
GCN2-Mediated eIF2α Phosphorylation Is Required for Central Nervous System Remyelination
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
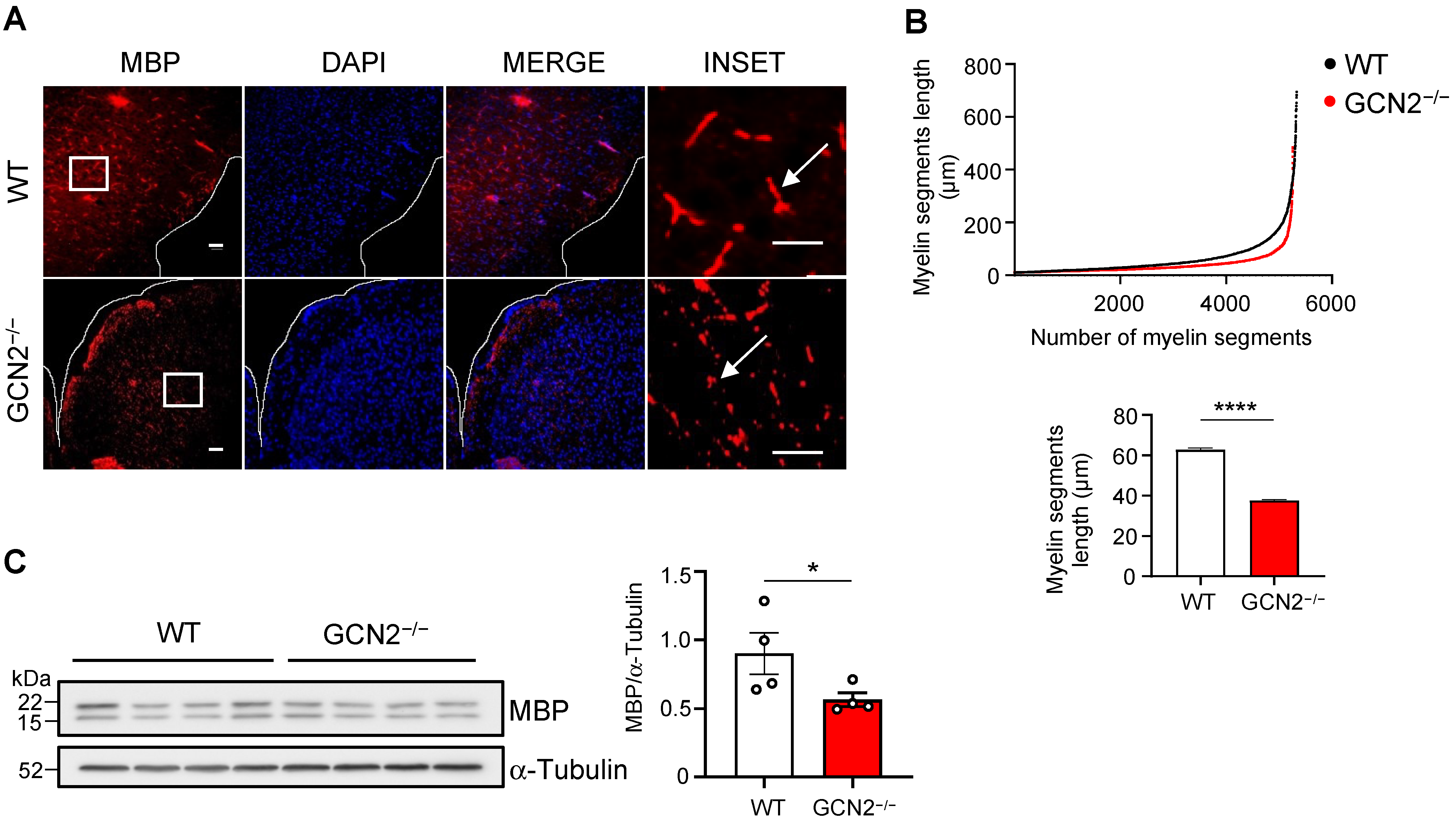
2.1. GCN2 Contributes to the Myelin Phenotype in the CNS
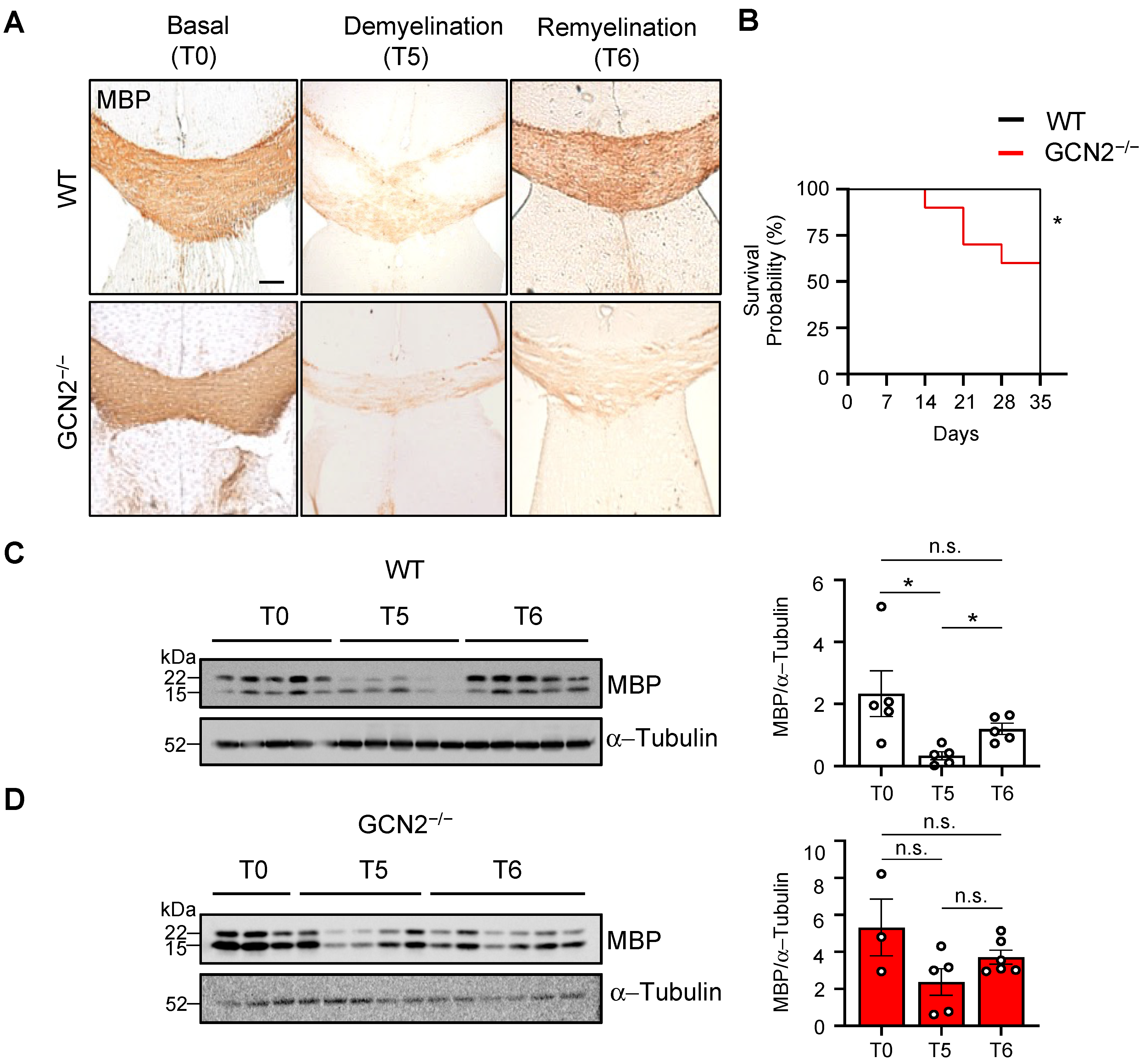
2.2. The Remyelination Process Is Impaired in GCN2-Deficient Mice
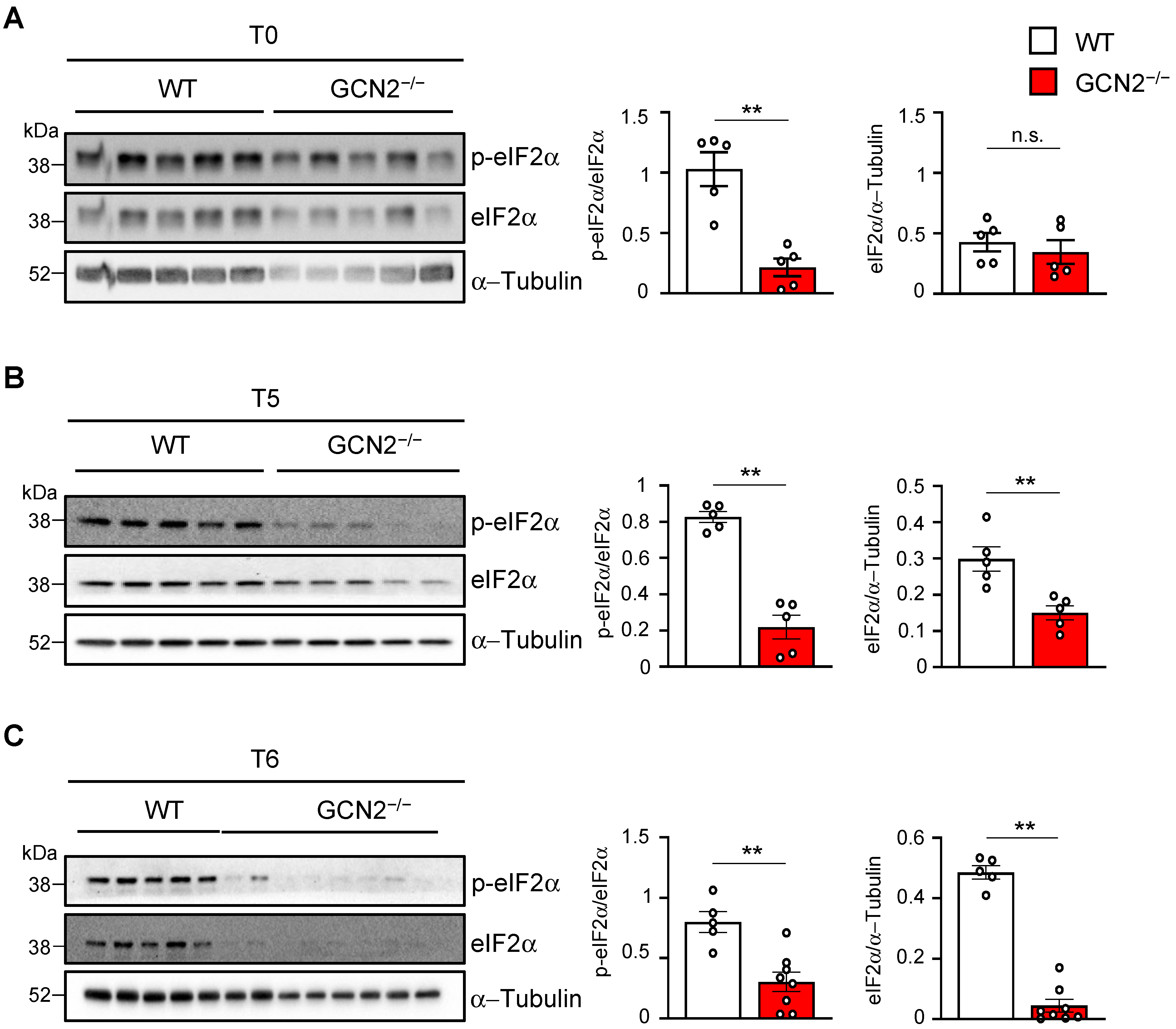
2.3. GCN2 Is Necessary for eIF2α Recovery and Phosphorylation During Remyelination
2.4. Oligodendrocyte Differentiation Markers Are Altered During the Demyelination/Remyelination Process in the GCN2 Deficient Mouse
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Cuprizone Intoxication Model
4.3. Tissue Processing for Immunofluorescence
4.4. Determination of MBP-Positive Myelin Fragments
4.5. Tissue Processing for Immunohistochemistry
4.6. Western Blot
4.7. Quantitative PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Costa-Mattioli, M.; Walter, P. The integrated stress response: From mechanism to disease. Science 2020, 368, eaat5314. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.D.; Harding, H.P.; Ron, D. Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J. Cell Biol. 2004, 167, 27–33. [Google Scholar] [CrossRef]
- Vattem, K.M.; Wek, R.C. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl. Acad. Sci. USA 2004, 101, 11269–11274. [Google Scholar] [CrossRef] [PubMed]
- Novoa, I.; Zeng, H.; Harding, H.P.; Ron, D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J. Cell Biol. 2001, 153, 1011–1022. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Qiu, H.; Garcia-Barrio, M.; Anderson, J.; Hinnebusch, A.G. Uncharged tRNA activates GCN2 by displacing the protein kinase moiety from a bipartite tRNA-binding domain. Mol. Cell 2000, 6, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.; Dong, J.; Hu, C.; Francklyn, C.S.; Hinnebusch, A.G. The tRNA-binding moiety in GCN2 contains a dimerization domain that interacts with the kinase domain and is required for tRNA binding and kinase activation. EMBO J. 2001, 20, 1425–1438. [Google Scholar] [CrossRef] [PubMed]
