
A Novel Pathogenic Variant of DICER1 Gene in a Young Greek Patient with 2 Different Sex-Cord Ovarian Tumors and Multinodular Goiter

 , , ,
, , ,  , and
, and 
Abstract
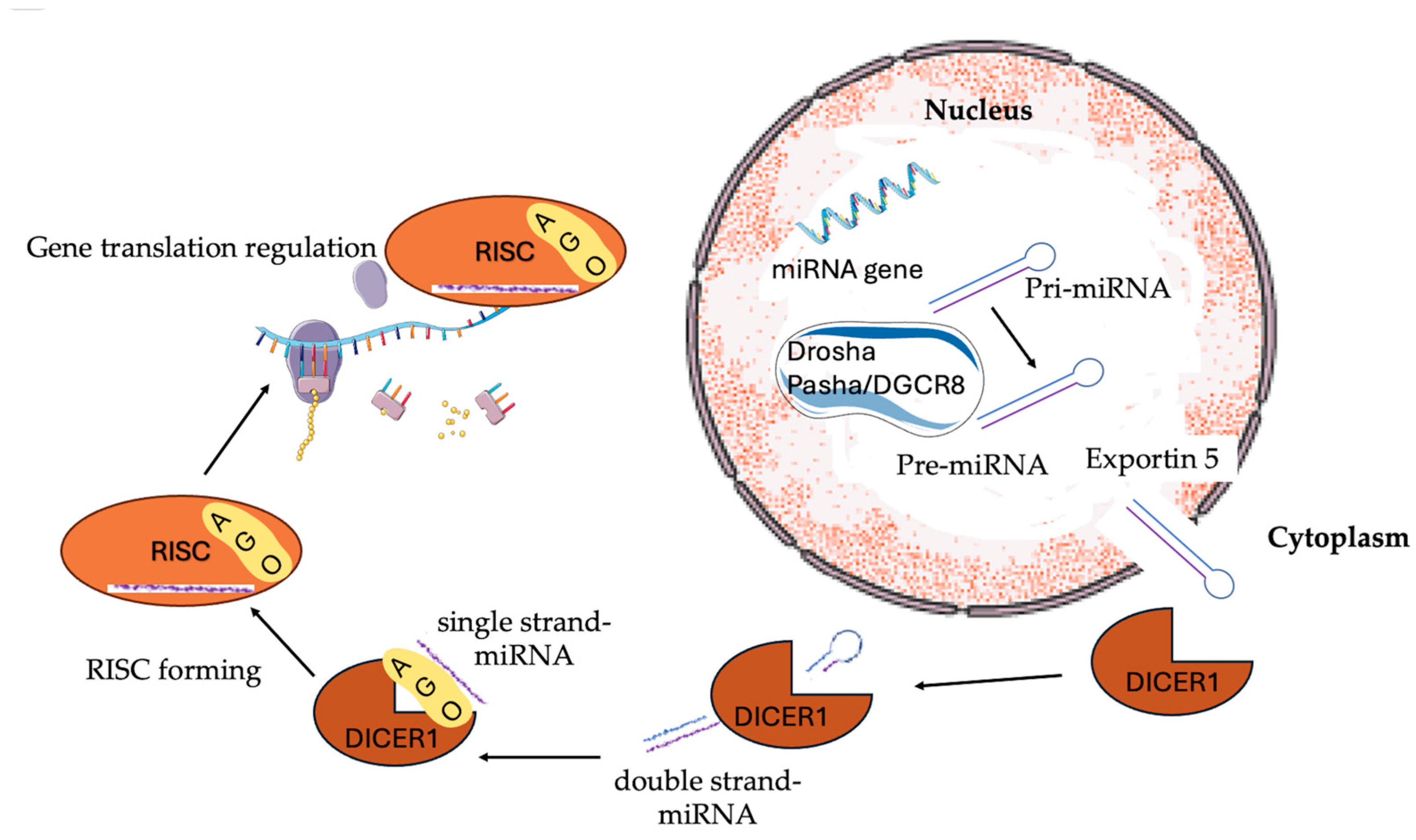
1. Introduction
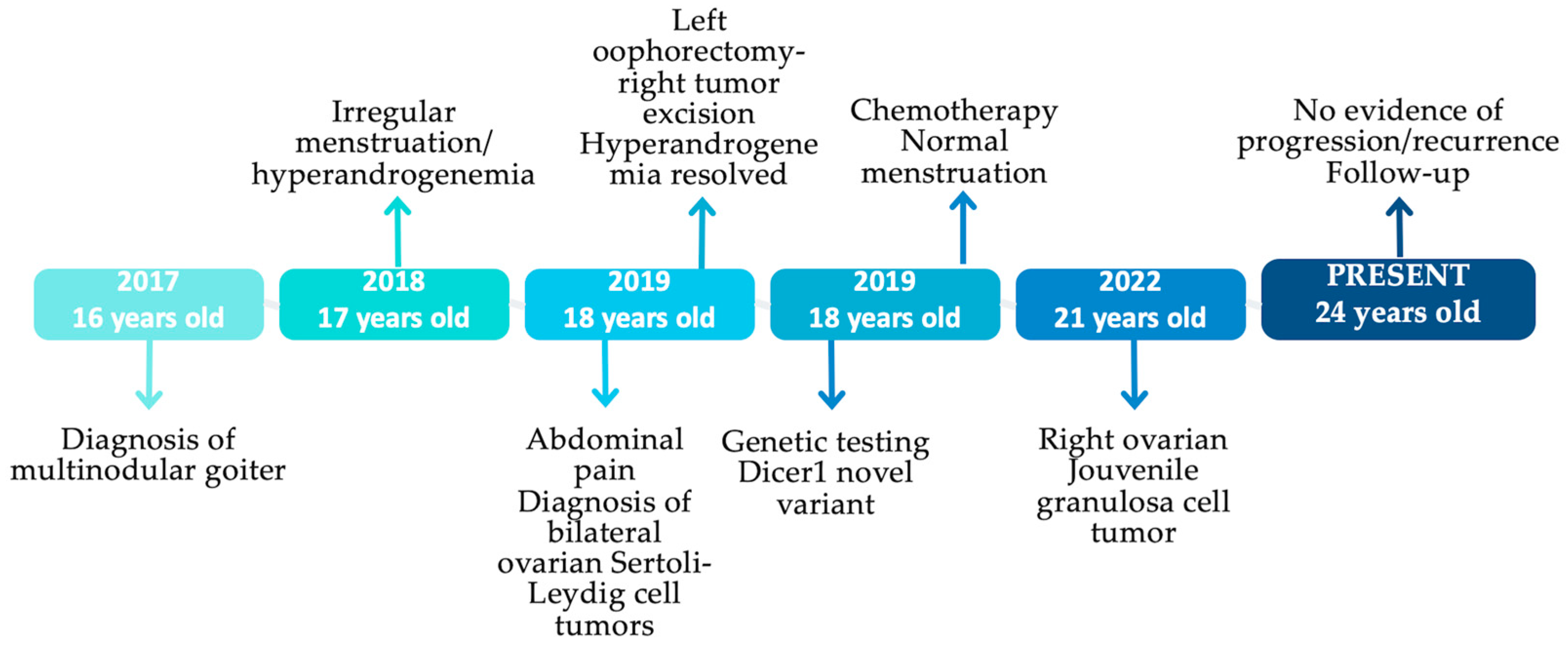
2. Case Presentation
2.1. Multinodular Goiter (MNG)
2.2. Irregular Menstruation-Hyperandrogenemia
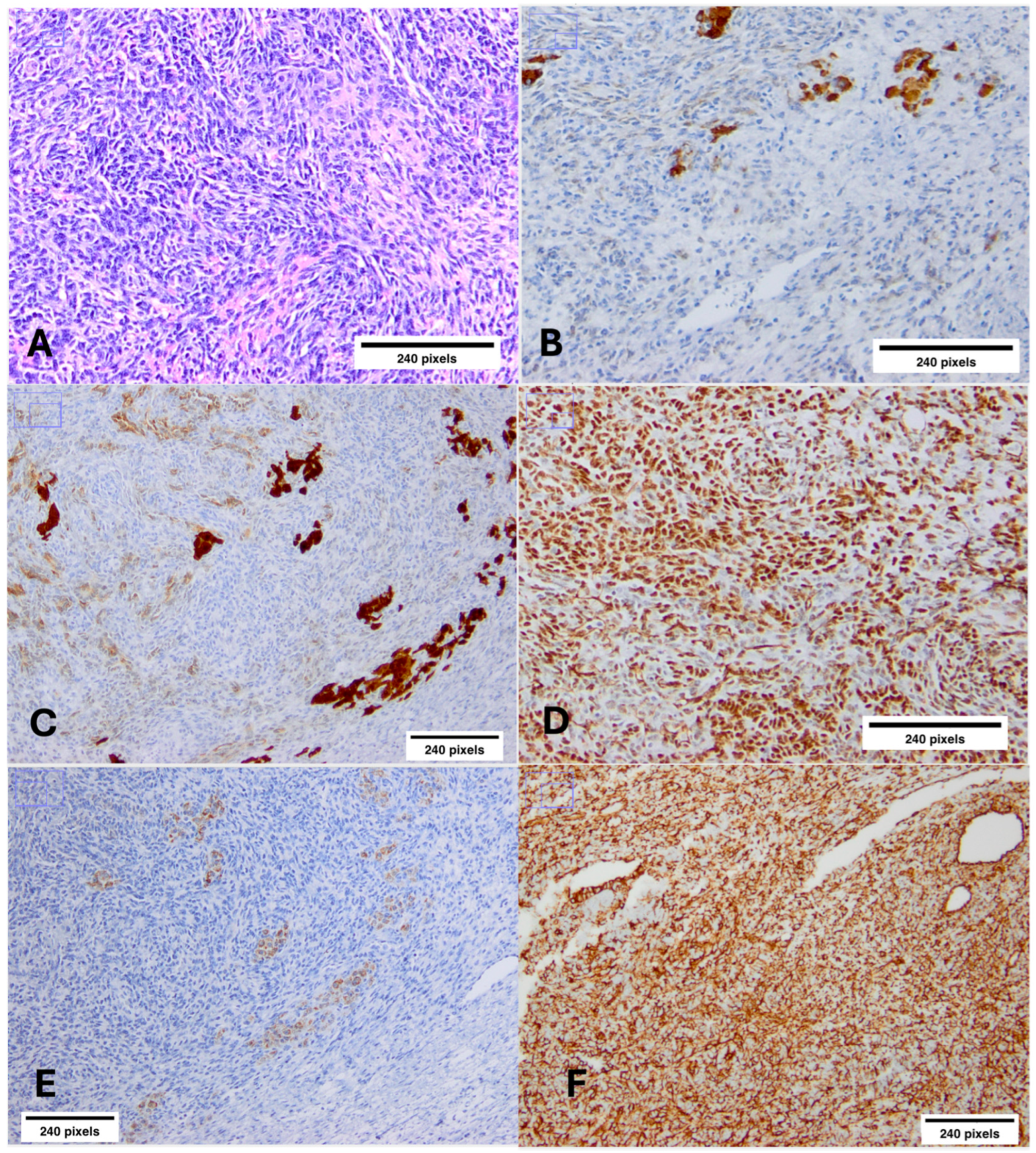
2.3. Bilateral Ovarian Sertoli–Leydig Cell Tumors (SLCTs)
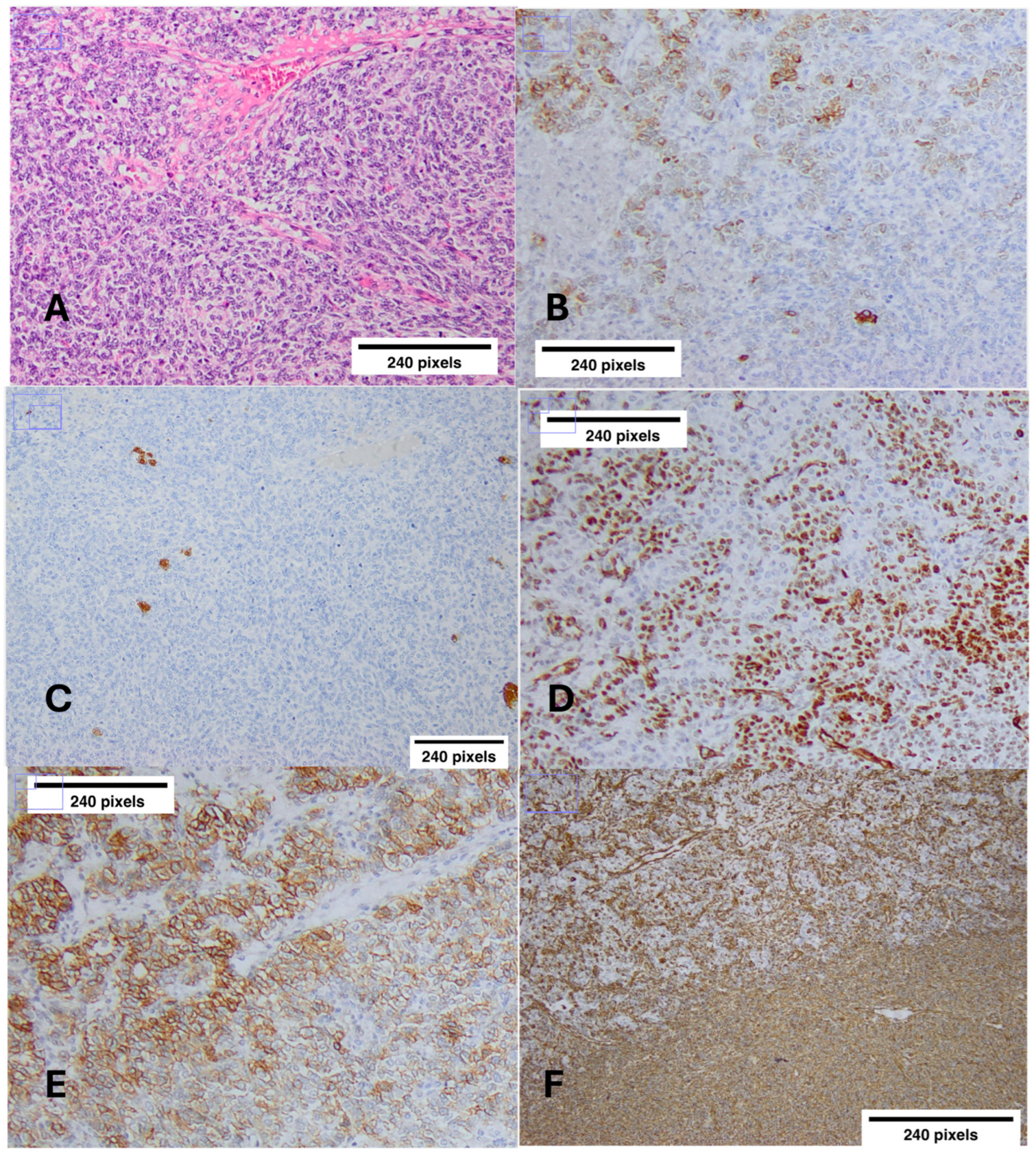
2.4. Jouvenile Granulosa Cell Tumor (JGCT)
2.5. Genetic Testing
2.6. Family History
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AFP | Alpha-fetoprotein |
| AGO | Argonaute protein |
| Ca 125 | Cancer antigen 125 |
| Ca 15-3 | Cancer antigen 15-3 |
| CEA | Carcinoembryonic antigen |
| CT | Computed tomography |
| DGCR85 | DiGeorge critical region 8 |
| DHEA-S | Dehydroepiandrosterone sulfate |
| DICER1s | DICER1 syndrome |
| FIGO | The International Federation of Gynecology and Obstetrics |
| FNA | Fine needle aspiration |
| FSH | Follicle-stimulating hormone |
| GLOW | Global developmental delay, Lung cysts, Overgrowth, Wilms tumor |
| IHC | Immunohistochemistry |
| JGCT | Juvenile granulosa cell tumor |
| LH | Luteinizing hormone |
| miRNA | Micro RNA |
| MNG | Multinodular goiter |
| MRI | Magnetic resonance imaging |
| NGS | Next-generation sequencing |
| PPB | Pleuropulmonary blastoma |
| RISC | RNA-induced silencing complex |
| SHBG | Sex hormone-binding globulin |
| SLCT | Sertoli–Leydig cell tumor |
| TBS | The Bethesda system |
| WES | Whole exome sequencing |
| WHO | World Health Organization |
References
- Schultz, K.A.P.; Harris, A.; Messinger, Y.; Sencer, S.; Baldinger, S.; Dehner, L.P.; Hill, D.A. Ovarian Tumors Related to Intronic Mutations in DICER1: A Report from the International Ovarian and Testicular Stromal Tumor Registry. Fam. Cancer 2016, 15, 105–110. [Google Scholar] [CrossRef]
