

Mitochondrial Dysfunction in Cardiovascular Diseases


Abstract
1. Introduction
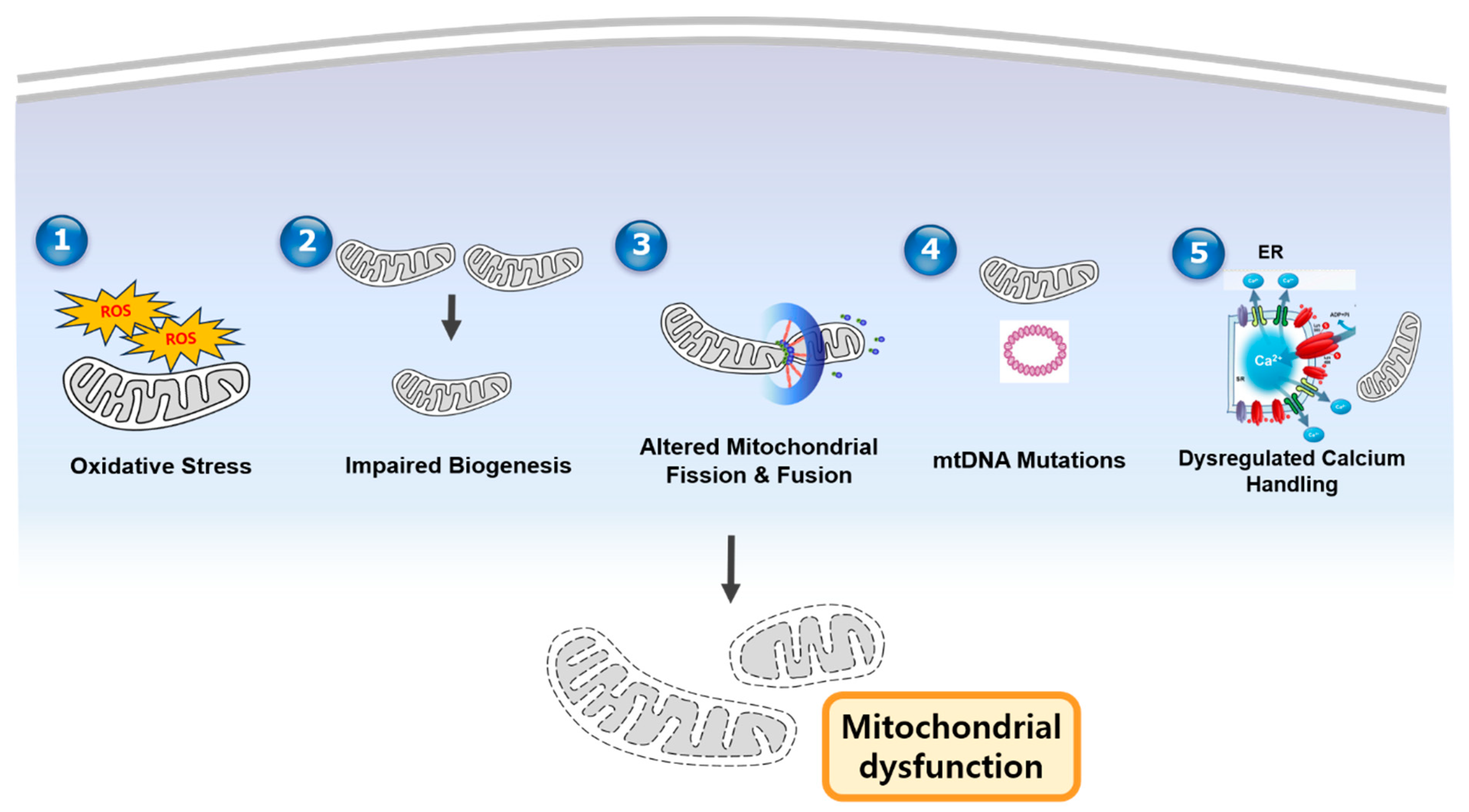
2. Mechanisms of Mitochondrial Dysfunction in CVD

{kind=link}
| Mechanism | Description | Associated CVDs | References |
|---|---|---|---|
| Impaired OXPHOS | Reduced ATP production due to defects in electron transport chain complexes | Heart failure, ischemic heart disease | [12,13,31,32,33,34,35] |
| mtDNA mutations | Accumulation of mutations leading to defective mitochondrial proteins | Cardiomyopathy, atherosclerosis | [20,21,22] |
| Excessive ROS production | Overproduction of ROS causing oxidative damage to lipids, proteins, and DNA | Hypertension, heart failure | [12,13] |
| Altered mitochondrial dynamics | Imbalance in fission/fusion and defective mitophagy | Myocardial infarction, hypertrophy | [16,17,18,19] |
| Calcium mishandling | Disrupted Ca2+ homeostasis leading to mitochondrial permeability transition | Arrhythmias, ischemia–reperfusion injury | [23,24,25] |
3. Mitochondrial Dysfunction in Specific Cardiovascular Diseases in Various Cell Types
4. Diagnostic Biomarkers of Mitochondrial Dysfunction in CVD
5. Therapeutic Strategies Targeting Mitochondrial Dysfunction in CVD
6. Discussion
7. Conclusions
Funding
Conflicts of Interest
Abbreviations
| CVD | Cardiovascular Disease |
| ROS | Reactive Oxygen Species |
| ATP | Adenosine Triphosphate |
| mtDNA | mitochondrial DNA |
| OXPHOS | Oxydative Phosphorylation |
| mPTP | mitochondrial Permeability Transition Pore |
| LDL | Low Density Lipoprotein |
| PGC-1α | Peroxisome proliferator-activated receptor Gamma Coactivator 1-alpha |
| NRF-1/2 | Nuclear Respiratory Factors–1/2 |
| TFAM | Transcription factor A, mitochondrial |
| Drp1 | Dynamin-related protein 1 |
| Mfn1/2 | Mitofusin 1/2 |
| OPA1 | Optic Atrophy 1 |
| ETC | Electron Transport Chain |
| DAMPs | Damage-Associated Molecular Patterns |
| MCU | Mitochondrial Calcium Uniporter |
| NCLX | Sodium–Calcium Exchanger |
| MICOS | Mitochondrial contact site and Cristae-Organizing System |
| TCA | Tricarboxylic acid |
| NADH | Nicotinamide Adenine Dinucleotide |
| FADH2 | Flavian Adenine Dinucleotide |
| VSMCs | Vascular Smooth Muscle Cells |
| FAO | Fatty Acid Oxidation |
| PPAR | Peroxisome Proliferator-Activated Receptor |
| SOD | Superoxide Dismutase |
| GPx | Glutathione Peroxidase |
| IRI | Ischemia–Reperfusion Injury |
| NAC | N-acetylcysteine |
| NO | Nitric Oxide |
| CoQ10 | Coenzyme Q10 |
| MOMP | Mitochondrial Outer Membrane Permeabilization |
| EV | Extracellular Vesicle |
| AMI | Acute Myocardial Infarction |
| MDA | Malondialdehyde |
| CAD | Coronary Artery Disease |
| GSH | Glutahione |
| PET | Positron Emission Tomography |
| MRI | Magnetic Resonance Imaging |
| PCr | Phosphocreatine |
| TMRM | Tetramethylrhodamine Methyl Ester |
| AAV | Adeno-Associated Viruses |
| MitoTALENs | Mitochondrially targeted Transcription Activator-like Effector Nucleases |
| CRISPR/Cas9 | Clustered Regularly Interspaced Short Palindromic Repeats/CRISPR-Associated Protein 9 |
| scRNA-seq | single-cell RNA sequencing |
| STED | Stimulated Emission Depletion |
| PALM | Photoactivated Localization Microscopy |
| FRET | Förster Resonance Energy Transfer |
References
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barasa, A.; Barquera, S.; Beaton, A.Z.; Benjamin, E.J.; et al. Global burden of cardiovascular diseases and risk factors, 1990–2019: Update from the GBD 2019 study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef]
- Brown, D.A.; Perry, J.B.; Allen, M.E.; Sabbah, H.N.; Stauffer, B.L.; Shaikh, S.R. Mitochondrial function as a therapeutic target in heart failure. Nat. Rev. Cardiol. 2017, 14, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Tian, R. Mitochondrial dysfunction in pathophysiology of heart failure. J. Clin. Investig. 2018, 128, 3716–3726. [Google Scholar] [CrossRef] [PubMed]
- Lesnefsky, E.J.; Chen, Q.; Hoppel, C.L. Mitochondrial metabolism in aging heart. Circ. Res. 2016, 118, 1593–1611. [Google Scholar] [CrossRef] [PubMed]
- Kaludercic, N.; Di Lisa, F. Mitochondrial ROS formation in the pathogenesis of diabetic cardiomyopathy. Front. Cardiovasc. Med. 2020, 7, 12. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijević, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef]
- Murphy, E.; Ardehali, H.; Balaban, R.S.; DiLisa, F.; Dorn, G.W.; Kitsis, R.N.; Otsu, K.; Ping, P.; Rizzuto, R.; Sack, M.N.; et al. Mitochondrial function, biology, and role in disease: A scientific statement from the American Heart Association. Circ. Res. 2016, 118, 1960–1991. [Google Scholar] [CrossRef]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef]
