
Rational Design and Optimization of Novel PDE5 Inhibitors for Targeted Colorectal Cancer Therapy: An In Silico Approach
 , and
, and
Abstract
1. Introduction
2. Results and Discussion
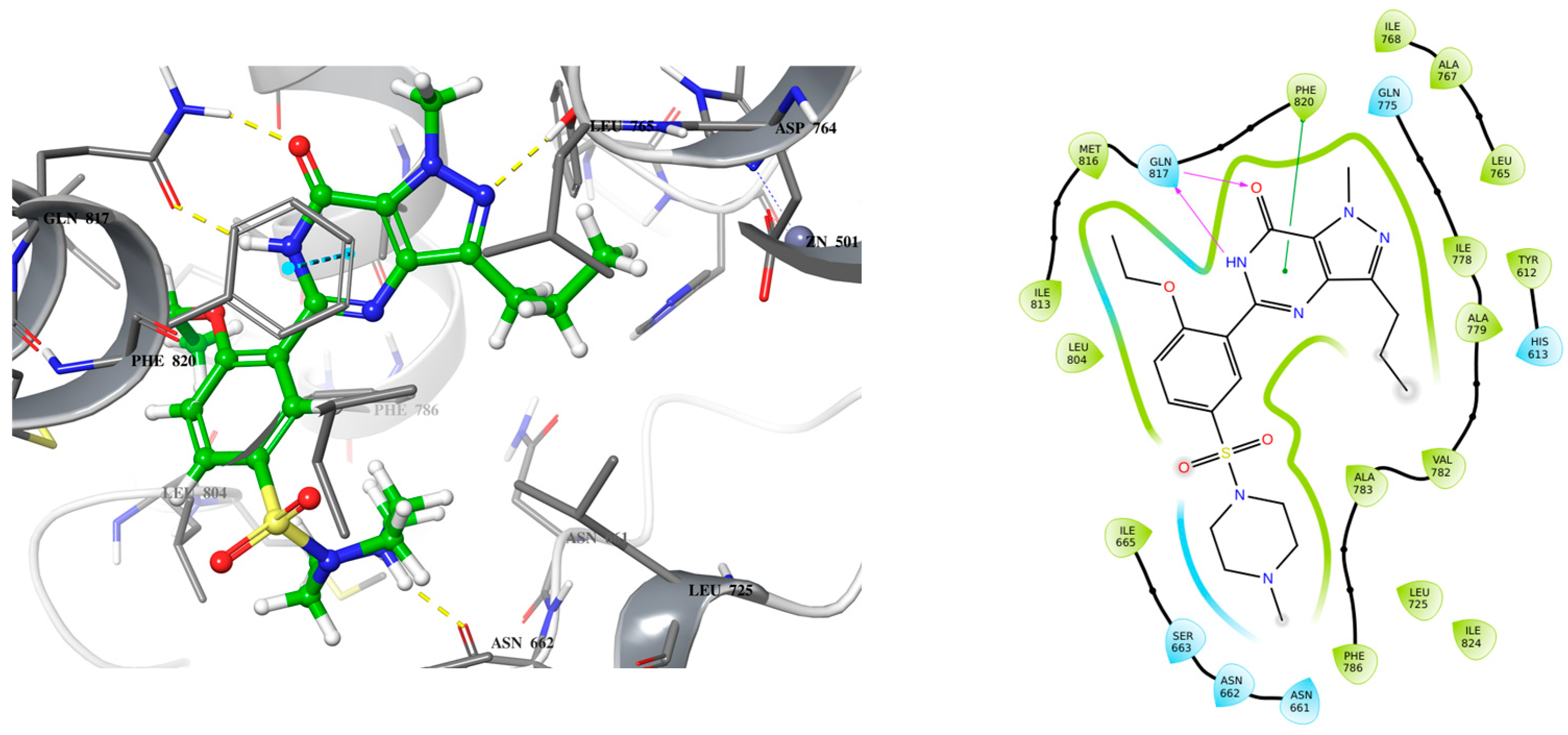
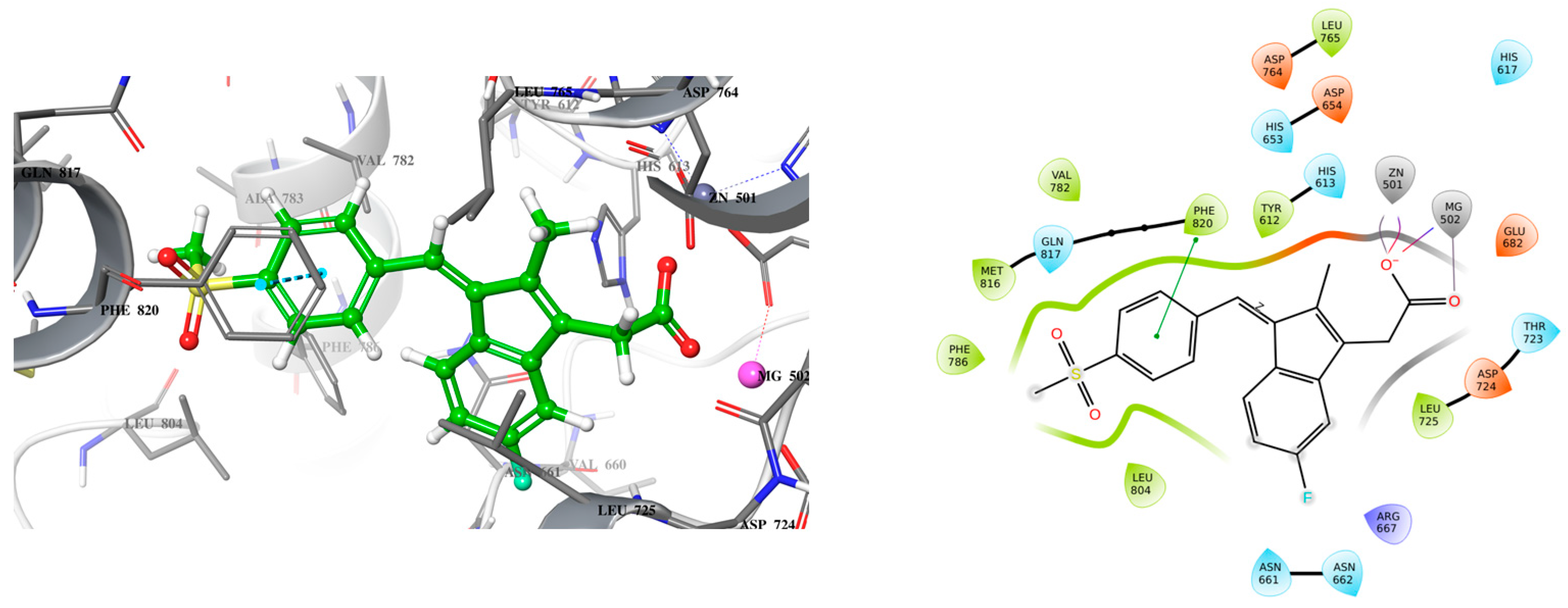
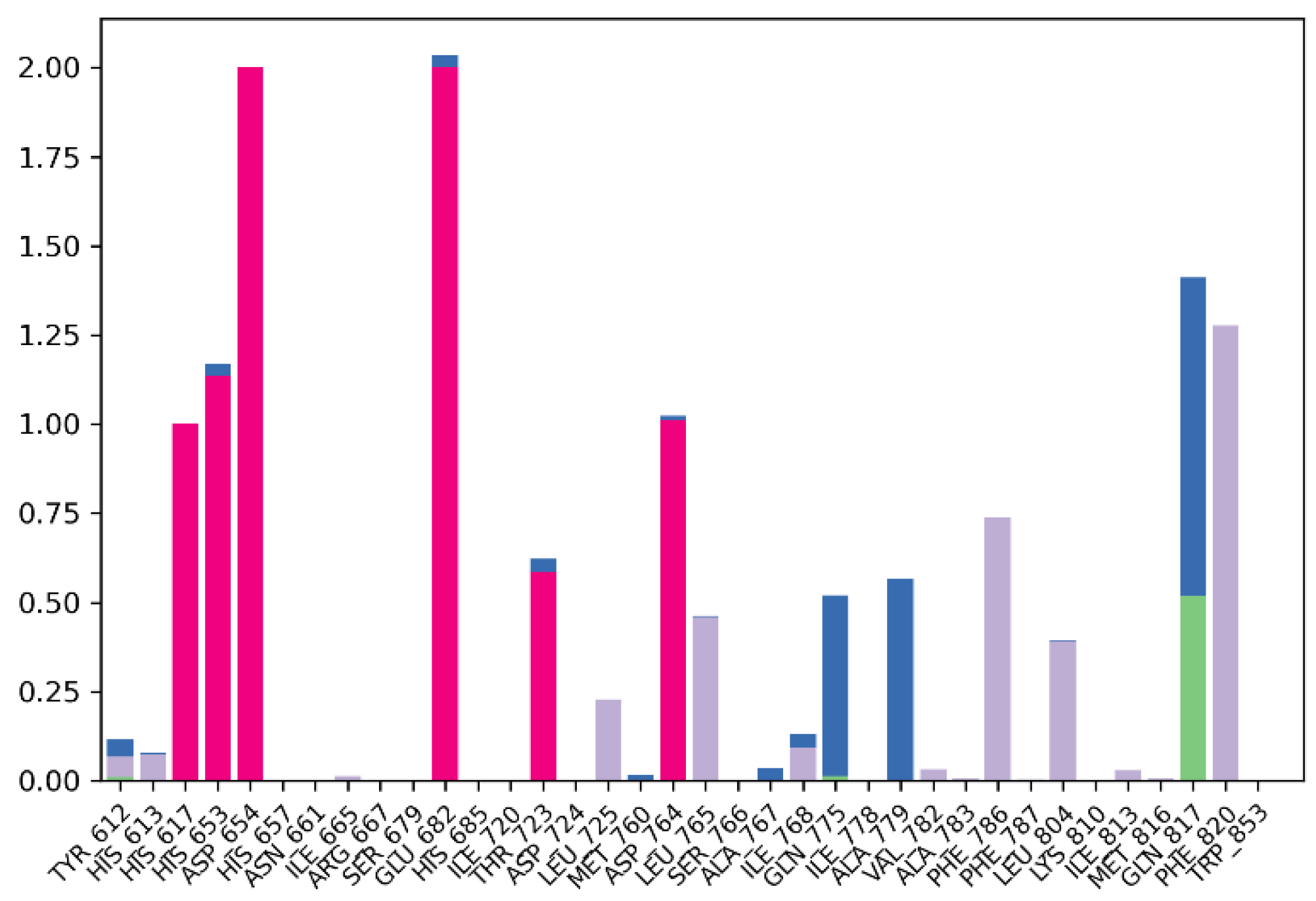
2.1. Lead Compound Development
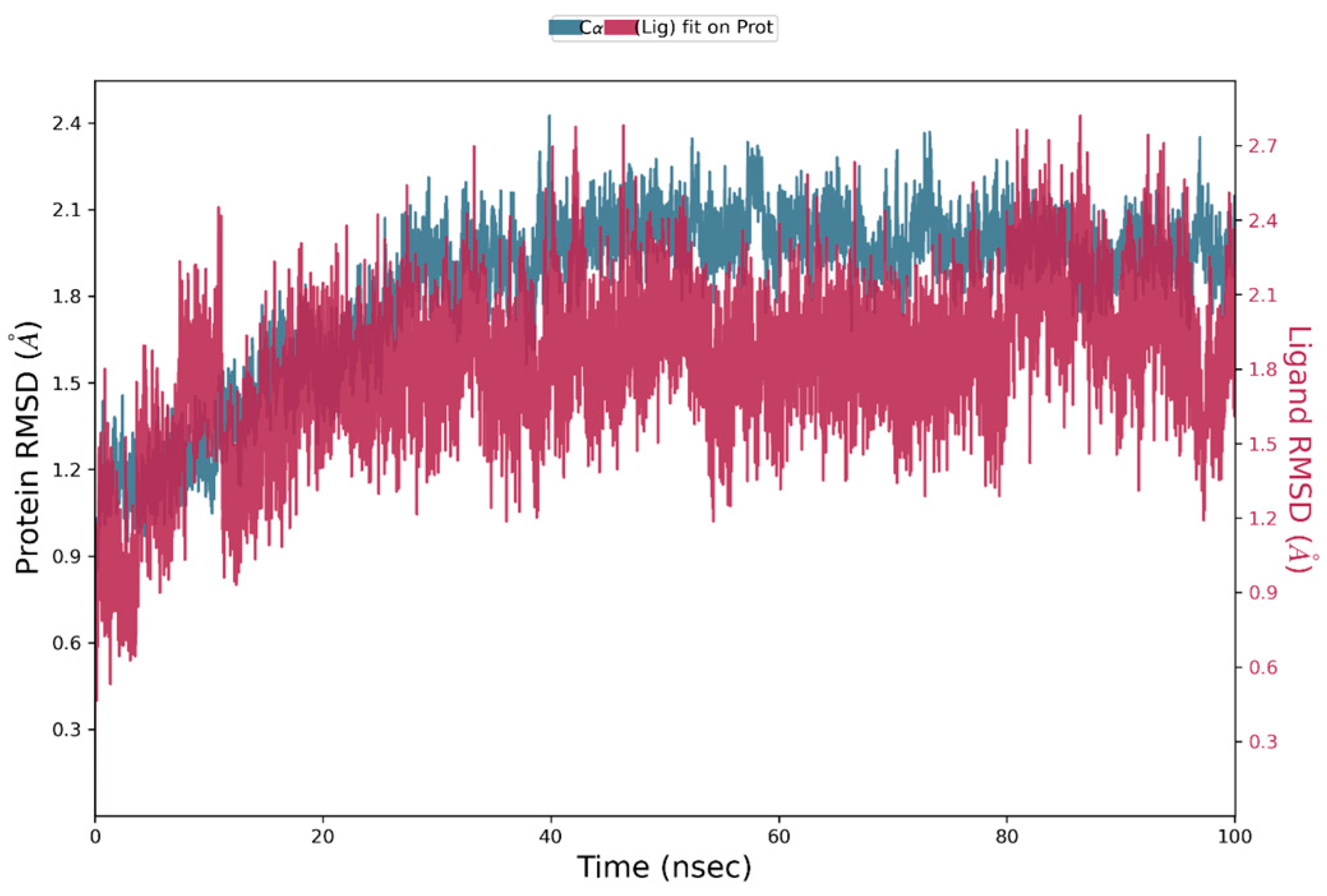
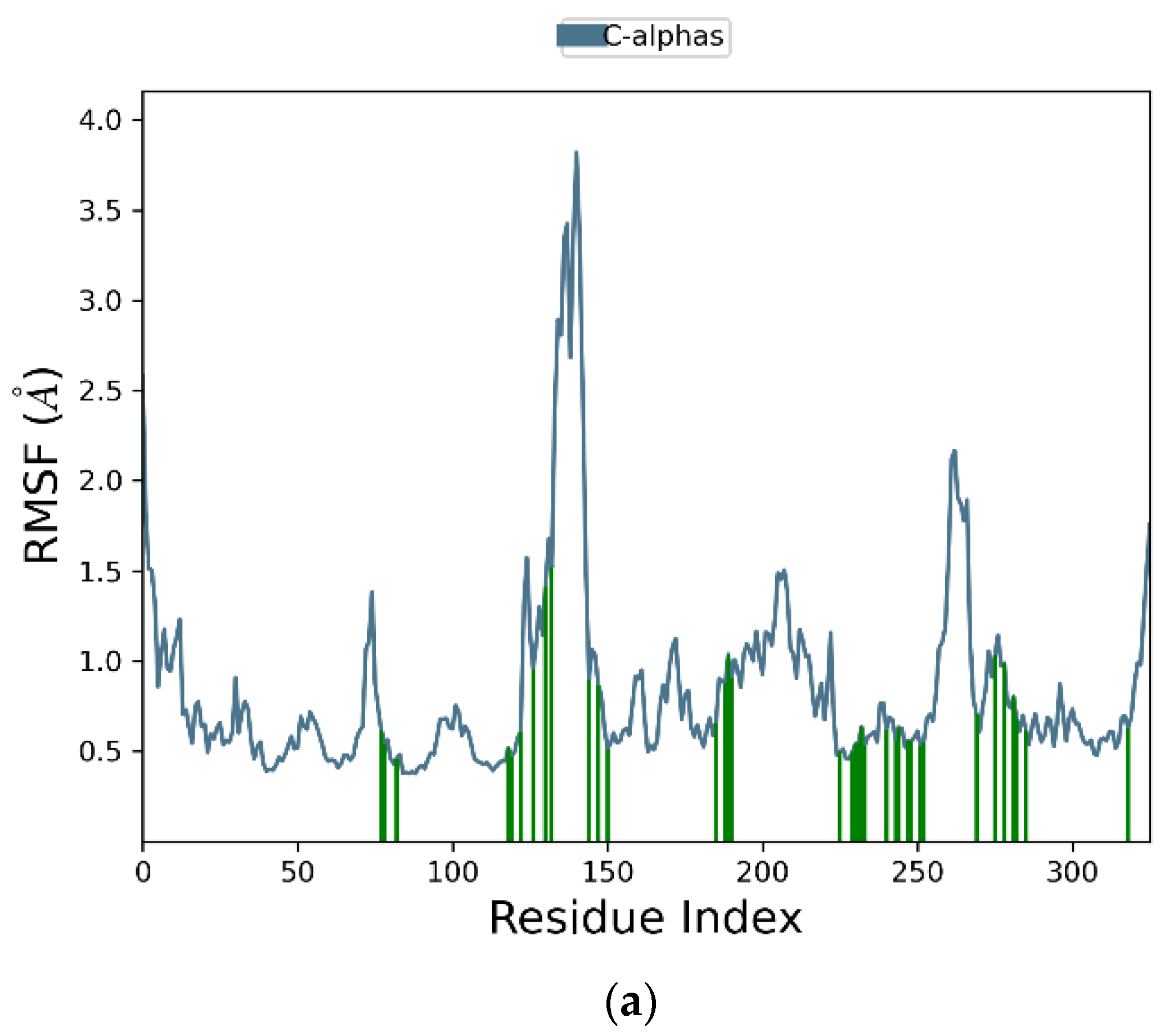
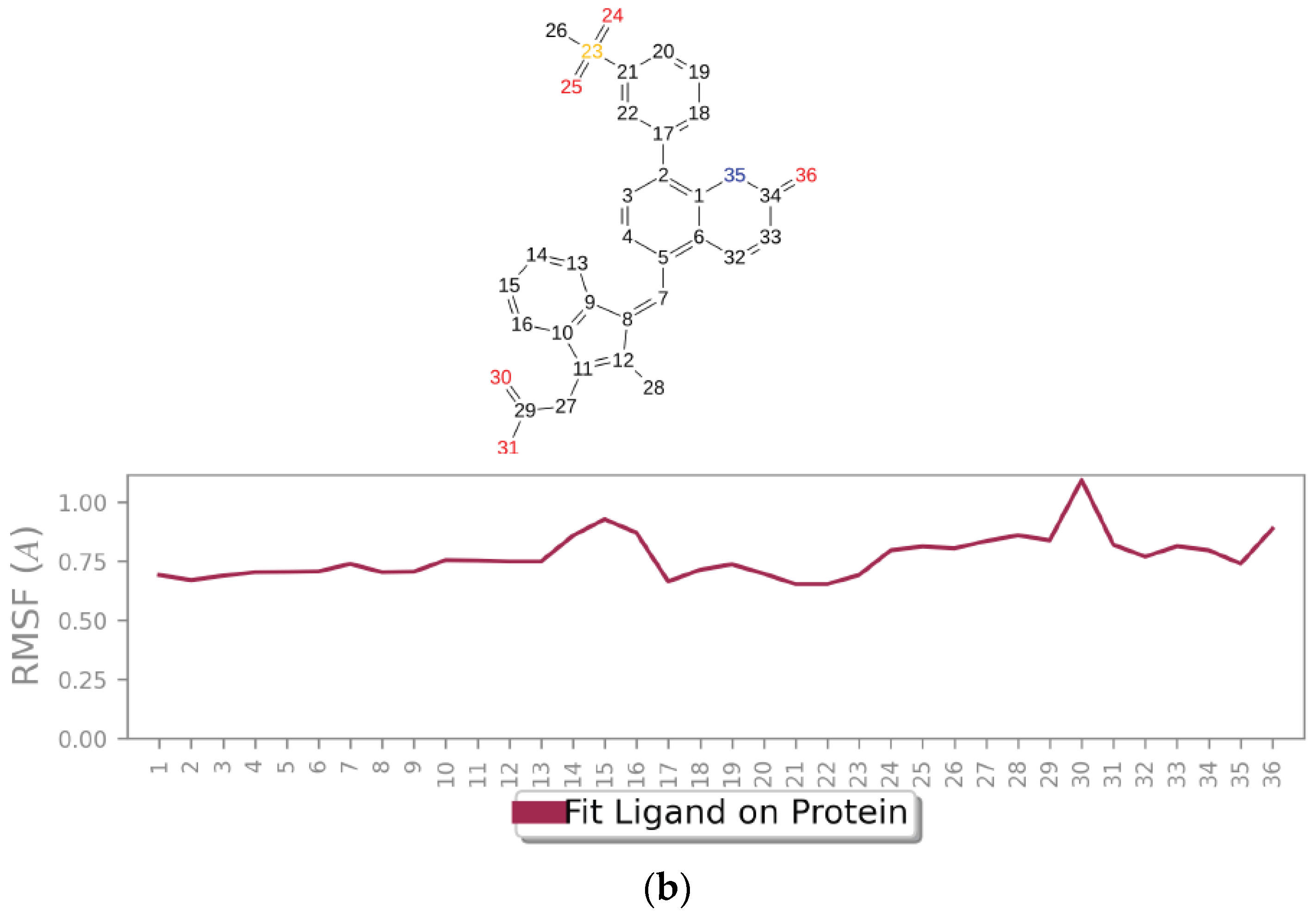
2.2. Molecular Dynamic Simulation
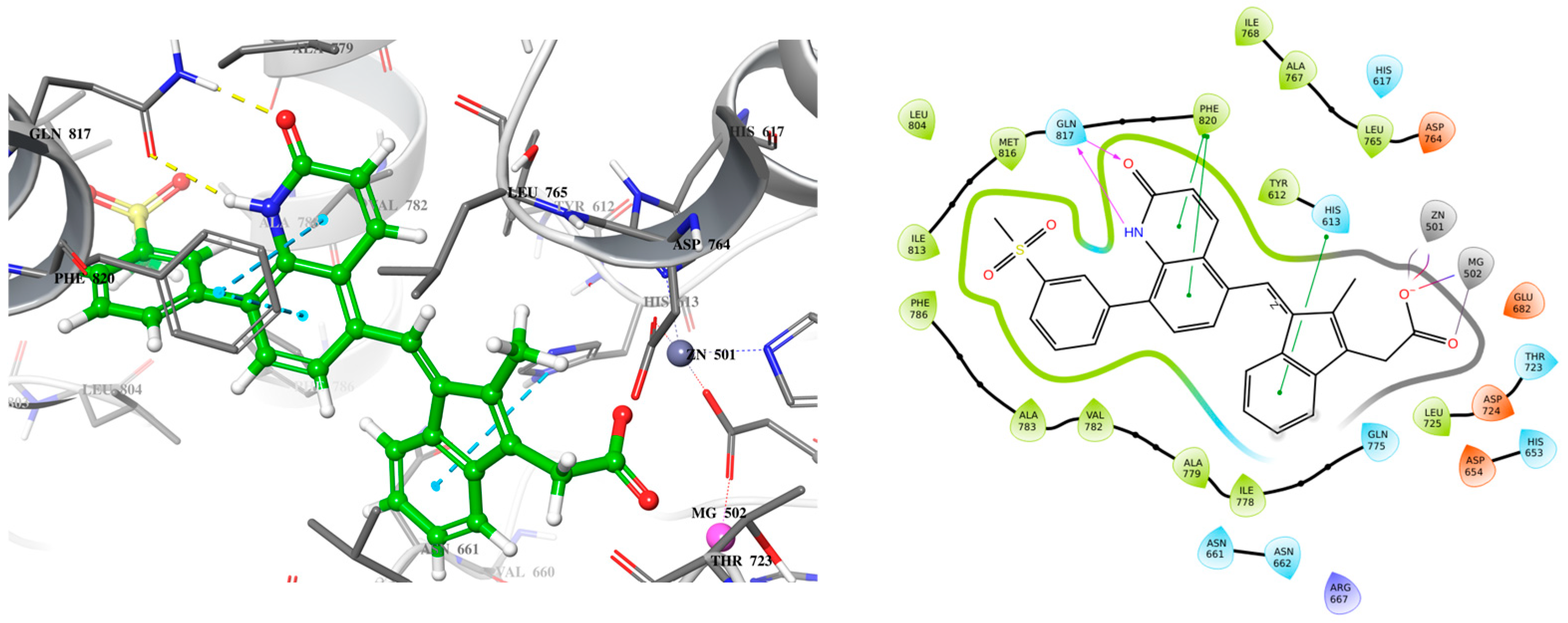
2.3. Lead Compound Optimization
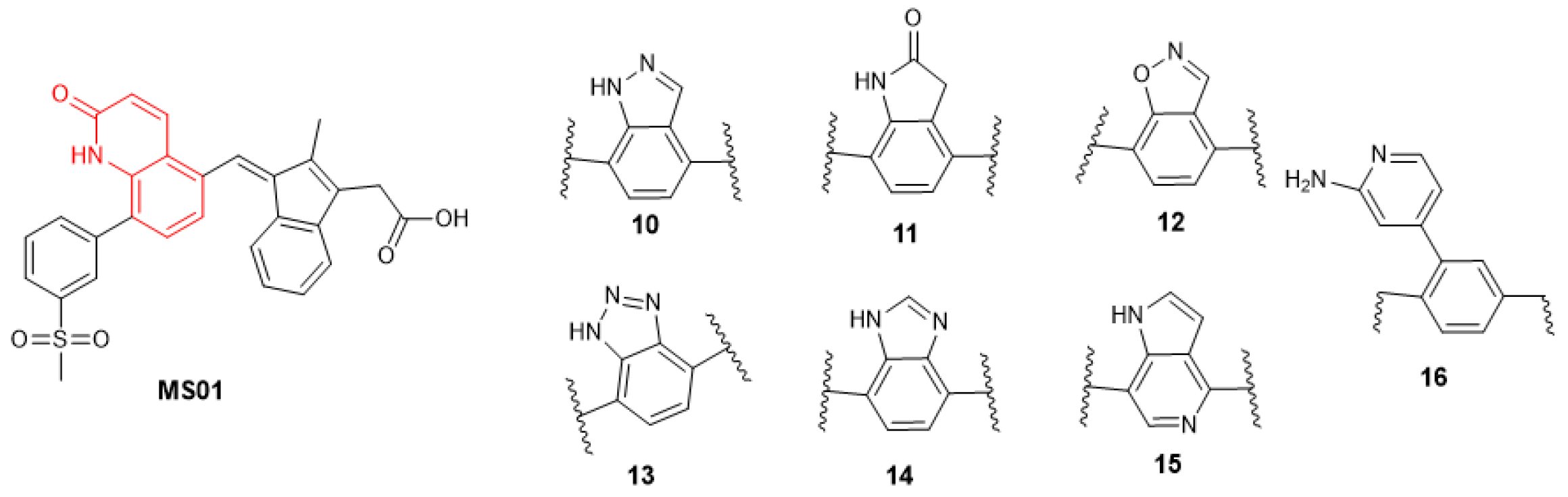
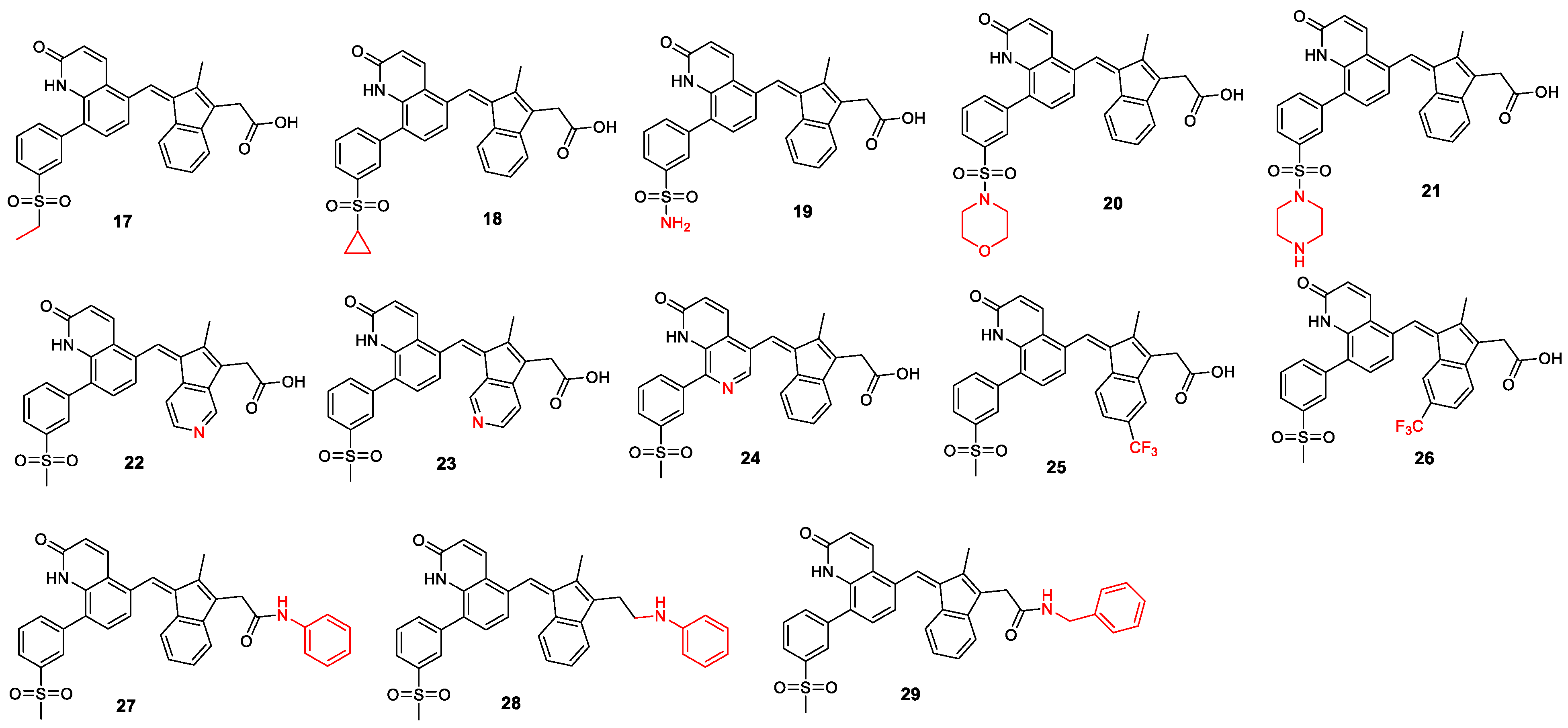
2.3.1. Changing the Quinolone Scaffold
2.3.2. Other Changes to Improve Drug-like Properties
2.4. ADMET Property Prediction
3. Materials and Methods
3.1. Ligand Preparation
3.2. Protein Preparation
3.3. Receptor Grid Generation
3.4. Molecular Docking
3.5. Induced Fit Docking (IFD)
3.6. Molecular Dynamic Simulation