- Dever, T.E.; Feng, L.; Wek, R.C.; Cigan, A.M.; Donahue, T.F.; Hinnebusch, A.G. Phosphorylation of initiation factor 2α by protein kinase GCN2 mediates gene-specific translational control of GCN4 in yeast. Cell 1992, 68, 585–596. [Google Scholar] [CrossRef] [PubMed]
- Kilberg, M.S.; Shan, J.; Su, N. ATF4-dependent transcription mediates signaling of amino acid limitation. Trends Endocrinol. Metab. 2009, 20, 436–443. [Google Scholar] [CrossRef]
- Zhang, P.; McGrath, B.C.; Reinert, J.; Olsen, D.S.; Lei, L.; Gill, S.; Wek, S.A.; Vattem, K.M.; Wek, R.C.; Kimball, S.R.; et al. The GCN2 eIF2alpha kinase is required for adaptation to amino acid deprivation in mice. Mol. Cell Biol. 2002, 22, 6681–6688. [Google Scholar] [CrossRef] [PubMed]
- Costa-Mattioli, M.; Gobert, D.; Harding, H.; Herdy, B.; Azzi, M.; Bruno, M.; Bidinosti, M.; Ben Mamou, C.; Marcinkiewicz, E.; Yoshida, M.; et al. Translational control of hippocampal synaptic plasticity and memory by the eIF2alpha kinase GCN2. Nature 2005, 436, 1166–1173. [Google Scholar] [CrossRef]
- Hao, S.; Sharp, J.W.; Ross-Inta, C.M.; McDaniel, B.J.; Anthony, T.G.; Wek, R.C.; Cavener, D.R.; McGrath, B.C.; Rudell, J.B.; Koehnle, T.J.; et al. Uncharged tRNA and sensing of amino acid deficiency in mammalian piriform cortex. Science 2005, 307, 1776–1778. [Google Scholar] [CrossRef] [PubMed]
- Maurin, A.C.; Benani, A.; Lorsignol, A.; Brenachot, X.; Parry, L.; Carraro, V.; Guissard, C.; Averous, J.; Jousse, C.; Bruhat, A.; et al. Hypothalamic eIF2alpha signaling regulates food intake. Cell Rep. 2014, 6, 438–444. [Google Scholar] [CrossRef] [PubMed]
- Maurin, A.C.; Jousse, C.; Averous, J.; Parry, L.; Bruhat, A.; Cherasse, Y.; Zeng, H.; Zhang, Y.; Harding, H.P.; Ron, D.; et al. The GCN2 kinase biases feeding behavior to maintain amino acid homeostasis in omnivores. Cell Metab. 2005, 1, 273–277. [Google Scholar] [CrossRef] [PubMed]
- She, P.; Bunpo, P.; Cundiff, J.K.; Wek, R.C.; Harris, R.A.; Anthony, T.G. General Control Nonderepressible 2 (GCN2) Kinase Protects Oligodendrocytes and White Matter during Branched-chain Amino Acid Deficiency in Mice. J Biol Chem 2013, 288, 31250–31260. [Google Scholar] [CrossRef]
- Simons, M.; Trajkovic, K. Neuron-glia communication in the control of oligodendrocyte function and myelin biogenesis. J. Cell Sci. 2006, 119, 4381–4389. [Google Scholar] [CrossRef]
