [PubMed]
- Yano, J.K.; Wester, M.R.; Schoch, G.A.; Griffin, K.J.; Stout, C.D.; Johnson, E.F. The Structure of Human Microsomal Cytochrome P450 3A4 Determined by X-Ray Crystallography to 2.05-Å Resolution *. J. Biol. Chem. 2004, 279, 38091–38094. [Google Scholar] [CrossRef]
- Scott, E.E.; Halpert, J.R. Structures of Cytochrome P450 3A4. Trends Biochem. Sci. 2005, 30, 5–7. [Google Scholar] [CrossRef]
- Zhao, Y.; White, M.A.; Muralidhara, B.K.; Sun, L.; Halpert, J.R.; Stout, C.D. Structure of Microsomal Cytochrome P450 2B4 Complexed with the Antifungal Drug Bifonazole: Insight into P450 Conformational Plasticity and Membrane Interaction *. J. Biol. Chem. 2006, 281, 5973–5981. [Google Scholar] [CrossRef] [PubMed]
- Ekroos, M.; Sjögren, T. Structural Basis for Ligand Promiscuity in Cytochrome P450 3A4. Proc. Natl. Acad. Sci. USA 2006, 103, 13682–13687. [Google Scholar] [CrossRef] [PubMed]
- Skopalík, J.; Anzenbacher, P.; Otyepka, M. Flexibility of Human Cytochromes P450: Molecular Dynamics Reveals Differences between CYPs 3A4, 2C9, and 2A6, Which Correlate with Their Substrate Preferences. J. Phys. Chem. B 2008, 112, 8165–8173. [Google Scholar] [CrossRef] [PubMed]
- Kandagatla, S.K.; Mack, T.; Simpson, S.; Sollenberger, J.; Helton, E.; Raner, G.M. Inhibition of Human Cytochrome P450 2E1 and 2A6 by Aldehydes: Structure and Activity Relationships. Chem. Biol. Interact. 2014, 219, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Porubsky, P.R.; Meneely, K.M.; Scott, E.E. Structures of Human Cytochrome P-450 2E1: Insights into the Binding of Inhibitors and both Small Molecular Weight and Fatty Acid Substrates *. J. Biol. Chem. 2008, 283, 33698–33707. [Google Scholar] [CrossRef]
- Keizers, P.H.J.; Lussenburg, B.M.A.; de Graaf, C.; Mentink, L.M.; Vermeulen, N.P.E.; Commandeur, J.N.M. Influence of Phenylalanine 120 on Cytochrome P450 2D6 Catalytic Selectivity and Regiospecificity: Crucial Role in 7-Methoxy-4-(Aminomethyl)-Coumarin Metabolism. Biochem. Pharmacol. 2004, 68, 2263–2271. [Google Scholar] [CrossRef]
- Keizers, P.H.J.; de Graaf, C.; de Kanter, F.J.J.; Oostenbrink, C.; Feenstra, K.A.; Commandeur, J.N.M.; Vermeulen, N.P.E. Metabolic Regio- and Stereoselectivity of Cytochrome P450 2D6 towards 3,4-Methylenedioxy-N-Alkylamphetamines: In Silico Predictions and Experimental Validation. J. Med. Chem. 2005, 48, 6117–6127. [Google Scholar] [CrossRef] [PubMed]
- Hsu, M.-H.; Johnson, E.F. Active-Site Differences between Substrate-Free and Ritonavir-Bound Cytochrome P450 (CYP) 3A5 Reveal Plasticity Differences between CYP3A5 and CYP3A4. J. Biol. Chem. 2019, 294, 8015–8022. [Google Scholar] [CrossRef] [PubMed]
- Hsu, M.-H.; Savas, U.; Johnson, E.F. The X-ray Crystal Structure of the Human Mono-Oxygenase Cytochrome P450 3A5-Ritonavir Complex Reveals Active Site Differences between P450s 3A4 and 3A5. Mol. Pharmacol. 2018, 93, 14–24. [Google Scholar] [CrossRef] [PubMed]