- Riascos, M.C.; Huynh, A.; Faquin, W.C.; Nosé, V. Expanding Our Knowledge of DICER1 Gene Alterations and Their Role in Thyroid Diseases. Cancers 2024, 16, 347. [Google Scholar] [CrossRef]
- Lee, Y.; Kim, M.; Han, J.; Yeom, K.-H.; Lee, S.; Baek, S.H.; Kim, V.N. MicroRNA Genes Are Transcribed by RNA Polymerase II. EMBO J. 2004, 23, 4051–4060. [Google Scholar] [CrossRef]
- Han, J.; Lee, Y.; Yeom, K.-H.; Kim, Y.-K.; Jin, H.; Kim, V.N. The Drosha-DGCR8 Complex in Primary MicroRNA Processing. Genes Dev. 2004, 18, 3016–3027. [Google Scholar] [CrossRef]
- Denli, A.M.; Tops, B.B.J.; Plasterk, R.H.A.; Ketting, R.F.; Hannon, G.J. Processing of Primary MicroRNAs by the Microprocessor Complex. Nature 2004, 432, 231–235. [Google Scholar] [CrossRef]
- Yi, R.; Qin, Y.; Macara, I.G.; Cullen, B.R. Exportin-5 Mediates the Nuclear Export of Pre-MicroRNAs and Short Hairpin RNAs. Genes Dev. 2003, 17, 3011–3016. [Google Scholar] [CrossRef]
- Bernstein, E.; Caudy, A.A.; Hammond, S.M.; Hannon, G.J. Role for a Bidentate Ribonuclease in the Initiation Step of RNA Interference. Nature 2001, 409, 363–366. [Google Scholar] [CrossRef]
- Hammond, S.M. Dicing and Slicing. FEBS Lett. 2005, 579, 5822–5829. [Google Scholar] [CrossRef]
- Anglesio, M.; Wang, Y.; Yang, W.; Senz, J.; Wan, A.; Heravi-Moussavi, A.; Salamanca, C.; Maines-Bandiera, S.; Huntsman, D.; Morin, G. Cancer-Associated Somatic DICER1 Hotspot Mutations Cause Defective MiRNA Processing and Reverse-Strand Expression Bias to Predominantly Mature 3p Strands through Loss of 5p Strand Cleavage. J. Pathol. 2013, 229, 400–409. [Google Scholar] [CrossRef]
- Foulkes, W.D.; Priest, J.R.; Duchaine, T.F. DICER1: Mutations, MicroRNAs and Mechanisms. Nat. Rev. Cancer 2014, 14, 662–672. [Google Scholar] [CrossRef]