- Baumann, N.; Pham-Dinh, D. Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol. Rev. 2001, 81, 871–927. [Google Scholar] [CrossRef]
- Bradl, M.; Lassmann, H. Oligodendrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 37–53. [Google Scholar] [CrossRef] [PubMed]
- D’Antonio, M.; Feltri, M.L.; Wrabetz, L. Myelin under stress. J. Neurosci. Res. 2009, 87, 3241–3249. [Google Scholar] [CrossRef] [PubMed]
- Rosko, L.; Smith, V.N.; Yamazaki, R.; Huang, J.K. Oligodendrocyte Bioenergetics in Health and Disease. Neuroscientist 2019, 25, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Philips, T.; Rothstein, J.D. Oligodendroglia: Metabolic supporters of neurons. J. Clin. Investig. 2017, 127, 3271–3280. [Google Scholar] [CrossRef]
- Funfschilling, U.; Supplie, L.M.; Mahad, D.; Boretius, S.; Saab, A.S.; Edgar, J.; Brinkmann, B.G.; Kassmann, C.M.; Tzvetanova, I.D.; Mobius, W.; et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 2012, 485, 517–521. [Google Scholar] [CrossRef]
- Lee, Y.; Morrison, B.M.; Li, Y.; Lengacher, S.; Farah, M.H.; Hoffman, P.N.; Liu, Y.; Tsingalia, A.; Jin, L.; Zhang, P.-W.; et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 2012, 487, 443–448. [Google Scholar] [CrossRef]
- Dulamea, A.O. Role of Oligodendrocyte Dysfunction in Demyelination, Remyelination and Neurodegeneration in Multiple Sclerosis. Adv. Exp. Med. Biol. 2017, 958, 91–127. [Google Scholar] [CrossRef]
- Orsini, H.; Araujo, L.P.; Maricato, J.T.; Guereschi, M.G.; Mariano, M.; Castilho, B.A.; Basso, A.S. GCN2 kinase plays an important role triggering the remission phase of experimental autoimmune encephalomyelitis (EAE) in mice. Brain Behav. Immun. 2014, 37, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Glatigny, S.; Bettelli, E. Experimental Autoimmune Encephalomyelitis (EAE) as Animal Models of Multiple Sclerosis (MS). Cold Spring Harb. Perspect. Med. 2018, 8, a028977. [Google Scholar] [CrossRef] [PubMed]