3.7. Pharmacology Parameters
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Dekker, E.; Tanis, P.J.; Vleugels, J.L.A.; Kasi, P.M.; Wallace, M.B. Colorectal Cancer. Lancet 2019, 394, 1467–1480. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2019. CA A Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef]
- Xie, Y.-H.; Chen, Y.-X.; Fang, J.-Y. Comprehensive Review of Targeted Therapy for Colorectal Cancer. Signal. Transduct. Target. Ther. 2020, 5, 22. [Google Scholar] [CrossRef]
- Nappi, A.; Berretta, M.; Romano, C.; Tafuto, S.; Cassata, A.; Casaretti, R.; Silvestro, L.; Divitiis, C.D.; Alessandrini, L.; Fiorica, F.; et al. Metastatic Colorectal Cancer: Role of Target Therapies and Future Perspectives. Curr. Cancer Drug Targets CCDT 2018, 18, 421–429. [Google Scholar] [CrossRef]
- Baraibar, I.; Ros, J.; Mulet, N.; Salvà, F.; Argilés, G.; Martini, G.; Cuadra, J.L.; Sardo, E.; Ciardiello, D.; Tabernero, J.; et al. Incorporating Traditional and Emerging Biomarkers in the Clinical Management of Metastatic Colorectal Cancer: An Update. Expert Rev. Mol. Diagn. 