- Filipowicz, W.; Jaskiewicz, L.; Kolb, F.A.; Pillai, R.S. Post-Transcriptional Gene Silencing by SiRNAs and MiRNAs. Curr. Opin. Struct. Biol. 2005, 15, 331–341. [Google Scholar] [CrossRef]
- Hammond, S.M.; Bernstein, E.; Beach, D.; Hannon, G.J. An RNA-Directed Nuclease Mediates Post-Transcriptional Gene Silencing in Drosophila Cells. Nature 2000, 404, 293–296. [Google Scholar] [CrossRef]
- Schultz, K.A.P.; Williams, G.M.; Kamihara, J.; Stewart, D.R.; Harris, A.K.; Bauer, A.J.; Turner, J.; Shah, R.; Schneider, K.; Schneider, K.W.; et al. DICER1 and Associated Conditions: Identification of At-Risk Individuals and Recommended Surveillance Strategies. Clin. Cancer Res. 2018, 24, 2251–2261. [Google Scholar] [CrossRef]
- Robertson, J.; Jorcyk, C.; Oxford, J. DICER1 Syndrome: DICER1 Mutations in Rare Cancers. Cancers 2018, 10, 143. [Google Scholar] [CrossRef]
- De Kock, L.; Wang, Y.C.; Revil, T.; Badescu, D.; Rivera, B.; Sabbaghian, N.; Wu, M.; Weber, E.; Sandoval, C.; Hopman, S.M.J.; et al. High-Sensitivity Sequencing Reveals Multi-Organ Somatic Mosaicism Causing DICER1 Syndrome. J. Med. Genet. 2016, 53, 43–52. [Google Scholar] [CrossRef]
- Han, L.M.; Weiel, J.J.; Longacre, T.A.; Folkins, A.K. DICER1-Associated Tumors in the Female Genital Tract: Molecular Basis, Clinicopathologic Features, and Differential Diagnosis. Adv. Anat. Pathol. 2022, 29, 297–308. [Google Scholar] [CrossRef]
- Kim, J.; Field, A.; Schultz, K.A.P.; Hill, D.A.; Stewart, D.R. The Prevalence of DICER1 Pathogenic Variation in Population Databases. Int. J. Cancer 2017, 141, 2030–2036. [Google Scholar] [CrossRef]
- Manivel, J.C.; Priest, J.R.; Watterson, J.; Steiner, M.; Woods, W.G.; Wick, M.R.; Dehner, L.P. Pleuropulmonary Blastoma. The so-Called Pulmonary Blastoma of Childhood. Cancer 1988, 62, 1516–1526. [Google Scholar] [CrossRef]
- Hill, D.A.; Ivanovich, J.; Priest, J.R.; Gurnett, C.A.; Dehner, L.P.; Desruisseau, D.; Jarzembowski, J.A.; Wikenheiser-Brokamp, K.A.; Suarez, B.K.; Whelan, A.J.; et al. DICER1 Mutations in Familial Pleuropulmonary Blastoma. Science 2009, 325, 965. [Google Scholar] [CrossRef]
