

Practical Preparation of (3S)-Hydroxy-5-Phenylpentanoic Acid: Asymmetric Synthesis of (S)-Daphneolone and (S)-Dihydroyashabushiketol, and Formal Synthesis of (3S,5S)-Yashabushidiol B
 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
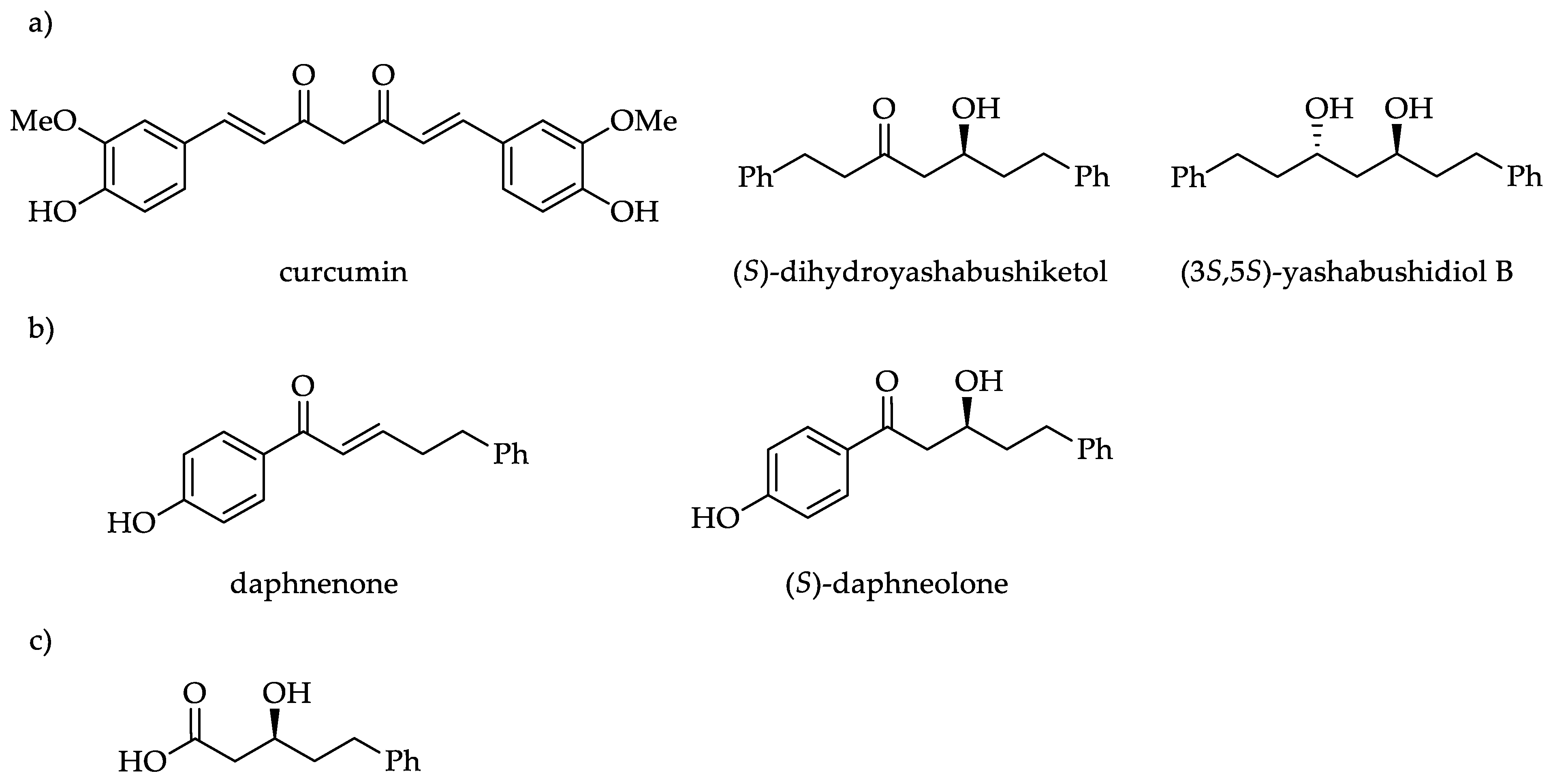
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. General
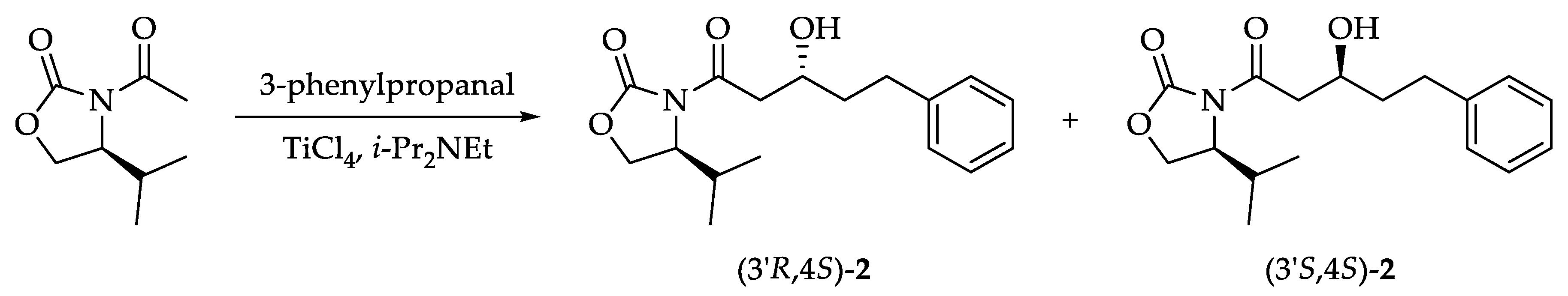
3.2. Preliminary Result
Aldol Addition of (S)-3-Acetyl-4-Isopropyl-2-Oxazolidinone with 3-Phenylpropanal (See, Scheme 1)
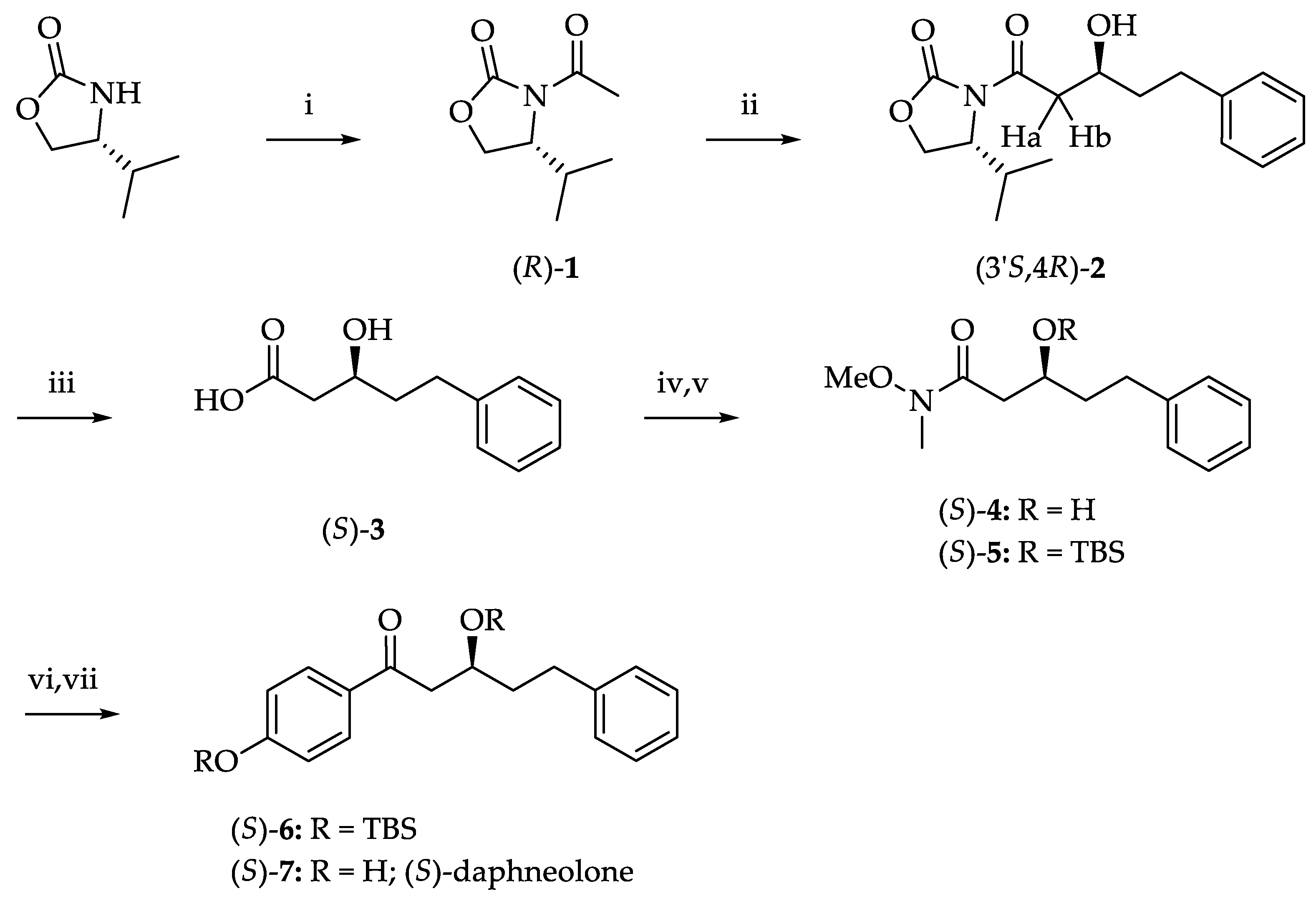
3.3. Synthesis of (S)-3-Hydroxy-5-Phenylpentanoic Acid
3.3.1. Preparation of (R)-3-Acetyl-4-Isopropyl-2-Oxazolidinone, (R)-1 [24]
3.3.2. Aldol Addition of (R)-1 with 3-Phenylpropanal
3.3.3. Synthesis of (S)-3-Hydroxy-5-Phenylpentanoic Acid, (S)-3 [20]
3.3.4. Synthesis of Weinreb Amide, (S)-4 [30]
3.3.5. Preparation of TBS Ether, (S)-5
3.4. Synthesis of (S)-Daphneolone
3.4.1. Preparation of Rac-Daphneolone [9]
3.4.2. Isolation of (S)-(+)-Daphneolone [10,11]
3.4.3. Asymmetric Synthesis of (S)-Daphneolone
Synthesis of β-Hydroxyketone, (S)-6
Synthesis of (S)-Daphneolone, (S)-7
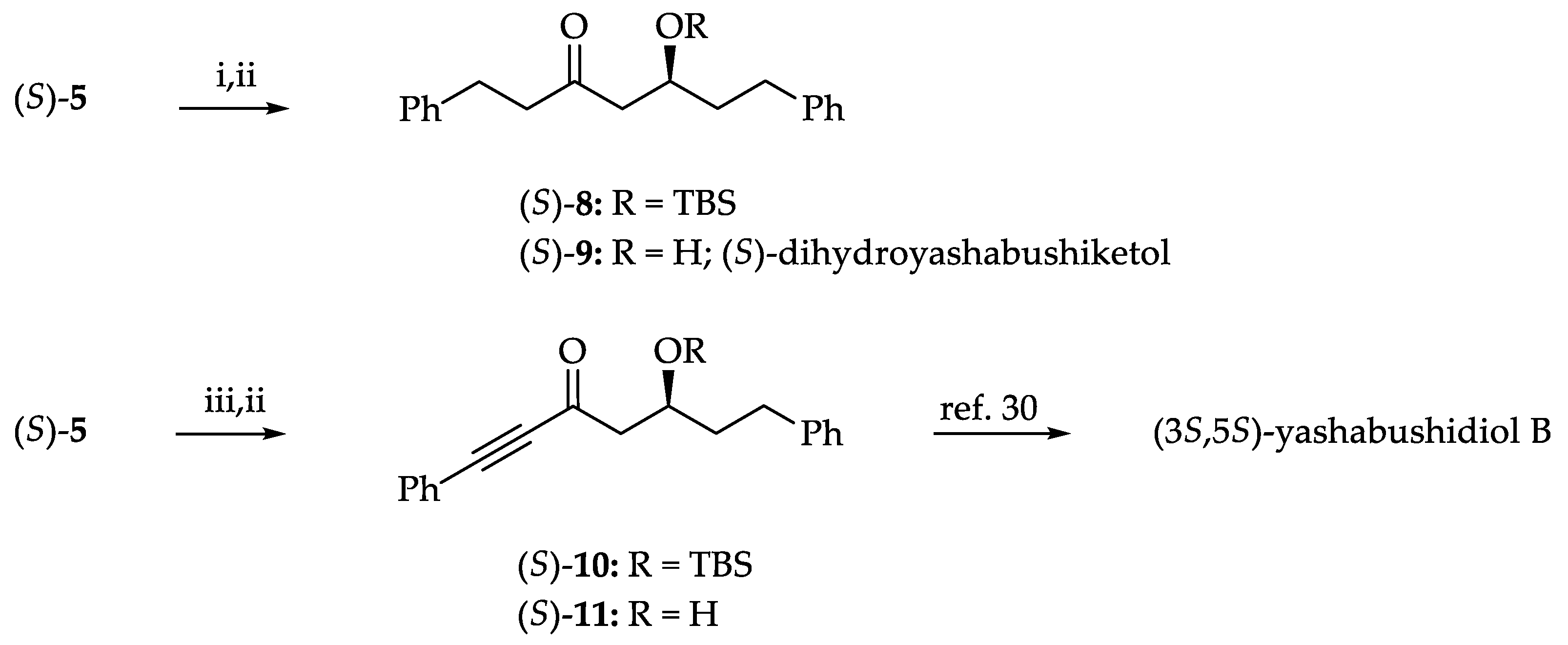
3.5. Synthesis of (S)-Dihydroyashachecked, OKbushiketol and Formal Synthesis of (3S,5S)-Yashabushidiol
3.5.1. Typical Procedure for the Synthesis of (S)-10