- de Visser, S.P.; Kumar, D.; Shaik, S. How Do Aldehyde Side Products Occur during Alkene Epoxidation by Cytochrome P450? Theory Reveals a State-Specific Multi-State Scenario Where the High-Spin Component Leads to All Side Products. J. Inorg. Biochem. 2004, 98, 1183–1193. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; de Visser, S.P.; Shaik, S. Multistate Reactivity in Styrene Epoxidation by Compound I of Cytochrome P450: Mechanisms of Products and Side Products Formation. Chem. A Eur. J. 2005, 11, 2825–2835. [Google Scholar] [CrossRef] [PubMed]
- de Visser, S.P.; Ogliaro, F.; Harris, N.; Shaik, S. Multi-State Epoxidation of Ethene by Cytochrome P450: A Quantum Chemical Study. J. Am. Chem. Soc. 2001, 123, 3037–3047. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.-N.; Zhao, Y.-X.; Xue, W.; Wang, Z.-C.; He, S.-G.; Ding, X.-L. Active Sites of Stoichiometric Cerium Oxide Cations (Ce m O 2m+) Probed by Reactions with Carbon Monoxide and Small Hydrocarbon Molecules. Phys. Chem. Chem. Phys. 2010, 12, 3984–3997. [Google Scholar] [CrossRef]
- Zhao, Y.-X.; Wu, X.-N.; Ma, J.-B.; He, S.-G.; Ding, X.-L. Experimental and Theoretical Study of the Reactions between Vanadium−Silicon Heteronuclear Oxide Cluster Anions with N-Butane. J. Phys. Chem. C 2010, 114, 12271–12279. [Google Scholar] [CrossRef]
- Ma, J.-B.; Wu, X.-N.; Zhao, X.-X.; Ding, X.-L.; He, S.-G. Methane Activation by V3PO10˙+ and V4O10˙+ Clusters: A Comparative Study. Phys. Chem. Chem. Phys. 2010, 12, 12223–12228. [Google Scholar] [CrossRef] [PubMed]
- Feyel, S.; Döbler, J.; Höckendorf, R.; Beyer, M.K.; Sauer, J.; Schwarz, H. Activation of Methane by Oligomeric (Al2O3)+ (X=3,4,5): The Role of Oxygen-Centered Radicals in Thermal Hydrogen-Atom Abstraction. Angew. Chem. Int. Ed. 2008, 47, 1946–1950. [Google Scholar] [CrossRef]
- Coleman, T.; Kirk, A.M.; Chao, R.R.; Podgorski, M.N.; Harbort, J.S.; Churchman, L.R.; Bruning, J.B.; Bernhardt, P.V.; Harmer, J.R.; Krenske, E.H.; et al. Understanding the Mechanistic Requirements for Efficient and Stereoselective Alkene Epoxidation by a Cytochrome P450 Enzyme. ACS Catal. 2021, 11, 1995–2010. [Google Scholar] [CrossRef]
- Shaik, S.; Cohen, S.; Wang, Y.; Chen, H.; Kumar, D.; Thiel, W. P450 Enzymes: Their Structure, Reactivity, and Selectivity—Modeled by QM/MM Calculations. Chem. Rev. 2010, 110, 949–1017. [Google Scholar] [CrossRef] [PubMed]
- Podgorski, M.N.; Coleman, T.; Chao, R.R.; De Voss, J.J.; Bruning, J.B.; Bell, S.G. Investigation of the Requirements for Efficient and Selective Cytochrome P450 Monooxygenase Catalysis across Different Reactions. J. Inorg. Biochem. 2020, 203, 110913. [Google Scholar] [CrossRef] [PubMed]
- Don, C.G.; Smieško, M. Deciphering Reaction Determinants of Altered-Activity CYP2D6 Variants by Well-Tempered Metadynamics Simulation and QM/MM Calculations. J. Chem. Inf. Model. 2020, 60, 6642–6653. [Google Scholar] [CrossRef]