- Apellaniz-Ruiz, M.; Segni, M.; Kettwig, M.; Glüer, S.; Pelletier, D.; Nguyen, V.-H.; Wagener, R.; López, C.; Muchantef, K.; Bouron-Dal Soglio, D.; et al. Mesenchymal Hamartoma of the Liver and DICER1 Syndrome. N. Engl. J. Med. 2019, 380, 1834–1842. [Google Scholar] [CrossRef]
- Solarski, M.; Rotondo, F.; Foulkes, W.D.; Priest, J.R.; Syro, L.V.; Butz, H.; Cusimano, M.D.; Kovacs, K. DICER1 Gene Mutations in Endocrine Tumors. Endocr. Relat. Cancer 2018, 25, R197–R208. [Google Scholar] [CrossRef] [PubMed]
- Saskin, A.; de Kock, L.; Sabbaghian, N.; Apellaniz-Ruiz, M.; Bozkurt, C.; Bouron-Dal Soglio, D.; Foulkes, W.D. A Case of Neuroblastoma in DICER1 Syndrome: Chance Finding or Noncanonical Causation? Pediatr. Blood Cancer 2018, 65, e26715. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Zhou, X.; Wu, L.; Wu, P.; Liu, Y.; Li, Y.; Cai, L.; Fu, X.; Zhang, C. Ovarian Sertoli-Leydig Cell Tumor, Multinodular Goiter, Cystic Nephromas and DICER1 Mutations: Case Report and Literature Review. Pharmgenomics Pers. Med. 2021, 14, 947–953. [Google Scholar] [CrossRef] [PubMed]
- González, I.A.; Stewart, D.R.; Schultz, K.A.P.; Field, A.P.; Hill, D.A.; Dehner, L.P. DICER1 Tumor Predisposition Syndrome: An Evolving Story Initiated with the Pleuropulmonary Blastoma. Mod. Pathol. 2022, 35, 4–22. [Google Scholar] [CrossRef]
- Schultz, K.A.P.; Rednam, S.P.; Kamihara, J.; Doros, L.; Achatz, M.I.; Wasserman, J.D.; Diller, L.R.; Brugières, L.; Druker, H.; Schneider, K.A.; et al. PTEN, DICER1, FH, and Their Associated Tumor Susceptibility Syndromes: Clinical Features, Genetics, and Surveillance Recommendations in Childhood. Clin. Cancer Res. 2017, 23, e76–e82. [Google Scholar] [CrossRef]