- Pietri, S.; Maurelli, E.; Drieu, K.; Culcasi, M.; Pietri, G.; Cozzone, P.J. Cardioprotective effects of a novel mitochondrial-targeted antioxidant in myocardial ischemia-reperfusion injury. Antioxidants 2021, 10, 1352. [Google Scholar]
- Szeto, H.H. Mitochondria-targeted peptide antioxidants: Novel neuroprotective agents. AAPS J. 2016, 18, 1021–1030. [Google Scholar] [CrossRef]
- Dai, D.F.; Rabinovitch, P.S.; Ungvari, Z. Mitochondria and cardiovascular aging. Circ. Res. 2018, 123, 943–959. [Google Scholar] [CrossRef] [PubMed]
- Peoples, J.N.; Saraf, A.; Ghazal, N.; Pham, T.T.; Kwong, J.Q. Mitochondrial dysfunction and oxidative stress in heart disease. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, J.; Chen, Q.; Guan, J.; Ma, C.; Geng, Y. Oxidative stress and mitochondrial dysfunction in atherosclerosis. Int. J. Mol. Sci. 2022, 23, 12145. [Google Scholar]
- Fernandez-Marcos, P.J.; Auwerx, J. Regulation of PGC-1α, a mitochondrial function modulator. Nat. Rev. Mol. Cell Biol. 2016, 17, 197–205. [Google Scholar]
- Suliman, H.B.; Piantadosi, C.A. Mitochondrial biogenesis: Regulation by endogenous gases during inflammation and injury. Antioxidants 2020, 9, 312. [Google Scholar]
- Dorn, G.W.; Vega, R.B.; Kelly, D.P. Mitochondrial dynamics and heart failure. Circ. Res. 2015, 116, 167–182. [Google Scholar] [CrossRef]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef]
- Ong, S.B.; Subrayan, S.; Lim, S.Y.; Yellon, D.M.; Davidson, S.M.; Hausenloy, D.J. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation 2010, 121, 2012–2022. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, Y.; Zhang, J.; Jia, D. Exercise alleviates cardiovascular diseases by improving mitochondrial homeostasis. J. Am. Heart Assoc. 2024, 13, e036555. [Google Scholar] [CrossRef]
- Wallace, D.C. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: A dawn for evolutionary medicine. Annu. Rev. Genet. 2005, 39, 359–407. [Google Scholar] [CrossRef]
- West, A.P.; Shadel, G.S.; Ghosh, S. Mitochondria in innate immune responses. Nat. Rev. Immunol. 2011, 11, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Bushra; Ahmed, S.I.; Begum, S.; Maaria; Habeeb, M.S.; Jameel, T.; Khan, A.A. Molecular basis of sepsis: A new insight into the role of mitochondrial DNA as a damage-associated molecular pattern. Mitochondrion 2024, 79, 101967. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 566–578. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, E.J.; Halestrap, A.P. Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochem. J. 1995, 307, 93–98. [Google Scholar] [CrossRef]
- Xiao, Z.; Li, S.; Wu, X.; Chen, X.; Yan, D.; He, J. GATA-4 overexpressing BMSC-derived exosomes suppress H/R-induced cardiomyocyte ferroptosis. iScience 2024, 27, 110784. [Google Scholar] [CrossRef]
- Gottlieb, R.A.; Thomas, A.; Gustafsson, Å.B. Mitochondrial morphology and function in cardiac health and disease. J. Mol. Cell. Cardiol. 2021, 158, 34–44. [Google Scholar]
- Shirakabe, A.; Zhai, P.; Ikeda, Y.; Saito, T.; Maejima, Y.; Hsu, C.P.; Nomura, M.; Egashira, K.; Levine, B.; Sadoshima, J. Drp1-dependent mitochondrial autophagy plays a protective role against pressure overload-induced mitochondrial dysfunction and heart failure. Circulation 2016, 133, 1249–1263. [Google Scholar] [CrossRef]
- Disatnik, M.H.; Ferreira, J.C.; Campos, J.C.; Gomes, K.S.; Dourado, P.M.; Qi, X.; Mochly-Rosen, D. Acute inhibition of excessive mitochondrial fission after myocardial infarction prevents cardiomyocyte death and improves cardiac function. J. Mol. Cell. Cardiol. 2019, 136, 1–10. [Google Scholar]
- Rampelt, H.; Wollweber, F.; Gerke, C.; van der Laan, M. Role of MICOS in mitochondrial cristae organization and function. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2022, 1869, 119168. [Google Scholar]
- Calo, L.; Dong, Y.; Kumar, R.; Przyklenk, K.; Sanderson, T.H. Mitochondrial Dynamics: An Emerging Paradigm in Ischemia-Reperfusion Injury. Curr. Pharm. Des. 2013, 19, 6848–6857. [Google Scholar] [CrossRef]
- Lesnefsky, E.J.; Chen, Q.; Hoppel, C.L. Mitochondrial dysfunction in heart failure: Morphology and function. Front. Cardiovasc. Med. 2022, 9, 892028. [Google Scholar]
- Kondadi, A.K.; Anand, R.; Reichert, A.S. Cristae junctions maintain mitochondrial function. Nat. Commun. 2020, 11, 5809. [Google Scholar]
- Chen, H.; Wang, Y.; Zhang, J.; Li, Q.; Zhao, Y. Cristae disruption and ROS production in cardiac mitochondria. Redox Biol. 2022, 50, 102256. [Google Scholar]
- Tong, M.; Zablocki, D.; Sadoshima, J. Mitophagy impairment in cardiovascular disease. Circ. Res. 2021, 129, 579–592. [Google Scholar]
- Sabbah, H.N.; Zhang, K.; Gupta, R.C.; Xu, J.; Singh-Gupta, V. Mitochondrial dysfunction in heart failure: Evidence from translational studies. JACC Basic Transl. Sci. 2020, 5, 844–857. [Google Scholar]