2020, 20, 653–664. [Google Scholar] [CrossRef] [PubMed]
- Jeanne, R.; Riddle, B.S.N.; Beverly Peeples, B.A.; Christine Alden, B.S.N.; Kathleen Gillaspy, B.S.N.; Hannah Brewer, R.N. Recognizing and Managing Side Effects Associated with Novel Targeted Therapies. Oncology 2006, 20, 1520. [Google Scholar]
- Baillie, G.S.; Tejeda, G.S.; Kelly, M.P. Therapeutic Targeting of 3′,5′-Cyclic Nucleotide Phosphodiesterases: Inhibition and Beyond. Nat. Rev. Drug Discov. 2019, 18, 770–796. [Google Scholar] [CrossRef] [PubMed]
- Fajardo, A.M.; Piazza, G.A.; Tinsley, H.N. The Role of Cyclic Nucleotide Signaling Pathways in Cancer: Targets for Prevention and Treatment. Cancers 2014, 6, 436–458. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-S. Tissue Expression, Distribution, and Regulation of PDE5. Int. J. Impot. Res. 2004, 16, S8–S10. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Durrant, D.; Salloum, F.N.; Xi, L.; Kukreja, R.C. PDE5 Inhibitors as Therapeutics for Heart Disease, Diabetes and Cancer. Pharmacol. Ther. 2015, 147, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, W.S.; Geethakumari, A.M.; Biswas, K.H. Phosphodiesterase 5 (PDE5): Structure-Function Regulation and Therapeutic Applications of Inhibitors. Biomed. Pharmacother. 2021, 134, 111128. [Google Scholar] [CrossRef] [PubMed]
- Haider, M.; Elsherbeny, A.; Pittalà, V.; Fallica, A.N.; Alghamdi, M.A.; Greish, K. The Potential Role of Sildenafil in Cancer Management through EPR Augmentation. J. Pers. Med. 2021, 11, 585. [Google Scholar] [CrossRef] [PubMed]
- Mei, X.-L.; Yang, Y.; Zhang, Y.-J.; Li, Y.; Zhao, J.-M.; Qiu, J.-G.; Zhang, W.-J.; Jiang, Q.-W.; Xue, Y.-Q.; Zheng, D.-W.; et al. Sildenafil Inhibits the Growth of Human Colorectal Cancer in Vitro and in Vivo. Am. J. Cancer Res. 2015, 5, 3311–3324. [Google Scholar]