- De Paolis, E.; Paragliola, R.; Concolino, P. Spectrum of DICER1 Germline Pathogenic Variants in Ovarian Sertoli–Leydig Cell Tumor. J. Clin. Med. 2021, 10, 1845. [Google Scholar] [CrossRef]
- Sauer, M.; Barletta, J.A. Proceedings of the North American Society of Head and Neck Pathology, Los Angeles, CA, March 20, 2022: DICER1-Related Thyroid Tumors. Head Neck Pathol. 2022, 16, 190–199. [Google Scholar] [CrossRef]
- De Kock, L.; Sabbaghian, N.; Soglio, D.B.-D.; Guillerman, R.P.; Park, B.-K.; Chami, R.; Deal, C.L.; Priest, J.R.; Foulkes, W.D. Exploring the Association Between DICER1 Mutations and Differentiated Thyroid Carcinoma. J. Clin. Endocrinol. Metab. 2014, 99, E1072–E1077. [Google Scholar] [CrossRef]
- Xiao, H.; Li, B.; Zuo, J.; Feng, X.; Li, X.; Zhang, R.; Wu, L. Ovarian Sertoli–Leydig Cell Tumor: A Report of Seven Cases and a Review of the Literature. Gynecol. Endocrinol. 2013, 29, 192–195. [Google Scholar] [CrossRef]
- Fuller, P.J.; Leung, D.; Chu, S. Genetics and Genomics of Ovarian Sex Cord-stromal Tumors. Clin. Genet. 2017, 91, 285–291. [Google Scholar] [CrossRef]
- Sigismondi, C.; Gadducci, A.; Lorusso, D.; Candiani, M.; Breda, E.; Raspagliesi, F.; Cormio, G.; Marinaccio, M.; Mangili, G. Ovarian Sertoli-Leydig Cell Tumors. A Retrospective MITO Study. Gynecol. Oncol. 2012, 125, 673–676. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, J.; Yang, W.; Mo, F.; Senz, J.; Yap, D.; Anglesio, M.S.; Gilks, B.; Morin, G.B.; Huntsman, D.G. The Oncogenic Roles of DICER1 RNase IIIb Domain Mutations in Ovarian Sertoli-Leydig Cell Tumors. Neoplasia 2015, 17, 650–660. [Google Scholar] [CrossRef] [PubMed]
- Conlon, N.; Schultheis, A.M.; Piscuoglio, S.; Silva, A.; Guerra, E.; Tornos, C.; Reuter, V.E.; Soslow, R.A.; Young, R.H.; Oliva, E.; et al. A Survey of DICER1 Hotspot Mutations in Ovarian and Testicular Sex Cord-Stromal Tumors. Mod. Pathol. 2015, 28, 1603–1612. [Google Scholar] [CrossRef] [PubMed]