- Fernández-Caggiano, M.; Eaton, P.; Rodríguez-Sinovas, A. Mitochondrial cristae remodeling post-reperfusion in ischemic heart disease. J. Am. Heart Assoc. 2022, 11, e023685. [Google Scholar]
- Zhou, H.; Toan, S.; Zhu, H.; Klionsky, D.J.; Zhang, Y. Therapeutic strategies targeting mitochondrial dynamics in cardiovascular diseases. Nat. Rev. Cardiol. 2022, 19, 523–538. [Google Scholar]
- Civiletto, G.; Vettiger, A.; De Rasmo, D.; Papa, S.; Scorrano, L. OPA1 modulation enhances mitochondrial function in preclinical models of heart failure. Nat. Commun. 2021, 12, 3490. [Google Scholar]
- Martinez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 650–666. [Google Scholar] [CrossRef]
- Fernie, A.R.; Zhang, Y.; Sampathkumar, A. Cytosolic metabolism is a key regulator of Arabidopsis immunity. Nat. Rev. Mol. Cell Biol. 2020, 21, 451–464. [Google Scholar]
- McCommis, K.S.; Finck, B.N.; Kovacs, A.; Bultman, S.J.; Kelly, D.P. Pyruvate dehydrogenase regulation of cardiac metabolism. Circ. Res. 2022, 130, 174–189. [Google Scholar]
- Zhang, Y.; Taufalele, P.V.; Fernandez, D.; Cai, H.; Wang, Z. PDH dysregulation in heart failure and diabetic cardiomyopathy. J. Clin. Investig. 2021, 131, e140555. [Google Scholar]
- Cluntun, A.A.; Lukey, M.J.; Cerione, R.A.; Locasale, J.W. Metabolic shifts in cardiac energy production under stress. Nat. Metab. 2021, 3, 182–198. [Google Scholar]
- Karwi, Q.G.; Uddin, G.M.; Ho, K.L.; Lopaschuk, G.D. TCA cycle impairment in heart failure: Effects on energy production and ROS. Cardiovasc. Res. 2020, 116, 2068–2079. [Google Scholar]
- Ryan, D.G.; Murphy, M.P.; Frezza, C.; Prag, H.A.; Chouchani, E.T.; O’Neill, L.A. TCA cycle and ETC interplay in redox balance. Nature 2021, 595, 724–729. [Google Scholar]
- Stacpoole, P.W.; McCall, C.E.; Martyniuk, C.J.; James, M.O.; Shroads, A.L. PDH activation restores TCA cycle flux in cardiovascular disease models. J. Clin. Investig. 2022, 132, e157839. [Google Scholar]
- Ventura-Clapier, R.; Garnier, A.; Veksler, V.; Joubert, F. Bioenergetics of the failing heart. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2011, 1813, 1360–1372. [Google Scholar]
- Gibb, A.A.; Hill, B.G. Metabolic coordination of cardiovascular cell types in health and disease. Circ. Res. 2020, 127, 1230–1243. [Google Scholar]
- Kolwicz, S.C.; Purohit, S.; Tian, R. Cardiac metabolism and its regulation in health and disease. Circ. Res. 2021, 129, 697–710. [Google Scholar]
- Bertero, E.; Maack, C. Metabolic remodelling in heart failure. Nat. Rev. Cardiol. 2018, 15, 457–470. [Google Scholar] [CrossRef]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. Endothelial cell metabolism relies on glycolysis for ROS signaling. Nat. Metab. 2022, 4, 672–685. [Google Scholar]
- Li, X.; Yang, Q.; Shi, X.; Chen, Y.; Zhao, J.; Uddin, M. Metabolic adaptability of vascular smooth muscle cells in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 2456–2469. [Google Scholar]
- O’Neill, L.A.J.; Kishton, R.J.; Rathmell, J. Metabolic reprogramming in macrophage activation. Nat. Rev. Immunol. 2021, 21, 684–696. [Google Scholar]
- Viola, A.; Munari, F.; Sánchez-Rodríguez, R.; Scolaro, T.; Castegna, A. Pro-inflammatory M1 macrophage metabolism mirrors the Warburg effect. Front. Immunol. 2022, 13, 856465. [Google Scholar]
- Zhang, X.; Wang, Y.; Yuan, J.; Li, N.; Pei, S.; Xu, J.; Luo, X.; Deng, Z.; Wu, J.; Liu, J. Glycolytic shift supports inflammatory mediator production in M1 macrophages. Immunity 2021, 54, 1234–1248. [Google Scholar]
- Huang, S.C.; Smith, A.M.; Everts, B.; Colonna, M.; Pearce, E.L.; Pearce, E.J. Oxidative phosphorylation drives M2 macrophage polarization. Nat. Metab. 2020, 2, 903–916. [Google Scholar]
- Lopaschuk, G.D.; Karwi, Q.G.; Tian, R.; Wende, A.R.; Abel, E.D. Pathological shifts in cardiovascular cell metabolism. Nat. Rev. Cardiol. 2021, 18, 805–823. [Google Scholar]
- Bertero, E.; Dudley, S.C.; Maack, C. Metabolic inflexibility in heart failure: From bench to bedside. JACC Basic Transl. Sci. 2022, 7, 934–947. [Google Scholar]
- Doenst, T.; Nguyen, T.D.; Abel, E.D. Cardiac metabolism in heart failure: Implications beyond ATP production. Circ. Res. 2017, 121, 740–757. [Google Scholar] [CrossRef]
- Arany, Z.; He, H.; Lin, J.; Hoyer, K.; Handschin, C.; Toka, O.; Ahmad, F.; Matsui, T.; Chin, S.; Wu, P.H.; et al. Transcriptional coactivator PGC-1 alpha controls the energy state and contractile function of cardiac muscle. Cell Metab. 2005, 1, 259–271. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, H.; Wang, Q.; Li, X.; Huang, Y.; Yan, B.; Li, Q.; Zhao, Y.; Cai, H.; Wang, Z. Increased mitochondrial FAO in endothelial cells impairs NO production in hypertension. Hypertension 2022, 79, 1456–1467. [Google Scholar]