- Islam, B.N.; Sharman, S.K.; Hou, Y.; Bridges, A.E.; Singh, N.; Kim, S.; Kolhe, R.; Trillo-Tinoco, J.; Rodriguez, P.C.; Berger, F.G.; et al. Sildenafil Suppresses Inflammation-Driven Colorectal Cancer in Mice. Cancer Prev. Res. 2017, 10, 377–388. [Google Scholar] [CrossRef]
- Goluboff, E.T. Exisulind, a Selective Apoptotic Antineoplastic Drug. Expert Opin. Investig. Drugs 2001, 10, 1875–1882. [Google Scholar] [CrossRef] [PubMed]
- Thompson, W.J.; Piazza, G.A.; Li, H.; Liu, L.; Fetter, J.; Zhu, B.; Sperl, G.; Ahnen, D.; Pamukcu, R. Exisulind Induction of Apoptosis Involves Guanosine 3′,5′-Cyclic Monophosphate Phosphodiesterase Inhibition, Protein Kinase G Activation, and Attenuated Beta-Catenin. Cancer Res. 2000, 60, 3338–3342. [Google Scholar]
- Li, H.; Liu, L.; David, M.L.; Whitehead, C.M.; Chen, M.; Fetter, J.R.; Sperl, G.J.; Pamukcu, R.; Thompson, W.J. Pro-Apoptotic Actions of Exisulind and CP461 in SW480 Colon Tumor Cells Involve β-Catenin and Cyclin D1 down-Regulation. Biochem. Pharmacol. 2002, 64, 1325–1336. [Google Scholar] [CrossRef] [PubMed]
- Adis Editorial. Exisulind. Drugs R D 2004, 5, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.X.; Hassell, A.M.; Vanderwall, D.; Lambert, M.H.; Holmes, W.D.; Luther, M.A.; Rocque, W.J.; Milburn, M.V.; Zhao, Y.; Ke, H.; et al. Atomic Structure of PDE4: Insights into Phosphodiesterase Mechanism and Specificity. Science 2000, 288, 1822–1825. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.Y.J.; Card, G.L.; Suzuki, Y.; Artis, D.R.; Fong, D.; Gillette, S.; Hsieh, D.; Neiman, J.; West, B.L.; Zhang, C.; et al. A Glutamine Switch Mechanism for Nucleotide Selectivity by Phosphodiesterases. Mol. Cell 2004, 15, 279–286. [Google Scholar] [CrossRef]
- Minimizing the DILI Potential of Carboxylic Acid-Containing Drugs: A Perspective|Medicinal Chemistry Research. Available online: https://link.springer.com/article/10.1007/s00044-023-03140-9 (accessed on 18 August 2024).