- Schultz, K.A.P.; Harris, A.K.; Finch, M.; Dehner, L.P.; Brown, J.B.; Gershenson, D.M.; Young, R.H.; Field, A.; Yu, W.; Turner, J.; et al. DICER1-Related Sertoli-Leydig Cell Tumor and Gynandroblastoma: Clinical and Genetic Findings from the International Ovarian and Testicular Stromal Tumor Registry. Gynecol. Oncol. 2017, 147, 521–527. [Google Scholar] [CrossRef]
- Goulvent, T.; Ray-Coquard, I.; Borel, S.; Haddad, V.; Devouassoux-Shisheboran, M.; Vacher-Lavenu, M.; Pujade-Laurraine, E.; Savina, A.; Maillet, D.; Gillet, G.; et al. DICER1 and FOXL 2 Mutations in Ovarian Sex Cord–Stromal Tumours: A GINECO Group Study. Histopathology 2016, 68, 279–285. [Google Scholar] [CrossRef]
- Karnezis, A.N.; Wang, Y.; Keul, J.; Tessier-Cloutier, B.; Magrill, J.; Kommoss, S.; Senz, J.; Yang, W.; Proctor, L.; Schmidt, D.; et al. DICER1 and FOXL2 Mutation Status Correlates With Clinicopathologic Features in Ovarian Sertoli-Leydig Cell Tumors. Am. J. Surg. Pathol. 2019, 43, 628–638. [Google Scholar] [CrossRef]
- Kato, N.; Kusumi, T.; Kamataki, A.; Tsunoda, R.; Fukase, M.; Kurose, A. DICER1 Hotspot Mutations in Ovarian Sertoli-Leydig Cell Tumors: A Potential Association with Androgenic Effects. Hum. Pathol. 2017, 59, 41–47. [Google Scholar] [CrossRef]
- Watanabe, T.; Soeda, S.; Endo, Y.; Okabe, C.; Sato, T.; Kamo, N.; Ueda, M.; Kojima, M.; Furukawa, S.; Nishigori, H.; et al. Rare Hereditary Gynecological Cancer Syndromes. Int. J. Mol. Sci. 2022, 23, 1563. [Google Scholar] [CrossRef]
- WHO. Classification of Tumours. Available online: https://tumourclassification.iarc.who.int/welcome/ (accessed on 8 September 2024).
- Haley, M.; Bindal, P.; McAuliffe, A.; Vredenburgh, J. A Family with Sertoli–Leydig Cell Tumour, Multinodular Goiter, and DICER1 Mutation. Curr. Oncol. 2019, 26, 183–185. [Google Scholar] [CrossRef]