- Shi, X.; Li, X.; Yang, Q.; Zhao, J.; Uddin, M.; Chen, Y. Glycolytic and FAO shifts in VSMCs drive vascular remodeling in hypertension. Hypertension 2021, 77, 1245–1257. [Google Scholar]
- Ussher, J.R.; Sutendra, G.; Jaswal, J.S.; Lopaschuk, G.D. Targeted therapies to restore metabolic balance in cardiovascular disease. Cardiovasc. Res. 2022, 118, 2345–2357. [Google Scholar]
- Nickel, A.G.; von Hardenberg, A.; Hohl, M.; Löffler, J.R.; Kohlhaas, M.; Becker, J.; Reil, J.C.; Kazakov, A.; Bonnekoh, J.; Stadelmaier, M.; et al. Reversal of mitochondrial transhydrogenase causes oxidative stress in heart failure. Cell Metab. 2015, 22, 472–484. [Google Scholar] [CrossRef]
- Narula, J.; Pandey, P.; Arbustini, E.; Haider, N.; Narula, N.; Kolodgie, F.D.; Dal Bello, B.; Semigran, M.J.; Bielsa-Masdeu, A.; Dec, G.W.; et al. Apoptosis in heart failure: Release of cytochrome c from mitochondria and activation of caspases. Circulation 1999, 100, 1470–1475. [Google Scholar]
- Sharov, V.G.; Todor, A.; Khanal, S.; Imai, M.; Sabbah, H.N. Cyclosporine A attenuates mitochondrial permeability transition and improves mitochondrial function in cardiomyocytes isolated from dogs with heart failure. J. Mol. Cell. Cardiol. 2005, 38, 473–483. [Google Scholar] [CrossRef]
- Dai, D.F.; Johnson, S.C.; Villarin, J.J.; Chin, M.T.; Nieves-Cintrón, M.; Chen, T.; Marcinek, D.J.; Dorn, G.W.; Kang, Y.J.; Prolla, T.A.; et al. Mitochondrial oxidative stress mediates angiotensin II–induced cardiac hypertrophy and Gαq overexpression–induced heart failure. Circ. Res. 2011, 108, 837–846. [Google Scholar] [CrossRef]
- Lesnefsky, E.J.; Moghaddas, S.; Tandler, B.; Kerner, J.; Hoppel, C.L. Mitochondrial dysfunction in cardiac disease: Ischemia–reperfusion, aging, and heart failure. J. Mol. Cell. Cardiol. 2001, 33, 1065–1089. [Google Scholar] [CrossRef]
- Ong, S.B.; Samangouei, P.; Kalkhoran, S.B.; Hausenloy, D.J. The mitochondrial permeability transition pore and its role in myocardial ischemia reperfusion injury. J. Mol. Cell. Cardiol. 2019, 136, 31–40. [Google Scholar] [CrossRef]
- Argaud, L.; Gateau-Roesch, O.; Raisky, O.; Loufouat, J.; Robert, D.; Ovize, M. Postconditioning inhibits mitochondrial permeability transition. Circulation 2005, 111, 194–197. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Pasdois, P. The role of the mitochondrial permeability transition pore in heart disease. Biochim. Biophys. Acta (BBA)-Bioenerg. 2009, 1787, 1402–1415. [Google Scholar] [CrossRef] [PubMed]
- Zweier, J.L.; Talukder, M.A.H. The role of oxidants and free radicals in reperfusion injury. Cardiovasc. Res. 2006, 70, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Kevin, L.G.; Camara, A.K.; Riess, M.L.; Novalija, E.; Stowe, D.F. Ischemic preconditioning alters real-time measure of myocardial ROS generation in the ischemic and reperfused heart. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1416–H1425. [Google Scholar]
- Adlam, V.J.; Harrison, J.C.; Porteous, C.M.; James, A.M.; Smith, R.A.; Murphy, M.P.; Sammut, I.A. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J. 2005, 19, 1088–1095. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Harrison, D.G. Endothelial dysfunction in cardiovascular diseases: The role of oxidant stress. Circ. Res. 2000, 87, 840–844. [Google Scholar] [CrossRef]
- Higashi, Y.; Sasaki, S.; Nakagawa, K.; Kimura, M.; Noma, K.; Hara, K.; Jitsuiki, D.; Goto, C.; Oshima, T.; Chayama, K.; et al. Endothelial function and oxidative stress in renovascular hypertension. N. Engl. J. Med. 2002, 346, 1954–1962. [Google Scholar] [CrossRef]
- Ashor, A.W.; Lara, J.; Mathers, J.C.; Siervo, M. Effect of vitamin C on endothelial function in health and disease: A systematic review and meta-analysis of randomised controlled trials. Atherosclerosis 2019, 290, 121–129. [Google Scholar] [CrossRef]
- Sawyer, D.B.; Siwik, D.A.; Xiao, L.; Pimentel, D.R.; Singh, K.; Colucci, W.S. Role of oxidative stress in myocardial hypertrophy and failure. J. Mol. Cell. Cardiol. 2002, 34, 379–388. [Google Scholar] [CrossRef]
- Montezano, A.C.; Touyz, R.M. Reactive oxygen species, vascular Noxs, and hypertension: Focus on translational and clinical research. Antioxid. Redox Signal. 2019, 31, 1217–1238. [Google Scholar] [CrossRef]
- Wilcox, C.S.; Pearlman, A.; Cardiology, P.; Road, M.; Nw, W.; Dc, W.; Welch, W.J. Effects of tempol on vascular and metabolic dysfunction in spontaneously hypertensive rats. Antioxidants 2020, 9, 925. [Google Scholar]
- Moore, K.J.; Sheedy, F.J.; Fisher, E.A. Macrophages in atherosclerosis: A dynamic balance. Nat. Rev. Immunol. 2013, 13, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; García-Cardeña, G.; Owens, G.K. Recent insights into the cellular biology of atherosclerosis. J. Cell Biol. 2018, 217, 1915–1927. [Google Scholar] [CrossRef] [PubMed]