3.5.2. Typical Procedure for the Synthesis of (S)-Dihydroyashabushiketol, (S)-9 [28]
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sun, D.J.; Zhu, L.J.; Zhao, Y.Q.; Zhen, Y.Q.; Zhang, L.; Lin, C.C.; Chen, L.X. Diarylheptanoid: A Privileged Structure in Drug Discovery. Fitoterapia 2020, 142, 104490. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Pan, D.B.; Pang, Q.Q.; Zhou, M.; Yao, X.J.; Yao, X.S.; Li, H.B.; Yu, Y. Diarylheptanoid analogues from the rhizomes of Zingiber officinale and their anti-tumour activity. RSC Adv. 2021, 11, 29376–29384. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yu, Y.Y.; Peng, F.; Duan, W.T.; Wu, C.H.; Li, H.T.; Zhang, X.F.; Shi, Y.S. Neolignans and diarylheptanoids with anti-inflammatory activity from the rhizomes of Alpinia zerumbet. J. Agr. Food Chem. 2021, 69, 9229–9237. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.F.; Cheng, G.G.; Zhao, L.Q.; Khan, A.; Yang, X.W.; Zhang, B.Y.; Li, M.C.; Liu, Y.P.; Luo, X.D. Antioxidant and cytoprotective effects of new diarylheptanoids from Rhynchanthus beesianus. J. Agric. Food Chem. 2021, 69, 6229–6239. [Google Scholar] [CrossRef]
- Sudarshan, K.; Yarlagadda, S.; Sengupta, S. Recent Advances in the Synthesis of Diarylheptanoids. Chem. Asian J. 2024, 19, e202400380. [Google Scholar] [CrossRef]