- Stone, S.; Yue, Y.; Stanojlovic, M.; Wu, S.; Karsenty, G.; Lin, W. Neuron-specific PERK inactivation exacerbates neurodegeneration during experimental autoimmune encephalomyelitis. JCI Insight 2019, 4, e124232. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Podojil, J.R.; Kunjamma, R.B.; Jones, J.; Weiner, M.; Lin, W.; Miller, S.D.; Popko, B. Sephin1, which prolongs the integrated stress response, is a promising therapeutic for multiple sclerosis. Brain 2019, 142, 344–361. [Google Scholar] [CrossRef]
- Das, I.; Krzyzosiak, A.; Schneider, K.; Wrabetz, L.; D’Antonio, M.; Barry, N.; Sigurdardottir, A.; Bertolotti, A. Preventing proteostasis diseases by selective inhibition of a phosphatase regulatory subunit. Science 2015, 348, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Kunjamma, R.B.; Weiner, M.; Chan, J.R.; Popko, B. Prolonging the integrated stress response enhances CNS remyelination in an inflammatory environment. Elife 2021, 10, 65469. [Google Scholar] [CrossRef]
- Chen, Y.; Quan, S.; Patil, V.; Kunjamma, R.B.; Tokars, H.M.; Leisten, E.D.; Joy, G.; Wills, S.; Chan, J.R.; Wong, Y.C.; et al. Insights into the mechanism of oligodendrocyte protection and remyelination enhancement by the integrated stress response. Glia 2023, 71, 2180–2195. [Google Scholar] [CrossRef] [PubMed]
- Zhan, J.; Mann, T.; Joost, S.; Behrangi, N.; Frank, M.; Kipp, M. The Cuprizone Model: Dos and Do Nots. Cells 2020, 9, 843. [Google Scholar] [CrossRef] [PubMed]
- Zirngibl, M.; Assinck, P.; Sizov, A.; Caprariello, A.V.; Plemel, J.R. Oligodendrocyte death and myelin loss in the cuprizone model: An updated overview of the intrinsic and extrinsic causes of cuprizone demyelination. Mol. Neurodegener. 2022, 17, 34. [Google Scholar] [CrossRef] [PubMed]
- Blakemore, W.F. Observations on oligodendrocyte degeneration, the resolution of status spongiosus and remyelination in cuprizone intoxication in mice. J. Neurocytol. 1972, 1, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Belkadi, A.; Darnall, L.; Hu, T.; Drescher, C.; Cotleur, A.C.; Padovani-Claudio, D.; He, T.; Choi, K.; Lane, T.E.; et al. CXCR2-positive neutrophils are essential for cuprizone-induced demyelination: Relevance to multiple sclerosis. Nat. Neurosci. 2010, 13, 319–326. [Google Scholar] [CrossRef]