- Yu, E.P.; Bennett, M.R. Mitochondrial DNA damage and atherosclerosis. Trends Endocrinol. Metab. 2019, 30, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Antoniades, C.; Bakogiannis, C.; Leeson, P.; Guzik, T.J.; Zhang, M.H.; Tousoulis, D.; Antonopoulos, A.S.; Demosthenous, M.; Marinou, K.; Hale, S.; et al. Rapid effects of rosuvastatin on arterial stiffness and endothelial function in healthy humans: Evidence for mitochondrial protection. Circulation 2016, 134, 136–147. [Google Scholar]
- Bentzon, J.F.; Otsuka, F.; Virmani, R.; Falk, E. Mechanisms of plaque formation and rupture. Circ. Res. 2020, 126, 1368–1383. [Google Scholar] [CrossRef]
- Azzi, A.; Stocker, A. Vitamin E: Non-antioxidant roles. Prog. Lipid Res. 2000, 39, 231–255. [Google Scholar] [CrossRef]
- Stephens, N.G.; Parsons, A.; Schofield, P.M.; Kelly, F.; Cheeseman, K.; Mitchinson, M.J. Randomised controlled trial of vitamin E in patients with coronary disease: Cambridge Heart Antioxidant Study (CHAOS). Lancet 1996, 347, 781–786. [Google Scholar] [CrossRef]
- Pinti, M.; Cevenini, E.; Nasi, M.; De Biasi, S.; Salvioli, S.; Monti, D.; Benatti, S.; Gibellini, L.; Cotichini, R.; Stazi, M.A.; et al. Circulating mitochondrial DNA increases with age and is a familiar trait: Implications for “inflamm-aging”. Eur. J. Immunol. 2014, 44, 1552–1562. [Google Scholar] [CrossRef]
- Nakahira, K.; Hisata, S.; Choi, A.M.K. The roles of mitochondrial damage-associated molecular patterns in diseases. Antioxid. Redox Signal. 2015, 23, 1329–1350. [Google Scholar] [CrossRef]
- Wang, L.; Yu, P.; Zhou, B.; Song, J.; Li, Z.; Zhang, M.; Guo, G.; Wang, Y.; Chen, X.; Han, L.; et al. Circulating mitochondrial DNA as a potential biomarker for acute myocardial infarction. J. Am. Coll. Cardiol. 2019, 73, 1895–1905. [Google Scholar]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Dhalla, N.S.; Temsah, R.M.; Netticadan, T. Role of oxidative stress in cardiovascular diseases. J. Hypertens. 2000, 18, 655–673. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.L.; Chiou, C.C.; Chang, P.Y.; Wu, J.T. Urinary 8-OHdG: A marker of oxidative stress to DNA and a risk factor for cancer, atherosclerosis and diabetics. Clin. Chim. Acta 2019, 492, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Dikalov, S.I.; Ungvari, Z. Role of mitochondrial oxidative stress in hypertension. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H1079–H1088. [Google Scholar] [CrossRef]
- Camici, P.G.; Crea, F. Coronary microvascular dysfunction. N. Engl. J. Med. 2007, 356, 830–840. [Google Scholar] [CrossRef]
- Schwarz, K.; Siddiqi, N.; Singh, S.; Neil, C.J.; Dawson, D.K.; Frenneaux, M.P. The role of imaging in the assessment of mitochondrial function in heart failure. JACC Cardiovasc. Imaging 2018, 11, 1868–1878. [Google Scholar]
- Hansson, N.H.; Tolbod, L.P.; Harms, H.J.; Wiggers, H.; Kim, W.Y.; Hansen, E.; Zaremba, T.; Frøkiær, J.; Jakobsen, S.; Sørensen, J.; et al. Myocardial oxygen consumption and efficiency in patients with cardiac amyloidosis measured with 11C-acetate PET. J. Nucl. Cardiol. 2019, 26, 1972–1981. [Google Scholar]
- Bottomley, P.A.; Panjrath, G.S.; Lai, S.; Hirsch, G.A.; Wu, K.; Najjar, S.S.; Steinberg, A.; Gerstenblith, G.; Weiss, R.G. Metabolic rates of ATP transfer through creatine kinase (CK flux) predict clinical heart failure events and death. Sci. Transl. Med. 2013, 5, 215re3. [Google Scholar] [CrossRef]
- Davidson, S.M.; Duchen, M.R. Imaging mitochondrial calcium signalling with fluorescent probes: From single organelles to whole hearts. Methods 2017, 121–122, 66–74. [Google Scholar]
- Zhang, J.; Li, X.; Mueller, M.; Wang, Y.; Zong, C.; Deng, N.; Vondriska, T.M.; Liem, D.A.; Yang, J.I.; Korge, P.; et al. Systematic characterization of the murine mitochondrial proteome using functionally validated cardiac mitochondria. Proteomics 2008, 8, 1564–1575. [Google Scholar] [CrossRef]
- Lau, E.; Huang, D.; Cao, Q.; Dincer, T.U.; Black, C.M.; Lin, A.J.; Lee, J.M.; Wang, Y.; Liem, D.A.; Kwan, K.; et al. Proteomic analysis of mitochondrial dysfunction in heart failure. Circ. Res. 2020, 127, 1217–1230. [Google Scholar]
- Hunter, W.G.; Kelly, J.P.; McGarrah, R.W.; Khouri, M.G.; Craig, D.; Haynes, C.; Ilkayeva, O.; Stevens, R.D.; Bain, J.R.; Newgard, C.B.; et al. Metabolomic profiling identifies novel circulating biomarkers of mitochondrial dysfunction in chronic heart failure. JACC Basic Transl. Sci. 2016, 1, 625–636. [Google Scholar]