- Dong, S.H.; Lian, M.Y.; Han, J.L.; Ai, Y.F.; Zhou, X.F.; Bai, M.; Huang, X.X.; Song, S.J. Rapid screening of diarylpentanoids from Daphne bholua. Phytochemistry 2023, 209, 113614. [Google Scholar] [CrossRef]
- Wahab, N.A.A.; Abas, F.; Othman, l.; Naidu, R. Diarylpentanoid (1,5-bis(4-hydroxy-3-methoxyphenyl)-1,4-pentadiene-3-one) (MS13) Exhibits Anti-proliferative, Apoptosis Induction and Anti-migration Properties on Androgen-independent Human Prostate Cancer by Targeting Cell Cycle–Apoptosis and PI3K Signalling Pathways. Front. Pharmacol. 2021, 12, 707335. [Google Scholar]
- Wang, X.-H.; Gao, B.-W.; Nakashima, Y.; Mori, T.; Zhang, Z.X.; Kodama, T.; Lee, Y.E.; Zhang, Z.K.; Wong, C.P.; Liu, Q.Q.; et al. Identification of a diarylpentanoid-producing polyketide synthase revealing an unusual biosynthetic pathway of 2-(2-phenylethyl)chromones in agarwood. Nat. Commun. 2022, 13, 348. [Google Scholar] [CrossRef]
- Denniff, P.; Macleod, I.; Whiting, D.A. Syntheses of the (±)-[n]-gingerols (pungent principles of ginger) and related compounds through regioselective aldol condensations: Relative pungency assays. J. Chem. Soc. Perkin Trans. 1981, 1, 82–87. [Google Scholar] [CrossRef]
- Kogiso, S.; Hosozawa, S.; Wada, K.; Munakata, K. Daphneolone in roots of Daphhte odora. Phytochemistry 1974, 13, 2332–2334. [Google Scholar] [CrossRef]