- Steelman, A.J.; Thompson, J.P.; Li, J. Demyelination and remyelination in anatomically distinct regions of the corpus callosum following cuprizone intoxication. Neurosci. Res. 2012, 72, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, G.K.; Morell, P. The neurotoxicant, cuprizone, as a model to study demyelination and remyelination in the central nervous system. Brain Pathol. 2001, 11, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.J.; Wang, H.; Zhang, Y.; Xiao, L.; Clough, R.W.; Browning, R.; Li, X.M.; Xu, H. Region-specific susceptibilities to cuprizone-induced lesions in the mouse forebrain: Implications for the pathophysiology of schizophrenia. Brain Res. 2009, 1270, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Dong, H.; Wu, L.; Kane, M.; Rowitch, D.H.; Stiles, C.D. Cross-repressive interaction of the Olig2 and Nkx2.2 transcription factors in developing neural tube associated with formation of a specific physical complex. J. Neurosci. 2003, 23, 9547–9556. [Google Scholar] [CrossRef]
- Sun, T.; Echelard, Y.; Lu, R.; Yuk, D.I.; Kaing, S.; Stiles, C.D.; Rowitch, D.H. Olig bHLH proteins interact with homeodomain proteins to regulate cell fate acquisition in progenitors of the ventral neural tube. Curr. Biol. 2001, 11, 1413–1420. [Google Scholar] [CrossRef]
- Scherer, S.S.; Braun, P.E.; Grinspan, J.; Collarini, E.; Wang, D.Y.; Kamholz, J. Differential regulation of the 2’,3’-cyclic nucleotide 3’-phosphodiesterase gene during oligodendrocyte development. Neuron 1994, 12, 1363–1375. [Google Scholar] [CrossRef]
- Hou, J.; Zhou, Y.; Cai, Z.; Terekhova, M.; Swain, A.; Andhey, P.S.; Guimaraes, R.M.; Ulezko Antonova, A.; Qiu, T.; Sviben, S.; et al. Transcriptomic atlas and interaction networks of brain cells in mouse CNS demyelination and remyelination. Cell Rep. 2023, 42, 112293. [Google Scholar] [CrossRef]
- Wegener, A.; Deboux, C.; Bachelin, C.; Frah, M.; Kerninon, C.; Seilhean, D.; Weider, M.; Wegner, M.; Nait-Oumesmar, B. Gain of Olig2 function in oligodendrocyte progenitors promotes remyelination. Brain 2015, 138, 120–135. [Google Scholar] [CrossRef]
- Falcon, P.; Escandon, M.; Brito, A.; Matus, S. Nutrient Sensing and Redox Balance: GCN2 as a New Integrator in Aging. Oxid. Med. Cell Longev. 2019, 2019, 5730532. [Google Scholar] [CrossRef] [PubMed]