- Dhiman, P.; Arora, N.; Thanikachalam, P.V.; Monga, V. Recent Advances in the Synthetic and Medicinal Perspective of Quinolones: A Review. Bioorg. Chem. 2019, 92, 103291. [Google Scholar] [CrossRef] [PubMed]
- Maris, A.S.; Mody, P.; Brewer, D.J.; Humphries, R.M. The Fluoroquinolones: An Update for the Clinical Microbiologist. Clin. Microbiol. Newsl. 2021, 43, 97–107. [Google Scholar] [CrossRef]
- Pennington, L.D.; Moustakas, D.T. The Necessary Nitrogen Atom: A Versatile High-Impact Design Element for Multiparameter Optimization. J. Med. Chem. 2017, 60, 3552–3579. [Google Scholar] [CrossRef] [PubMed]
- Pearson, T.J.; Shimazumi, R.; Driscoll, J.L.; Dherange, B.D.; Park, D.-I.; Levin, M.D. Aromatic Nitrogen Scanning by Ipso-Selective Nitrene Internalization. Science 2023, 381, 1474–1479. [Google Scholar] [CrossRef]
- Yale, H.L. The Trifluoromethyl Group in Medical Chemistry. J. Med. Chem. 1959, 1, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Whitt, J.D.; Li, N.; Tinsley, H.N.; Chen, X.; Zhang, W.; Li, Y.; Gary, B.D.; Keeton, A.B.; Xi, Y.; Abadi, A.H.; et al. A Novel Sulindac Derivative That Potently Suppresses Colon Tumor Cell Growth by Inhibiting cGMP Phosphodiesterase and β-Catenin Transcriptional Activity. Cancer Prev. Res. 2012, 5, 822–833. [Google Scholar] [CrossRef] [PubMed]
- Piazza, G.A.; Keeton, A.B.; Tinsley, H.N.; Gary, B.D.; Whitt, J.D.; Mathew, B.; Thaiparambil, J.; Coward, L.; Gorman, G.; Li, Y.; et al. A Novel Sulindac Derivative That Does Not Inhibit Cyclooxygenases but Potently Inhibits Colon Tumor Cell Growth and Induces Apoptosis with Antitumor Activity. Cancer Prev. Res. 2009, 2, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Mannhold, R.; Poda, G.I.; Ostermann, C.; Tetko, I.V. Calculation of Molecular Lipophilicity: State-of-the-Art and Comparison of Log P Methods on More than 96,000 Compounds. J. Pharm. Sci. 2009, 98, 861–893. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Prasanna, S.; Doerksen, R.J. Topological Polar Surface Area: A Useful Descriptor in 2D-QSAR. Curr. Med. Chem. 2009, 16, 21–41. [Google Scholar] [CrossRef]
- Jia, C.-Y.; Li, J.-Y.; Hao, G.-F.; Yang, G.-F. A Drug-Likeness Toolbox Facilitates ADMET Study in Drug Discovery. Drug Discov. Today 2020, 25, 248–258. [Google Scholar] [CrossRef] [PubMed]
- The Biochemistry of Drug Metabolism—An Introduction—Testa—2009—Chemistry & Biodiversity—Wiley Online Library. Available online: https://onlinelibrary.wiley.com/doi/10.1002/cbdv.200900022 (accessed on 20 September 2024).
- Ursu, O.; Rayan, A.; Goldblum, A.; Oprea, T.I. Understanding Drug-Likeness. WIREs Comput. Mol. Sci. 2011, 1, 760–781. [Google Scholar] [CrossRef]
- P-Glycoprotein Substrate Assessment in Drug Discovery: Application of Modeling to Bridge Differential Protein Expression Across In Vitro Tools—ScienceDirect. Available online: https://www.sciencedirect.com/science/article/pii/S0022354920305153?via%3Dihub (accessed on 28 September 2024).
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Zhao, M.; Ma, J.; Li, M.; Zhang, Y.; Jiang, B.; Zhao, X.; Huai, C.; Shen, L.; Zhang, N.; He, L.; et al. Cytochrome P450 Enzymes and Drug Metabolism in Humans. Int. J. Mol. Sci. 2021, 22, 12808. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.; Kemmler, E.; Dunkel, M.; Preissner, R. ProTox 3.0: A Webserver for the Prediction of Toxicity of Chemicals. Nucleic Acids Res. 2024, 52, W513–W520. [Google Scholar] [CrossRef]
- Søndergaard, C.R.; Olsson, M.H.M.; Rostkowski, M.; Jensen, J.H. Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKa Values. J. Chem. Theory Comput. 2011, 7, 2284–2295. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | IFD Score |
|---|---|
| Sildenafil | −765.04 |
| Exisulind | −756.8 |
| cGMP | −756.22 |
| MS01 | −761.16 |
| Compound ID | ΔG Energy (Kcal/mol) |
|---|---|
| MS25 | −14.729 |
| MS18 | −14.441 |
| MS23 | −13.984 |
| MS01-Lead | −13.798 |
| MS21 | −13.092 |
| MS16 | −13.073 |
| MS17 | −12.772 |
| MS20 | −12.666 |
| MS26 | −12.618 |
| MS22 | −12.560 |
| MS24 | −12.082 |
| MS12 | −11.992 |
| MS19 | −11.913 |
| MS15 | −11.892 |
| MS11 | −11.608 |
| MS29 | −11.164 |
| MS10 | −11.060 |
| MS13 | −10.850 |
| MS14 | −10.763 |
| Sildenafil | −10.383 |
| Exisulind | −9.895 |
| cGMP | −9.798 |
| MS28 | −8.749 |
| MS27 | −7.952 |
| Molecule | MW (g/mol) | Consensus Log p | Log S (ESOL) | TPSA | Fraction Csp3 | Verber #Violations | Lipinski #Violations | Bioavailability Score |
|---|---|---|---|---|---|---|---|---|
| MS01 | 497.56 | 4.62 | −5.26 | 112.68 | 0.1 | 0 | 0 | 0.56 |
| MS10 | 470.54 | 4.35 | −5.23 | 108.5 | 0.11 | 0 | 0 | 0.56 |
| MS11 | 485.55 | 4.04 | −4.76 | 108.92 | 0.14 | 0 | 0 | 0.56 |
| MS12 | 471.52 | 4.57 | −5.34 | 105.85 | 0.11 | 0 | 0 | 0.56 |
| MS13 | 471.53 | 3.98 | −4.96 | 121.39 | 0.12 | 0 | 0 | 0.56 |
| MS14 | 470.54 | 4.31 | −5.20 | 108.50 | 0.11 | 0 | 0 | 0.55 |
| MS15 | 469.55 | 4.83 | −5.54 | 95.61 | 0.11 | 0 | 0 | 0.56 |
| MS16 | 522.61 | 4.86 | −5.87 | 118.73 | 0.10 | 1 | 0 | 0.56 |
| MS17 | 511.59 | 4.96 | −5.5 | 112.68 | 0.13 | 0 | 2 | 0.56 |
| MS18 | 523.6 | 5.05 | −5.68 | 112.68 | 0.16 | 0 | 2 | 0.56 |
| MS19 | 498.55 | 3.81 | −4.84 | 138.7 | 0.07 | 0 | 0 | 0.56 |
| MS20 | 568.64 | 4.24 | −5.29 | 125.15 | 0.19 | 0 | 1 | 0.56 |