- Williams, J.A.; Ring, B.J.; Cantrell, V.E.; Jones, D.R.; Eckstein, J.; Ruterbories, K.; Hamman, M.A.; Hall, S.D.; Wrighton, S.A. Comparative Metabolic Capabilities of CYP3A4, CYP3A5, and CYP3A7. Drug Metab. Dispos. 2002, 30, 883–891. [Google Scholar] [CrossRef]
- Bakken, G.V.; Rudberg, I.; Christensen, H.; Molden, E.; Refsum, H.; Hermann, M. Metabolism of Quetiapine by CYP3A4 and CYP3A5 in Presence or Absence of Cytochrome B5. Drug Metab. Dispos. 2009, 37, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Towles, J.K.; Clark, R.N.; Wahlin, M.D.; Uttamsingh, V.; Rettie, A.E.; Jackson, K.D. Cytochrome P450 3A4 and CYP3A5-Catalyzed Bioactivation of Lapatinib. Drug Metab. Dispos. 2016, 44, 1584–1597. [Google Scholar] [CrossRef]
- Kamdem, L.K.; Meineke, I.; Gödtel-Armbrust, U.; Brockmöller, J.; Wojnowski, L. Dominant Contribution of P450 3A4 to the Hepatic Carcinogenic Activation of Aflatoxin B1. Chem. Res. Toxicol. 2006, 19, 577–586. [Google Scholar] [CrossRef]
- De Vore, N.M.; Meneely, K.M.; Bart, A.G.; Stephens, E.S.; Battaile, K.P.; Scott, E.E. Structural Comparison of Cytochromes P450 2A6, 2A13, and 2E1 with Pilocarpine. FEBS J. 2012, 279, 1621–1631. [Google Scholar] [CrossRef] [PubMed]
- Uno, T.; Obe, Y.; Ogura, C.; Goto, T.; Yamamoto, K.; Nakamura, M.; Kanamaru, K.; Yamagata, H.; Imaishi, H. Metabolism of 7-Ethoxycoumarin, Safrole, Flavanone and Hydroxyflavanone by Cytochrome P450 2A6 Variants. Biopharm. Drug Dispos. 2013, 34, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Le Gal, A. Diversity of Selective Environmental Substrates for Human Cytochrome P450 2A6: Alkoxyethers, Nicotine, Coumarin, N-Nitrosodiethylamine, and N-Nitrosobenzylmethylamine. Toxicol. Lett. 2003, 144, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Hosono, H.; Kumondai, M.; Maekawa, M.; Yamaguchi, H.; Mano, N.; Oda, A.; Hirasawa, N.; Hiratsuka, M. Functional Characterization of 34 CYP2A6 Allelic Variants by Assessment of Nicotine C-Oxidation and Coumarin 7-Hydroxylation Activities. Drug Metab. Dispos. 2017, 45, 279–285. [Google Scholar] [CrossRef]
- Manikandan, P.; Nagini, S. Cytochrome P450 Structure, Function and Clinical Significance: A Review. Curr. Drug Target 2018, 19, 1225–1232. [Google Scholar] [CrossRef]
- Zanger, U.M.; Schwab, M. Cytochrome P450 Enzymes in Drug Metabolism: Regulation of Gene Expression, Enzyme Activities, and Impact of Genetic Variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef] [PubMed]
- Sansen, S.; Yano, J.K.; Reynald, R.L.; Schoch, G.A.; Griffin, K.J.; Stout, C.D.; Johnson, E.F. Adaptations for the Oxidation of Polycyclic Aromatic Hydrocarbons Exhibited by the Structure of Human P450 1A2. J. Biol. Chem. 2007, 282, 14348–14355. [Google Scholar] [CrossRef] [PubMed]
- Yano, J.K.; Hsu, M.-H.; Griffin, K.J.; Stout, C.D.; Johnson, E.F. Structures of Human Microsomal Cytochrome P450 2A6 Complexed with Coumarin and Methoxsalen. Nat. Struct. Mol. Biol. 2005, 12, 822–823. [Google Scholar] [CrossRef]