- Stadelmann, C.; Timmler, S.; Barrantes-Freer, A.; Simons, M. Myelin in the Central Nervous System: Structure, Function, and Pathology. Physiol. Rev. 2019, 99, 1381–1431. [Google Scholar] [CrossRef] [PubMed]
- Boggs, J.M. Myelin basic protein: A multifunctional protein. Cell Mol. Life Sci. 2006, 63, 1945–1961. [Google Scholar] [CrossRef] [PubMed]
- Readhead, C.; Hood, L. The dysmyelinating mouse mutations shiverer (shi) and myelin deficient (shimld). Behav. Genet. 1990, 20, 213–234. [Google Scholar] [CrossRef]
- Rivers, L.E.; Young, K.M.; Rizzi, M.; Jamen, F.; Psachoulia, K.; Wade, A.; Kessaris, N.; Richardson, W.D. PDGFRA/NG2 glia generate myelinating oligodendrocytes and piriform projection neurons in adult mice. Nat. Neurosci. 2008, 11, 1392–1401. [Google Scholar] [CrossRef] [PubMed]
- Richardson, W.D.; Young, K.M.; Tripathi, R.B.; McKenzie, I. NG2-glia as multipotent neural stem cells: Fact or fantasy? Neuron 2011, 70, 661–673. [Google Scholar] [CrossRef]
- Mares, V.; Lodin, Z.; Jilek, M. An estimate of the number of cells arising by division in mouse cerebral hemispheres from age one to 12 months: An autoradiographic study of DNA synthesis. J. Comp. Neurol. 1975, 161, 471–482. [Google Scholar] [CrossRef]
- Elbaz, B.; Popko, B. Molecular Control of Oligodendrocyte Development. Trends Neurosci. 2019, 42, 263–277. [Google Scholar] [CrossRef]
- Zhou, Q.; Wang, S.; Anderson, D.J. Identification of a novel family of oligodendrocyte lineage-specific basic helix-loop-helix transcription factors. Neuron 2000, 25, 331–343. [Google Scholar] [CrossRef]
- Fernandez-Castaneda, A.; Gaultier, A. Adult oligodendrocyte progenitor cells—Multifaceted regulators of the CNS in health and disease. Brain Behav. Immun. 2016, 57, 1–7. [Google Scholar] [CrossRef]
- Goldberg, J.; Daniel, M.; van Heuvel, Y.; Victor, M.; Beyer, C.; Clarner, T.; Kipp, M. Short-term cuprizone feeding induces selective amino acid deprivation with concomitant activation of an integrated stress response in oligodendrocytes. Cell Mol. Neurobiol. 2013, 33, 1087–1098. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Bailey, S.L.; Ho, H.; Harding, H.P.; Ron, D.; Miller, S.D.; Popko, B. The integrated stress response prevents demyelination by protecting oligodendrocytes against immune-mediated damage. J. Clin. Investig. 2007, 117, 448–456. [Google Scholar] [CrossRef]