- Lewis, G.D.; Asnani, A.; Gerszten, R.E. Application of metabolomics to cardiovascular biomarker and pathway discovery. J. Am. Coll. Cardiol. 2008, 52, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, S.A.; Rosenfeldt, F.; Kumar, A.; Dolliner, P.; Filipiak, K.J.; Pella, D.; Alehagen, U.; Steurer, G.; Littarru, G.P.; Q-SYMBIO Study Investigators. The effect of coenzyme Q10 on morbidity and mortality in chronic heart failure: Results from Q-SYMBIO: A randomized controlled trial. JACC Heart Fail. 2014, 2, 641–649. [Google Scholar] [CrossRef]
- Rossman, M.J.; Santos-Parker, J.R.; Steward, C.A.; Bispham, N.Z.; Cuevas, L.M.; Rosenberg, H.L.; Woodward, K.A.; Chonchol, M.; Gioscia-Ryan, R.A.; Murphy, M.P.; et al. Chronic supplementation with a mitochondrial antioxidant (MitoQ) improves vascular function in healthy older adults. Hypertension 2019, 73, 1055–1063. [Google Scholar] [CrossRef]
- Talasaz, A.H.; Khalili, H.; Jenab, Y.; Salarifar, M.; Broumand, M.A.; Darabi, F.; Jafari, M.; Sharif-Kashani, B.; Maleki, M.; Etezadi, F.; et al. N-acetylcysteine effects on oxidative stress and inflammatory biomarkers in patients with acute coronary syndrome. J. Cardiovasc. Pharmacol. 2020, 76, 171–177. [Google Scholar]
- Song, M.; Mihara, K.; Chen, Y.; Scaduto, R.C.; Dorn, G.W. Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab. 2015, 21, 273–286. [Google Scholar] [CrossRef]
- Franco, A.; Kitsis, R.N.; Fleischer, J.A.; Gavathiotis, E.; Kornfeld, O.S.; Gong, G.; Biris, N.; Benz, A.; Qvit, N.; Donnelly, S.K.; et al. Correcting mitochondrial fusion by manipulating mitofusin conformations. Nature 2021, 598, 645–650. [Google Scholar] [CrossRef]
- Piot, C.; Croisille, P.; Staat, P.; Thibault, H.; Rioufol, G.; Mewton, N.; Elbelghiti, R.; Cung, T.T.; Bonnefoy, E.; Angoulvant, D.; et al. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N. Engl. J. Med. 2008, 359, 473–481. [Google Scholar] [CrossRef]
- Atar, D.; Arheden, H.; Berdeaux, A.; Bonnet, J.L.; Carlsson, M.; Clemmensen, P.; Cuvier, V.; Danchin, N.; Dubois-Randé, J.L.; Engblom, H.; et al. Effect of intravenous TRO40303 as an adjunct to primary percutaneous coronary intervention for acute ST-elevation myocardial infarction: MITOCARE study results. Eur. Heart J. 2016, 37, 112–121. [Google Scholar] [CrossRef]
- Tucker, W.J.; Beaudry, R.I.; Liang, Y.; Clark, A.M.; Tompkins, C.; Nelson, M.D.; Ellsworth, S.; Wright, S.P.; Haykowsky, M.J.; Angadi, S.S.; et al. Meta-analysis of exercise training on left ventricular ejection fraction in heart failure with reduced ejection fraction. Am. J. Cardiol. 2020, 125, 1398–1405. [Google Scholar] [CrossRef] [PubMed]
- Haykowsky, M.J.; Tomczak, C.R.; Scott, J.M.; Paterson, D.I.; Kitzman, D.W. Determinants of exercise intolerance in patients with heart failure and reduced or preserved ejection fraction. J. Appl. Physiol. 2015, 119, 739–744. [Google Scholar] [CrossRef] [PubMed]
- Fontana, L.; Partridge, L.; Longo, V.D. Extending healthy life span—From yeast to humans. Science 2010, 328, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Estruch, R.; Ros, E.; Salas-Salvadó, J.; Covas, M.I.; Corella, D.; Arós, F.; Gómez-Gracia, E.; Ruiz-Gutiérrez, V.; Fiol, M.; Lapetra, J.; et al. Primary prevention of cardiovascular disease with a Mediterranean diet supplemented with extra-virgin olive oil or nuts. N. Engl. J. Med. 2018, 378, e34. [Google Scholar] [CrossRef]
- Sciarretta, S.; Maejima, Y.; Zablocki, D.; Sadoshima, J. Emerging therapies for mitochondrial dysfunction in cardiovascular disease. Circ. Res. 2021, 129, 405–420. [Google Scholar]
- Perry, H.M.; Huang, L.; Wilson, R.J.; Bajwa, A.; Jeschke, M.G.; Klein, G.L.; Herndon, D.N.; Porter, C.; Hundeshagen, G.; Olvera, H.; et al. Gene therapy approaches for cardiovascular diseases. Nat. Rev. Cardiol. 2022, 19, 611–628. [Google Scholar]
- Xue, W.; Zhang, S.; Wang, H.; Zhang, X.; Hu, R.; Liu, C.; Wang, Y.; Zhou, B.; Chen, X.; Li, Z.; et al. AAV-mediated gene therapy for mitochondrial disorders. Nat. Med. 2021, 27, 1234–1242. [Google Scholar]
- Ruan, L.; Zhang, X.; Li, Y.; Chen, Y.; Liu, C.; Han, W.; Zhao, J.; Wang, Q.; Huang, Y.; Yan, B.; et al. PGC-1α targeting improves mitochondrial function in heart failure models. J. Clin. Investig. 2020, 130, 4321–4335. [Google Scholar]
- Lai, Y.; Zhang, H.; Chen, Q.; Li, X.; Wang, Z.; Huang, Y.; Zhao, J.; Yan, B.; Cai, H.; Liu, C.; et al. Targeting PGC-1α enhances cardiac mitochondrial function in preclinical models. Cardiovasc. Res. 2023, 119, 1567–1580. [Google Scholar]
- Moraes, C.T.; Bacman, S.R.; Williams, S.L.; Pereira, C.V. Mitochondrial genome editing: Challenges and opportunities. Nat. Rev. Genet. 2021, 22, 735–750. [Google Scholar]
- Reddy, P.; Ocampo, A.; Suzuki, K.; Luo, J.; Bacman, S.R.; Williams, S.L.; Sugawara, T.; Okamura, D.; Tsunoda, T.; Hayashi, Y.; et al. CRISPR-based mitochondrial DNA editing reduces mutation load in preclinical models. Science 2022, 375, eabf0589. [Google Scholar]