- Gay, S.C.; Shah, M.B.; Talakad, J.C.; Maekawa, K.; Roberts, A.G.; Wilderman, P.R.; Sun, L.; Yang, J.Y.; Huelga, S.C.; Hong, W.-X.; et al. Crystal Structure of a Cytochrome P450 2B6 Genetic Variant in Complex with the Inhibitor 4-(4-Chlorophenyl)Imidazole at 2.0-Å Resolution. Mol. Pharmacol. 2010, 77, 529–538. [Google Scholar] [CrossRef]
- Brändén, G.; Sjögren, T.; Schnecke, V.; Xue, Y. Structure-Based Ligand Design to Overcome CYP Inhibition in Drug Discovery Projects. Drug Discov. Today 2014, 19, 905–911. [Google Scholar] [CrossRef]
- Butler, C.R.; Ogilvie, K.; Martinez-Alsina, L.; Barreiro, G.; Beck, E.M.; Nolan, C.E.; Atchison, K.; Benvenuti, E.; Buzon, L.; Doran, S.; et al. Aminomethyl-Derived Beta Secretase (BACE1) Inhibitors: Engaging Gly230 without an Anilide Functionality. J. Med. Chem. 2017, 60, 386–402. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chen, L.; Crichlow, G.V.; Christie, C.H.; Dalenberg, K.; Di Costanzo, L.; Duarte, J.M.; et al. RCSB Protein Data Bank: Powerful New Tools for Exploring 3D Structures of Biological Macromolecules for Basic and Applied Research and Education in Fundamental Biology, Biomedicine, Biotechnology, Bioengineering and Energy Sciences. Nucleic Acids Res. 2021, 49, D437–D451. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Fu, T.; Zheng, Q.; Zhang, H. Investigation of the Molecular and Mechanistic Basis for the Regioselective Metabolism of Midazolam by Cytochrome P450 3A4. Phys. Chem. Chem. Phys. 2022, 24, 8104–8112. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Protein Structure Modeling with MODELLER. In Structural Genomics; Chen, Y.W., Yiu, C.-P.B., Eds.; Springer: New York, NY, USA, 2021; Volume 2199, pp. 239–255. ISBN 978-1-07-160891-3. [Google Scholar]
- Monk, B.C.; Tomasiak, T.M.; Keniya, M.V.; Huschmann, F.U.; Tyndall, J.D.A.; O’Connell, J.D.; Cannon, R.D.; McDonald, J.G.; Rodriguez, A.; Finer-Moore, J.S.; et al. Architecture of a Single Membrane Spanning Cytochrome P450 Suggests Constraints That Orient the Catalytic Domain Relative to a Bilayer. Proc. Natl. Acad. Sci. USA 2014, 111, 3865–3870. [Google Scholar] [CrossRef] [PubMed]
- Dennington, R.; Todd, A.K.; John, M. Millam GaussView, Version 6. Available online: https://gaussian.com/gaussview6/ (accessed on 13 August 2024).
- Cantú Reinhard, F.G.; De Visser, S.P. Biodegradation of Cosmetics Products: A Computational Study of Cytochrome P450 Metabolism of Phthalates. Inorganics 2017, 5, 77. [Google Scholar] [CrossRef]
- Bolton, E.E.; Chen, J.; Kim, S.; Han, L.; He, S.; Shi, W.; Simonyan, V.; Sun, Y.; Thiessen, P.A.; Wang, J.; et al. PubChem3D: A New Resource for Scientists. J. Cheminformatics 2011, 3, 32. [Google Scholar] [CrossRef]
- Kim, S.; Bolton, E.E.; Bryant, S.H. PubChem3D: Conformer Ensemble Accuracy. J. Cheminformatics 2013, 5, 1. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01. Available online: https://gaussian.com/ (accessed on 13 August 2024).