- Wang, L.-B.; Dong, N.-W.; Wu, Z.-H.; Wu, L.-J. Two new compounds with cytotoxic activity on the human melanoma A375-S2 cells from Daphne giraldii callus cells. J. Asian Nat. Prod. Res. 2012, 14, 1020–1026. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.A.; Bartroli, J.; Shih, T.L. Enantioselective Aldol Condensations. 2. Erythro-Selective Chiral Aldol Condensations via Boron Enolates. J. Am. Chem. Soc. 1981, 103, 2127–2129. [Google Scholar] [CrossRef]
- Yamada, Y.M.A.; Yoshikawa, N.; Sasai, H.; Shibasaki, M. Direct Catalytic Asymmetric Aldol Reactions of Aldehydes with Unmodified Ketones. Angew. Chem. Int. Ed. Engl. 1997, 36, 1871–1873. [Google Scholar] [CrossRef]
- Sodeoka, M.; Ohrai, K.; Shibasaki, M. Catalytic Asymmetric Aldol Reaction via Chiral Pd(II) Enolate in Wet DMF. J. Org. Chem. 1995, 60, 2648–2649. [Google Scholar] [CrossRef]
- Kiyooka, S.-i.; Hosokawa, S.; Tsukasa, S. Dicationic (BINAP)palladium-catalyzed enantioselective aldol reaction of aldehydes with a silyl enol ether: A simplified practical procedure. Tetrahedron Lett. 2006, 47, 3959–3962. [Google Scholar] [CrossRef]
- Tsutsumi, R.; Taguchi, R.; Yamanaka, M. Chiral Bipyridine Ligand with Flexible Molecular Recognition Site: Development and Application to Copper-Catalyzed Asymmetric Borylation of α,β-Unsaturated Ketones. ChemCatChem 2022, 14, e202101278. [Google Scholar] [CrossRef]