- Ray-Coquard, I.; Morice, P.; Lorusso, D.; Prat, J.; Oaknin, A.; Pautier, P.; Colombo, N. Non-Epithelial Ovarian Cancer: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2018, 29, iv1–iv18. [Google Scholar] [CrossRef]
- de Kock, L.; Terzic, T.; McCluggage, W.G.; Stewart, C.J.R.; Shaw, P.; Foulkes, W.D.; Clarke, B.A. DICER1 Mutations Are Consistently Present in Moderately and Poorly Differentiated Sertoli-Leydig Cell Tumors. Am. J. Surg. Pathol. 2017, 41, 1178–1187. [Google Scholar] [CrossRef] [PubMed]
- Inada, Y.; Nakai, G.; Yamamoto, K.; Yamada, T.; Hirose, Y.; Terai, Y.; Ohmichi, M.; Narumi, Y. Rapidly Growing Juvenile Granulosa Cell Tumor of the Ovary Arising in Adult: A Case Report and Review of the Literature. J. Ovarian Res. 2018, 11, 100. [Google Scholar] [CrossRef] [PubMed]
- Roth, L.M. Recent Advances in the Pathology and Classification of Ovarian Sex Cord-Stromal Tumors. Int. J. Gynecol. Pathol. 2006, 25, 199–215. [Google Scholar] [CrossRef] [PubMed]
- Outwater, E.K.; Wagner, B.J.; Mannion, C.; McLarney, J.K.; Kim, B. Sex Cord-Stromal and Steroid Cell Tumors of the Ovary. Radio Graphics 1998, 18, 1523–1546. [Google Scholar] [CrossRef]
- Riker, D.; Goba, D. Ovarian Mass, Pleural Effusion, and Ascites. J. Bronchol. Interv. Pulmonol. 2013, 20, 48–51. [Google Scholar] [CrossRef]
- Prat, J. Staging Classification for Cancer of the Ovary, Fallopian Tube, and Peritoneum. Int. J. Gynecol. Obstet. 2014, 124, 1–5. [Google Scholar] [CrossRef]
- Gershenson, D.M. Current Advances in the Management of Malignant Germ Cell and Sex Cord-Stromal Tumors of the Ovary. Gynecol. Oncol. 2012, 125, 515–517. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. ClinVar; [VCV000929411.1]. Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000929411.1 (accessed on 10 September 2024).
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Orphanet: DICER1 Tumor-Predisposition Syndrome. Available online: https://www.orpha.net/en/disease/detail/284343 (accessed on 16 May 2023).