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Tirado-Rives, J.; Jorgensen, W.L. Performance of B3LYP Density Functional Methods for a Large Set of Organic Molecules. J. Chem. Theory Comput. 2008, 4, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Anandakrishnan, R.; Aguilar, B.; Onufriev, A.V. H++ 3.0: Automating pK Prediction and the Preparation of Biomolecular Structures for Atomistic Molecular Modeling and Simulations. Nucleic Acids Res. 2012, 40, W537–W541. [Google Scholar] [CrossRef]
- Gordon, J.C.; Myers, J.B.; Folta, T.; Shoja, V.; Heath, L.S.; Onufriev, A. H++: A Server for Estimating pKas and Adding Missing Hydrogens to Macromolecules. Nucleic Acids Res. 2005, 33, W368–W371. [Google Scholar] [CrossRef] [PubMed]
- Myers, J.; Grothaus, G.; Narayanan, S.; Onufriev, A. A Simple Clustering Algorithm Can Be Accurate Enough for Use in Calculations of pKs in Macromolecules. Proteins Struct. Funct. Bioinform. 2006, 63, 928–938. [Google Scholar] [CrossRef]
- Goodsell, D.S.; Morris, G.M.; Olson, A.J. Automated Docking of Flexible Ligands: Applications of Autodock. J. Mol. Recognit. 1996, 9, 1–5. [Google Scholar] [CrossRef]
- Case, D.; Betz, R.; Cerutti, D.S.; Cheatham, T.; Darden, T.; Duke, R.; Giese, T.J.; Gohlke, H.; Götz, A.; Homeyer, N.; et al. Amber 2016; University of California: San Francisco, CA, USA, 2016. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Shahrokh, K.; Orendt, A.; Yost, G.S.; Cheatham, T.E. Quantum Mechanically Derived AMBER-compatible Heme Parameters for Various States of the Cytochrome P450 Catalytic Cycle. J. Comput. Chem. 2012, 33, 119–133. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic Atom Type and Bond Type Perception in Molecular Mechanical Calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Uberuaga, B.P.; Anghel, M.; Voter, A.F. Synchronization of Trajectories in Canonical Molecular-Dynamics Simulations: Observation, Explanation, and Exploitation. J. Chem. Phys. 2004, 120, 6363–6374. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; Di Nola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N⋅log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Tsui, V.; Case, D.A. Theory and Applications of the Generalized Born Solvation Model in Macromolecular Simulations. Biopolymers 2000, 56, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Hay, P.J.; Wadt, W.R. Ab Initio Effective Core Potentials for Molecular Calculations. Potentials for the Transition Metal Atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- Fukui, K. Formulation of the Reaction Coordinate. J. Phys. Chem. 1970, 74, 4161–4163. [Google Scholar] [CrossRef]
- Fukui, K. The Path of Chemical Reactions—The IRC Approach. Acc. Chem. Res. 1981, 14, 363–368. [Google Scholar] [CrossRef]
- Hratchian, H.P.; Schlegel, H.B. Accurate Reaction Paths Using a Hessian Based Predictor–Corrector Integrator. J. Chem. Phys. 2004, 120, 9918–9924. [Google Scholar] [CrossRef] [PubMed]