- Way, S.W.; Podojil, J.R.; Clayton, B.L.; Zaremba, A.; Collins, T.L.; Kunjamma, R.B.; Robinson, A.P.; Brugarolas, P.; Miller, R.H.; Miller, S.D.; et al. Pharmaceutical integrated stress response enhancement protects oligodendrocytes and provides a potential multiple sclerosis therapeutic. Nat. Commun. 2015, 6, 6532. [Google Scholar] [CrossRef] [PubMed]
- Cohen, R.B.; Boal, T.R.; Safer, B. Increased eIF-2 alpha expression in mitogen-activated primary T lymphocytes. EMBO J. 1990, 9, 3831–3837. [Google Scholar] [CrossRef]
- Xu, H.; Dzhashiashvili, Y.; Shah, A.; Kunjamma, R.B.; Weng, Y.L.; Elbaz, B.; Fei, Q.; Jones, J.S.; Li, Y.I.; Zhuang, X.; et al. m(6)A mRNA Methylation Is Essential for Oligodendrocyte Maturation and CNS Myelination. Neuron 2020, 105, 293–309.e5. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Falcón, P.; Brito, Á.; Escandón, M.; Roa, J.F.; Martínez, N.W.; Tapia-Godoy, A.; Farfán, P.; Matus, S. GCN2-Mediated eIF2α Phosphorylation Is Required for Central Nervous System Remyelination. Int. J. Mol. Sci. 2025, 26, 1626. https://doi.org/10.3390/ijms26041626
Falcón P, Brito Á, Escandón M, Roa JF, Martínez NW, Tapia-Godoy A, Farfán P, Matus S. GCN2-Mediated eIF2α Phosphorylation Is Required for Central Nervous System Remyelination. International Journal of Molecular Sciences. 2025; 26(4):1626. https://doi.org/10.3390/ijms26041626
Chicago/Turabian StyleFalcón, Paulina, Álvaro Brito, Marcela Escandón, Juan Francisco Roa, Nicolas W. Martínez, Ariel Tapia-Godoy, Pamela Farfán, and Soledad Matus. 2025. "GCN2-Mediated eIF2α Phosphorylation Is Required for Central Nervous System Remyelination" International Journal of Molecular Sciences 26, no. 4: 1626. https://doi.org/10.3390/ijms26041626
APA StyleFalcón, P., Brito, Á., Escandón, M., Roa, J. F., Martínez, N. W., Tapia-Godoy, A., Farfán, P., & Matus, S. (2025). GCN2-Mediated eIF2α Phosphorylation Is Required for Central Nervous System Remyelination. International Journal of Molecular Sciences, 26(4), 1626. https://doi.org/10.3390/ijms26041626