| MS21 | 567.65 | 3.41 | −3.67 | 127.95 | 0.19 | 0 | 1 | 0.55 |
| MS22 | 498.55 | 3.82 | −4.59 | 125.57 | 0.11 | 0 | 0 | 0.56 |
| MS23 | 498.55 | 3.9 | −4.59 | 125.57 | 0.11 | 0 | 0 | 0.56 |
| MS24 | 498.55 | 3.96 | −4.62 | 125.57 | 0.11 | 0 | 0 | 0.56 |
| MS25 | 565.56 | 5.65 | −6.13 | 112.68 | 0.13 | 0 | 2 | 0.56 |
| MS26 | 565.56 | 5.62 | −6.13 | 112.68 | 0.13 | 0 | 2 | 0.56 |
| MS27 | 572.67 | 5.75 | −6.46 | 104.48 | 0.09 | 0 | 2 | 0.17 |
| MS28 | 558.69 | 6.28 | −7.12 | 87.41 | 0.11 | 0 | 2 | 0.17 |
| MS29 | 586.7 | 5.79 | −6.43 | 104.48 | 0.11 | 0 | 2 | 0.17 |
| Molecule | GI Absorption | BBB Permeant | Pgp Substrate | CYP1A2 Inhibitor | CYP2C19 Inhibitor | CYP2C9 Inhibitor | CYP2D6 Inhibitor | CYP3A4 Inhibitor | Log Kp (cm/s) |
|---|---|---|---|---|---|---|---|---|---|
| MS01 | Low | No | No | No | Yes | Yes | No | No | −6.84 |
| MS10 | Low | No | No | No | Yes | Yes | No | No | −6.52 |
| MS11 | Low | No | No | Yes | Yes | Yes | No | No | −7.17 |
| MS12 | Low | No | No | No | Yes | Yes | No | No | −6.41 |
| MS13 | Low | No | No | No | Yes | No | No | No | −6.85 |
| MS14 | Low | No | No | No | Yes | Yes | No | No | −6.56 |
| MS15 | Low | No | No | No | Yes | Yes | No | No | −6.16 |
| MS16 | Low | No | No | No | No | No | No | No | −6.43 |
| MS17 | Low | No | No | Yes | Yes | Yes | No | No | −6.67 |
| MS18 | Low | No | No | Yes | No | Yes | No | No | −6.6 |
| MS19 | Low | No | No | No | Yes | No | No | No | −7.32 |
| MS20 | Low | No | No | No | No | Yes | No | No | −7.6 |
| MS21 | Low | No | Yes | No | No | Yes | No | No | −9.41 |
| MS22 | Low | No | No | No | Yes | Yes | No | No | −7.61 |
| MS23 | Low | No | No | No | Yes | Yes | No | No | −7.61 |
| MS24 | Low | No | No | No | Yes | Yes | No | No | −7.58 |
| MS25 | Low | No | No | Yes | No | No | No | No | −6.63 |
| MS26 | Low | No | No | Yes | No | No | No | No | −6.63 |
| MS27 | Low | No | No | No | No | No | No | No | −6.37 |
| MS28 | Low | No | No | No | No | No | No | No | −5.46 |
| MS29 | Low | No | No | Yes | No | No | No | Yes | −6.5 |
| Molecule | LD50 mg/kg | Toxicity Class | Hepatotoxicity | Respiratory Toxicity | Carcinogenicity | Immunotoxicity | Mutagenicity | Cytotoxicity |
|---|---|---|---|---|---|---|---|---|
| MS01 | 800 | 4 | Active | Active | Inactive | Inactive | Inactive | Inactive |
| MS10 | 1000 | 4 | Active | Active | Inactive | Inactive | Inactive | Inactive |
| MS11 | 5000 | 5 | Active | Active | Inactive | Inactive | Inactive | Inactive |
| MS12 | 1000 | 4 | Active | Active | Inactive | Inactive | Inactive | Inactive |
| MS13 | 1000 | 4 | Active | Active | Inactive | Inactive | Inactive | Inactive |
| MS14 | 264 | 3 | Active | Active | Inactive | Inactive | Inactive | Inactive |
| MS15 | 264 | 3 | Active | Active | Inactive | Inactive | Inactive | Inactive |
| MS16 | 264 | 3 | Active | Active | Inactive | Inactive | Inactive | Inactive |
| MS17 | 800 | 4 | Active | Active | Inactive | Active | Inactive | Inactive |
| MS18 | 800 | 4 | Active | Active | Inactive | Inactive | Inactive | Inactive |
| MS19 | 800 | 4 | Active | Active | Inactive | Inactive | Inactive | Inactive |
| MS20 | 300 | 3 | Inactive | Active | Inactive | Inactive | Inactive | Inactive |
| MS21 | 300 | 3 | Inactive | Active | Inactive | Inactive | Inactive | Inactive |
| MS22 | 264 | 3 | Active | Active | Inactive | Active | Inactive | Inactive |
| MS23 | 264 | 3 | Active | Active | Inactive | Active | Inactive | Inactive |
| MS24 | 800 | 4 | Active | Active | Inactive | Inactive | Inactive | Inactive |
| MS25 | 264 | 3 | Active | Active | Inactive | Active | Inactive | Inactive |
| MS26 | 264 | 3 | Active | Active | Inactive | Active | Inactive | Inactive |
| MS27 | 20,000 | 6 | Active | Active | Inactive | Inactive | Inactive | Inactive |
| MS28 | 264 | 3 | Inactive | Active | Inactive | Active | Inactive | Inactive |
| MS29 | 200 | 3 | Inactive | Active | Inactive | Active | Inactive | Inactive |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oladeji, S.M.; Conteh, D.N.; Bello, L.A.; Adegboyega, A.E.; Shokunbi, O.S. Rational Design and Optimization of Novel PDE5 Inhibitors for Targeted Colorectal Cancer Therapy: An In Silico Approach. Int. J. Mol. Sci. 2025, 26, 1937. https://doi.org/10.3390/ijms26051937
Oladeji SM, Conteh DN, Bello LA, Adegboyega AE, Shokunbi OS. Rational Design and Optimization of Novel PDE5 Inhibitors for Targeted Colorectal Cancer Therapy: An In Silico Approach. International Journal of Molecular Sciences. 2025; 26(5):1937. https://doi.org/10.3390/ijms26051937
Chicago/Turabian StyleOladeji, Samson Marvellous, Deborah Ngozi Conteh, Lukman Abidemi Bello, Abayomi Emmanuel Adegboyega, and Oluwatosin Sarah Shokunbi. 2025. "Rational Design and Optimization of Novel PDE5 Inhibitors for Targeted Colorectal Cancer Therapy: An In Silico Approach" International Journal of Molecular Sciences 26, no. 5: 1937. https://doi.org/10.3390/ijms26051937
APA StyleOladeji, S. M., Conteh, D. N., Bello, L. A., Adegboyega, A. E., & Shokunbi, O. S. (2025). Rational Design and Optimization of Novel PDE5 Inhibitors for Targeted Colorectal Cancer Therapy: An In Silico Approach. International Journal of Molecular Sciences, 26(5), 1937. https://doi.org/10.3390/ijms26051937