- Hratchian, H.P.; Schlegel, H.B. Using Hessian Updating To Increase the Efficiency of a Hessian Based Predictor-Corrector Reaction Path Following Method. J. Chem. Theory Comput. 2005, 1, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Shaik, S.; Kumar, D.; de Visser, S.P.; Altun, A.; Thiel, W. Theoretical Perspective on the Structure and Mechanism of Cytochrome P450 Enzymes. Chem. Rev. 2005, 105, 2279–2328. [Google Scholar] [CrossRef] [PubMed]
- Jedrzejewski, M.; Bełza, B.; Lewandowska, I.; Sadlej, M.; Perlinska, A.P.; Augustyniak, R.; Christian, T.; Hou, Y.; Kalek, M.; Sulkowska, J.I. Nucleolar Essential Protein 1 (Nep1): Elucidation of Enzymatic Catalysis Mechanism by Combined Molecular Dynamics Simulation and Quantum Chemical Calculations. Comput. Struct. Biotechnol. J. 2023, 21, 3999–4008. [Google Scholar] [CrossRef]
- Feng, S.; Zheng, Q. Mechanism of 7H-Dibenzo[c,g]Carbazole Metabolism in Cytochrome P450 1A1: Insights from Computational Studies. J. Hazard. Mater. 2024, 476, 134933. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zheng, Q.; Zhang, J.; Xie, M.; Zhan, J.; Zhang, H. How Mutations Affecting the Ligand-Receptor Interactions: A Combined MD and QM/MM Calculation on CYP2E1 and Its Two Mutants. Chem. Res. Chin. Univ. 2015, 31, 1029–1038. [Google Scholar] [CrossRef]
- Feng, S.; Li, Y.; Zhang, R.; Zhang, Q.; Wang, W. Origin of Metabolites Diversity and Selectivity of P450 Catalyzed Benzo[a]Pyrene Metabolic Activation. J. Hazard. Mater. 2022, 435, 129008. [Google Scholar] [CrossRef]
- Mulashkina, T.; Kulakova, A.; Nemukhin, A.; Khrenova, M. Molecular Basis of the Substrate Specificity of the Pd-PTE Phosphotriesterase: A Combined QM/MM MD and Electron Density Study; Chemistry: Bhadohi, India, 2023. [Google Scholar]
- Zhou, J.; Zhang, X.; Li, Y.; Feng, S.; Zhang, Q.; Wang, W. Endocrine-Disrupting Metabolic Activation of 2-Nitrofluorene Catalyzed by Human Cytochrome P450 1A1: A QM/MM Approach. Environ. Int. 2022, 166, 107355. [Google Scholar] [CrossRef]
- Ma, G.; Yu, H.; Han, C.; Jia, Y.; Wei, X.; Wang, Z. Binding and Metabolism of Brominated Flame Retardant β-1,2-Dibromo-4-(1,2-Dibromoethyl)Cyclohexane in Human Microsomal P450 Enzymes: Insights from Computational Studies. Chem. Res. Toxicol. 2020, 33, 1487–1496. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Chen, H.; Fu, W.; Garcia-Borràs, M.; Yang, Y.; Liu, P. Engineered P450 Atom-Transfer Radical Cyclases Are Bifunctional Biocatalysts: Reaction Mechanism and Origin of Enantioselectivity. J. Am. Chem. Soc. 2022, 144, 13344–13355. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CYPs | O-C2 | O-C3 | At Least One |
|---|---|---|---|
| CYP1A2 | 0% | 0% | 0% |
| CYP2A6 | 82.43% | 58.31% | 92.47% |
| CYP2B6 | 0.74% | 0.01% | 0.75% |
| CYP2C9 | 27.4% | 8.26% | 27.78% |
| CYP2D6 | 0% | 0% | 0% |
| CYP2E1 | 0.01% | 0.94% | 0.94% |
| CYP3A4 | 42.74% | 9.58% | 46.99% |
| CYP3A5 | 39.21% | 30.32% | 63.36% |