- Su, Z.; Zhang, H.; Zhang, Y.; Li, X.; Wang, Q.; Huang, Y.; Yan, B.; Zhao, J.; Cai, H.; Liu, C.; et al. Allotopic expression rescues mitochondrial function in preclinical models. Nat. Commun. 2021, 12, 4890. [Google Scholar]
- McCully, J.D.; Levitsky, S.; Del Nido, P.J.; Cowan, D.B.; Emani, S.M. Mitochondrial transplantation for cardioprotection in preclinical models. J. Thorac. Cardiovasc. Surg. 2020, 160, e159–e168. [Google Scholar]
- Cowan, D.B.; Yao, R.; Akurathi, V.; Snay, E.R.; Thedsanamoorthy, J.K.; Zurakowski, D.; Ericsson, M.; Friehs, I.; Wu, Y.; Levitsky, S.; et al. Mitochondrial transfer enhances cardiac repair in preclinical models. Circ. Res. 2022, 130, 186–199. [Google Scholar]
- Shin, B.; Saeidi, N.; Almeida, A.; Weissleder, R.; Langer, R.; Lee, H.; Pu, K.; Lee, K.; Kim, D.; Kim, Y.; et al. Peptide-mediated mitochondrial delivery enhances therapeutic outcomes. Nat. Biomed. Eng. 2021, 5, 1018–1029. [Google Scholar]
- Emani, S.M.; Piekarski, B.L.; Harrild, D.; Del Nido, P.J.; McCully, J.D. Early-phase clinical trials of mitochondrial transplantation in ischemia-reperfusion injury. J. Thorac. Cardiovasc. Surg. 2023, 165, e45–e55. [Google Scholar]
- Preble, J.M.; Pacak, C.A.; Kondo, H.; McCully, J.D.; Cowan, D.B.; Levitsky, S. Preclinical studies of mitochondrial transplantation for ischemia-reperfusion injury. JACC Basic Transl. Sci. 2021, 6, 815–827. [Google Scholar]
- Zhang, J.; Bolli, R.; Garry, D.J.; Marbán, E.; Menasché, P.; Zimmermann, W.H.; Kamp, T.J.; Wu, J.C.; Dzau, V.J.; Weissman, I.L.; et al. Stem cell therapies for cardiac repair: Current state and future directions. Nat. Rev. Cardiol. 2022, 19, 531–546. [Google Scholar]
- Nakamura, Y.; Kita, S.; Tanaka, Y.; Fukuda, S.; Obata, Y.; Okita, Y.; Kawachi, K.; Tsugawa-Shimizu, Y.; Fujishima, Y.; Nishizawa, H.; et al. Paracrine effects of stem cell-derived cardiomyocytes enhance cardiac repair. Circulation 2021, 144, 1795–1808. [Google Scholar]
- Forte, M.; Palmerio, S.; Bianchi, F.; Volpe, M.; Sadoshima, J.; Sciarretta, S. Challenges in translating mitochondrial therapies for cardiovascular disease. Cardiovasc. Res. 2021, 117, 654–668. [Google Scholar]
- Weissig, V.; Elbayoumi, T.A.; Torchilin, V.P. Mitochondrial drug delivery: Challenges and opportunities. Adv. Drug Deliv. Rev. 2022, 181, 114087. [Google Scholar]
- Murphy, M.P.; Hartley, R.C.; Smith, R.A.J.; Krieg, T.; Szabadkai, G.; Peers, C.; Hartley, R.; Duchen, M.R.; James, A.M.; Gamble, S.; et al. Advances in mitochondrial-targeted therapies: From bench to bedside. Nat. Rev. Drug Discov. 2023, 22, 343–362. [Google Scholar]
- Rossignol, D.A.; Frye, R.E. Mitochondrial dysfunction in autism spectrum disorders: A systematic review and meta-analysis. Mol. Psychiatry 2012, 17, 290–314. [Google Scholar] [CrossRef] [PubMed]
- Ni, R.; Cao, T.; Xiong, S.; Ma, J.; Fan, G.C.; Lacefield, J.C.; Lu, Y.; Li, T.; Peng, T. Therapeutic inhibition of mitochondrial reactive oxygen species with mito-TEMPO reduces diabetic cardiomyopathy. Free Radic. Biol. Med. 2020, 152, 374–384. [Google Scholar]
- Lopaschuk, G.D.; Karwi, Q.G.; Tian, R.; Wende, A.R.; Abel, E.D. Cardiac energy metabolism in heart failure. Circ. Res. 2021, 128, 1487–1513. [Google Scholar] [CrossRef]
- Ryan, J.J.; Archer, S.L. The right ventricle in pulmonary arterial hypertension: Disorders of metabolism, angiogenesis and adrenergic signaling in right ventricular failure. Circ. Res. 2019, 124, 153–164. [Google Scholar] [CrossRef]
- Viscomi, C.; Zeviani, M. MtDNA-maintenance defects: Syndromes and genes. J. Inherit. Metab. Dis. 2017, 40, 587–599. [Google Scholar] [CrossRef]
- Gammage, P.A.; Moraes, C.T.; Minczuk, M. Mitochondrial genome engineering: The revolution may not be CRISPR-ized. Trends Genet. 2018, 34, 101–110. [Google Scholar] [CrossRef]
- Maneechote, C.; Palee, S.; Chattipakorn, S.C.; Chattipakorn, N. Roles of mitochondrial dynamics modulators in cardiac ischemia/reperfusion injury. J. Cell. Mol. Med. 2017, 21, 2642–2652. [Google Scholar] [CrossRef]
- Hernández-Reséndiz, S.; Prunier, F.; Girão, H.; Dorn, G.W.; Hausenloy, D.J.; EU-CARDIOPROTECTION COST Action (CA16225). Targeting mitochondrial fusion and fission proteins for cardioprotection. J. Cell. Mol. Med. 2020, 24, 6571–6585. [Google Scholar] [CrossRef]
- Dhingra, R.; Kirshenbaum, L.A. Regulation of mitochondrial dynamics and cell fate by mitophagy in cardiovascular diseases. Antioxidants 2021, 10, 1125. [Google Scholar]
- Nah, J.; Miyamoto, S.; Sadoshima, J. Mitophagy as a protective mechanism against myocardial stress. Compr. Physiol. 2017, 7, 1407–1424. [Google Scholar]
- Picard, M.; Wallace, D.C.; Burelle, Y. Advances in mitochondrial research: From genetics to therapeutics. Nat. Rev. Genet. 2021, 22, 709–728. [Google Scholar]