- Onder, S.; Hurdogan, O.; Bayram, A.; Yilmaz, I.; Sozen, H.; Yavuz, E. The Role of FOXL2, SOX9, and β-Catenin Expression and DICER1 Mutation in Differentiating Sex Cord Tumor with Annular Tubules from Other Sex Cord Tumors of the Ovary. Virchows Archiv. 2021, 479, 317–324. [Google Scholar] [CrossRef]
- Stewart, D.R.; Best, A.F.; Williams, G.M.; Harney, L.A.; Carr, A.G.; Harris, A.K.; Kratz, C.P.; Dehner, L.P.; Messinger, Y.H.; Rosenberg, P.S.; et al. Neoplasm Risk Among Individuals With a Pathogenic Germline Variant in DICER1. J. Clin. Oncol. 2019, 37, 668–676. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Most Frequent | Moderate Frequent—Rare | Very Rare |
|---|---|---|
| Pleuropulmonary blastoma | Differentiated thyroid cancer | Anaplastic sarcoma of kidney |
| Multinodular goiter | Wilms tumor | Medulloblastoma |
| Cystic nephroma | Juvenile hamartomatous intestinal polyps | Embryonal rhabdomyosarcoma of the bladder |
| Ovarian Sertoli–Leydig cell tumor | Ciliary body medulloepithelioma | Embryonal rhabdomyosarcoma of the ovary |
| Nasal chondromesenchymal hamartoma | Neuroblastoma | |
| Pituitary blastoma | Congenital phthisis bulbi | |
| Pineoblastoma | Juvenile granulosa cell tumor | |
| Cervical embryonal rhabdomyosarcoma | Gynandroblastoma | |
| Cervix primitive neuroectodermal tumor |
| Neoplastic Conditions | Non-Neoplastic Conditions | ||
|---|---|---|---|
| Malignant Neoplasms | Benign Neoplasms | ||
| Pleuropulmonary blastoma | Juvenile granulosa cell tumor | Pleuropulmonary blastoma | Macrocephaly |
| Pineal blastoma | Gynandroblastoma | Multinodular goiter | Kidney structural abnormalities |
| Ovarian Sertoli–Leydig cell tumor | Embryonal rhabdomyosarcoma (bladder, ovary) | Cystic nephroma | Retinal abnormalities |
| Cervical embryonal rhabdomyosarcoma | Neuroblastoma | Juvenile hamartomatous intestinal polyps | Dental perturbations |
| Wilms tumor | Medulloblastoma | Nasal chondromesenchymal hamartoma | GLOW syndrome * |
| Sarcomas (uterine cervix, kidney, brain) | Ciliary body medulloepithelioma | Ciliary body medulloepithelioma | Congenital phthisis bulbi |
| Thyroid carcinoma | Mesenchymal hamartoma of the liver | ||
| Laboratory Test | Preoperatively | Postoperatively | Reference Range |
|---|---|---|---|
| FSH | 1.3 | 7.6 | 3–8.1 mIU/mL |
| LH | 2.3 | 5.8 | 1.8–11.8 mIU/mL |
| E2 | 45 | 24 | 21–251 pg/mL |
| Testosterone | 3.1 | 0.4 | 0.1–0.5 ng/mL |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roumpou, A.; Ieronimaki, A.-I.; Manta, A.; Panayiotides, I.G.; Stratakis, C.A.; Kalantaridou, S.; Peppa, M. A Novel Pathogenic Variant of DICER1 Gene in a Young Greek Patient with 2 Different Sex-Cord Ovarian Tumors and Multinodular Goiter. Int. J. Mol. Sci. 2025, 26, 1990. https://doi.org/10.3390/ijms26051990
Roumpou A, Ieronimaki A-I, Manta A, Panayiotides IG, Stratakis CA, Kalantaridou S, Peppa M. A Novel Pathogenic Variant of DICER1 Gene in a Young Greek Patient with 2 Different Sex-Cord Ovarian Tumors and Multinodular Goiter. International Journal of Molecular Sciences. 2025; 26(5):1990. https://doi.org/10.3390/ijms26051990
Chicago/Turabian StyleRoumpou, Afroditi, Argyro-Ioanna Ieronimaki, Aspasia Manta, Ioannis G. Panayiotides, Constantine A. Stratakis, Sophia Kalantaridou, and Melpomeni Peppa. 2025. "A Novel Pathogenic Variant of DICER1 Gene in a Young Greek Patient with 2 Different Sex-Cord Ovarian Tumors and Multinodular Goiter" International Journal of Molecular Sciences 26, no. 5: 1990. https://doi.org/10.3390/ijms26051990
APA StyleRoumpou, A., Ieronimaki, A.-I., Manta, A., Panayiotides, I. G., Stratakis, C. A., Kalantaridou, S., & Peppa, M. (2025). A Novel Pathogenic Variant of DICER1 Gene in a Young Greek Patient with 2 Different Sex-Cord Ovarian Tumors and Multinodular Goiter. International Journal of Molecular Sciences, 26(5), 1990. https://doi.org/10.3390/ijms26051990