| Energy Contribution | CYP2B6-HEP | CYP3A5-HEP | CYP2A6-HEP | CYP2E1-HEP | CYP3A4-HEP | CYP1A2-HEP | CYP2D6-HEP | CYP2C9-HEP |
|---|---|---|---|---|---|---|---|---|
| ΔEvdW | −137.76 | −137.18 | −153.59 | −168.57 | −133.03 | −142.24 | −140.40 | −129.05 |
| ΔEelec | −22.40 | −0.25 | 3.10 | −12.14 | −5.11 | −3.60 | −5.27 | −11.51 |
| ΔGGB | 49.81 | 21.18 | 23.89 | 30.73 | 31.48 | 29.34 | 36.50 | 35.29 |
| ΔGSA | −15.24 | −12.85 | −15.56 | −17.04 | −12.85 | −15.36 | −15.45 | −13.69 |
| ΔGbinding | −137.76 | −137.18 | −142.17 | −168.57 | −133.03 | −142.24 | −140.40 | −129.05 |
| Residues | CYP2B6-HEP | CYP3A5-HEP | CYP2A6-HEP | CYP2E1-HEP | CYP3A4-HEP | CYP1A2-HEP | CYP2D6-HEP | CYP2C9-HEP | |
|---|---|---|---|---|---|---|---|---|---|
| P1/P4 | Res278 | −4.31 | −3.51 | −6.07 | −7.53 | −6.15 | −2.43 | −1.30 | −1.13 |
| P1 | Res93 | −3.43 | −1.72 | −3.18 | −4.69 | −1.26 | −2.64 | −5.56 | −2.34 |
| Res275 | −0.29 | −4.23 | −2.93 | −0.02 | −1.72 | −2.80 | −0.84 | −0.13 | |
| Res279 | −1.63 | −3.35 | −2.64 | −4.52 | −5.82 | −5.15 | −5.98 | −5.48 | |
| P1/P2 | Res283 | −1.09 | −1.55 | −2.30 | −5.44 | −1.72 | −1.38 | −1.30 | −2.01 |
| P2 | Res343 | −5.52 | −3.10 | −5.06 | −5.82 | −2.26 | −0.79 | −0.21 | −4.81 |
| Res347 | −3.39 | −0.33 | −5.19 | −3.47 | −0.42 | −2.13 | −5.19 | −4.14 | |
| P2/P3 | Res455 | −5.61 | −4.90 | −1.38 | −4.23 | −4.99 | −4.94 | −6.15 | −0.75 |
| P3 | Res188 | −5.06 | 0.03 | −3.64 | −1.34 | −0.08 | −1.30 | −5.06 | −5.69 |
| P3/P4 | Res185 | −6.78 | −3.18 | −4.69 | −6.19 | −0.13 | −4.60 | −4.10 | −8.12 |
| P4 | Res184 | 0.00 | −0.96 | −1.59 | −0.54 | −0.08 | −4.31 | −2.26 | −2.13 |
| R | TS1 | IM2 | TS2 | P | |
|---|---|---|---|---|---|
| 2A6_1 (33.78%) | 0.33 | 57.10 | −11.32 | −6.09 | −175.98 |
| 3A5_2 (32.80%) | |||||
| 3A4_1 (32.80%) | 3.31 | 58.99 | −13.62 | −18.77 | −159.29 |
| 3A5_1 (37.01%) | |||||
| 2A6_2 (25.77%) | 0 | 81.06 | ____ | ____ | −159.29 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, X.; Zhang, H.; Shen, X.; Zheng, Q.; Wang, S. Theoretical Study on the Metabolic Mechanism of Heptachlor in Human Cytochrome P450 Enzymes. Int. J. Mol. Sci. 2025, 26, 2021. https://doi.org/10.3390/ijms26052021
Zhao X, Zhang H, Shen X, Zheng Q, Wang S. Theoretical Study on the Metabolic Mechanism of Heptachlor in Human Cytochrome P450 Enzymes. International Journal of Molecular Sciences. 2025; 26(5):2021. https://doi.org/10.3390/ijms26052021
Chicago/Turabian StyleZhao, Xuerui, Hao Zhang, Xiaoli Shen, Qingchuan Zheng, and Song Wang. 2025. "Theoretical Study on the Metabolic Mechanism of Heptachlor in Human Cytochrome P450 Enzymes" International Journal of Molecular Sciences 26, no. 5: 2021. https://doi.org/10.3390/ijms26052021
APA StyleZhao, X., Zhang, H., Shen, X., Zheng, Q., & Wang, S. (2025). Theoretical Study on the Metabolic Mechanism of Heptachlor in Human Cytochrome P450 Enzymes. International Journal of Molecular Sciences, 26(5), 2021. https://doi.org/10.3390/ijms26052021