- Wang, Y.; Jasper, H.; Toan, S.; Zhou, H.; Klionsky, D.J.; Zhang, Y.; Zhao, J.; Yan, B.; Cai, H.; Liu, C.; et al. Single-cell RNA sequencing reveals heterogeneity of mitochondrial function in cardiomyocytes during heart failure. Circ. Res. 2022, 130, 1088–1104. [Google Scholar]
- Li, X.; Sun, X.; Zhang, Y.; Huang, Y.; Wang, Z.; Chen, Q.; Zhao, J.; Yan, B.; Cai, H.; Liu, C.; et al. Single-cell omics reveal mitochondrial heterogeneity in cardiovascular disease. Cell Metab. 2023, 35, 789–803. [Google Scholar]
- Zhang, Y.; Murugesan, P.; Huang, K.; Cai, H. Single-cell transcriptomics reveals heterogeneity in mitochondrial function across cardiovascular cell types. Circulation 2021, 144, 1678–1691. [Google Scholar]
- Jakobs, S.; Wurm, C.A.; Eggeling, C. Super-resolution microscopy reveals mitochondrial structure and dynamics. Nat. Methods 2020, 17, 1077–1086. [Google Scholar]
- Stephan, T.; Roesch, A.; Riedel, D.; Jakobs, S. Cristae remodeling insights via STED microscopy in cardiovascular models. Nat. Commun. 2021, 12, 4946. [Google Scholar]
- De la Fuente, S.; Fernández-Martínez, M.; Trabulo, S.; Santos, J.; Aicher, A.; López-Rodríguez, M.; Sánchez-Cabo, F.; Lara-Pezzi, E.; García-Pavía, P.; Vázquez, J.; et al. FRET-based sensors reveal real-time mitochondrial dynamics in living cardiomyocytes. Cell Rep. 2022, 39, 110874. [Google Scholar]
- Din, S.; Konstandin, M.H.; Mason, B.; Volkers, M.; Brown, J.H.; Sussman, M.A.; De Windt, L.J.; Dorn, G.W.; Gottlieb, R.A.; Mentzer, R.M.; et al. PGC-1α and mitochondrial dysfunction in heart failure. J. Mol. Cell. Cardiol. 2019, 133, 193–203. [Google Scholar]
- Stocker, R.; Keaney, J.F., Jr. Role of oxidative modifications in atherosclerosis. Physiol. Rev. 2004, 84, 1381–1478. [Google Scholar] [CrossRef] [PubMed]
- Madan, S.; Kron, B.; Thai, N.; Do, E.; Cudkowicz, M.; Mootha, V.K.; Schon, E.A.; Paganoni, S.; Atassi, N.; Berry, J.D.; et al. Multi-omics profiling identifies pathways associated with mitochondrial dysfunction in heart failure. Cells 2020, 9, 2145. [Google Scholar]
- Akanda, M.K.; Mehjabin, S.; Mosaddik, A. Quinones in the Treatment of Cardiovascular Diseases. In Quinone-Based Compounds in Drug Discovery; Academic Press: Cambridge, MA, USA, 2025; Chapter 6; pp. 117–134. [Google Scholar]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed]
- Briston, T.; Roberts, M.; Lewis, S.; Powney, B.; Staddon, J.M.; Szabadkai, G.; Duchen, M.R. Mitochondrial permeability transition pore: Sensitivity to opening and mechanistic dependence on substrate availability. Sci. Rep. 2017, 7, 10492. [Google Scholar] [CrossRef]
- Battogtokh, G.; Choi, Y.S.; Kang, D.S.; Park, S.J.; Shim, M.S.; Kim, Y.I.; Yong, Y.; Kang, S.; Yoon, Y.R.; Lee, J.; et al. Mitochondria-targeting drug conjugates for cytotoxic, anti-oxidizing and sensing purposes: Current strategies and future perspectives. Acta Pharm. Sin. B 2018, 8, 862–880. [Google Scholar] [CrossRef]
- Bratic, A.; Larsson, N.G. The role of mitochondria in aging. J. Clin. Investig. 2013, 123, 951–957. [Google Scholar] [CrossRef]
- Milane, L.; Trivedi, M.; Singh, A.; Talekar, M.; Amiji, M. Mitochondrial biology, targets, and drug delivery. J. Control Release 2015, 207, 40–58. [Google Scholar] [CrossRef]
| Therapeutic Approach | Mechanism of Action | Clinical Applications | References |
|---|---|---|---|
| Mitochondrial antioxidants | Scavenge ROS and reduce oxidative stress | Heart failure, ischemic heart disease | [133,134] |
| Metabolic modulators | Shift metabolism from glycolysis to OXPHOS | Pulmonary hypertension, heart failure | [135,136] |
| Gene therapy | Correct mtDNA mutations or enhance mitochondrial biogenesis | Cardiomyopathy, ischemic heart disease | [137,138] |
| Mitochondrial fission/fusion modulators | Restore balance in mitochondrial dynamics | Myocardial infarction, hypertrophy | [139,140] |
| Mitophagy enhancers | Promote clearance of damaged mitochondria | Ischemia–reperfusion injury | [141,142] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, H.-M. Mitochondrial Dysfunction in Cardiovascular Diseases. Int. J. Mol. Sci. 2025, 26, 1917. https://doi.org/10.3390/ijms26051917
Yang H-M. Mitochondrial Dysfunction in Cardiovascular Diseases. International Journal of Molecular Sciences. 2025; 26(5):1917. https://doi.org/10.3390/ijms26051917
Chicago/Turabian StyleYang, Han-Mo. 2025. "Mitochondrial Dysfunction in Cardiovascular Diseases" International Journal of Molecular Sciences 26, no. 5: 1917. https://doi.org/10.3390/ijms26051917
APA StyleYang, H.-M. (2025). Mitochondrial Dysfunction in Cardiovascular Diseases. International Journal of Molecular Sciences, 26(5), 1917. https://doi.org/10.3390/ijms26051917