- Paladhi, S.; Hwang, I.-S.; Yoo, E.J.; Ryu, D.H.; Song, C.E. Kinetic Resolution of β-Hydroxy Carbonyl Compounds via Enantioselective Dehydration Using a Cation-Binding Catalyst: Facile Access to Enantiopure Chiral Aldols. Org. Lett. 2018, 20, 2003–2006. [Google Scholar] [CrossRef]
- Jiang, H.; Gschwend, B.; Albrecht, L.; Jorgensen, K.A. Organocatalytic Preparation of Simple β-Hydroxy and β-Amino Esters: Low Catalyst Loadings and Gram-Scale Synthesis. Org. Lett. 2010, 12, 5052–5055. [Google Scholar] [CrossRef]
- Carreira, E.M.; Singer, R.A.; Lee, W. Catalytic, Enantioselective Aldol Additions with Methyl and Ethyl Acetate O-Silyl Enolates: A Chiral Tridentate Chelate as a Ligand for Titanium(IV). J. Am. Chem. Soc. 1994, 116, 8837–8838. [Google Scholar] [CrossRef]
- Tian, B.; Li, X.; Chen, P.; Liu, G. Asymmetric Palladium-Catalyzed Oxycarbonylation of Terminal Alkenes: Efficient Access to β-Hydroxy Alkylcarboxylic Acids. Angew. Chem. Int. Ed. 2021, 60, 14881–14886. [Google Scholar] [CrossRef]
- Smith, S.M.; Uteuliyev, M.; Takacs, J.M. Catalytic asymmetric hydroboration of β,γ-unsaturated Weinreb amides: Striking influence of the borane. Chem. Commun. 2011, 47, 7812–7814. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, S.; Tambo, M.; Yamazaki, Y.; Muramatsu, T.; Kanda, Y.; Hirose, T.; Kodama, K. Enantioseparation of 3-Hydroxycarboxylic Acids via Diastereomeric Salt Formation by 2-Amino-1,2-diphenylethanol (ADPE) and Cinchonidine. Molecules 2023, 28, 114. [Google Scholar] [CrossRef] [PubMed]
- Nahm, S.; Weinreb, S.M. N-methoxy-n-methylamides as effective acylating agents. Tetrahedron Lett. 1981, 22, 3815. [Google Scholar] [CrossRef]
- Shibahara, F.; Fukunaga, T.; Murai, T. Synthesis of Chiral Selenazolines from N-Acyloxazolidinones via a Selenative Rearrangement of Chiral Cyclic Skeletons. Org. Lett. 2018, 20, 5826–5830. [Google Scholar] [CrossRef]
- Le Sann, C.; Muñoz, D.M.; Saunders, N.; Simpson, T.J.; Smith, D.I.; Soulas, F.; Watts, P.; Willis, C.L. Assembly Intermediates in Polyketide Biosynthesis: Enantioselective Syntheses of b-Hydroxycarbonyl Compounds. Org. Biomol. Chem. 2005, 3, 1719–1728. [Google Scholar] [CrossRef]
- Le Sann, C.; Simpson, T.J.; Smith, D.I.; Watts, P.; Willis, C.L. A General Synthesis of Homochiral β-Hydroxy N-Acetylcysteamine Thioesters. Tetrahedron Lett. 1999, 40, 4093–4096. [Google Scholar] [CrossRef]
- Beutner, G.L.; Cohen, B.M.; DelMonte, A.J.; Dixon, D.D.; Fraunhoffer, K.J.; Glace, A.W.; Lo, E.; Stevens, J.M.; Vanyo, D.; Wilbert, C. Revisiting the Cleavage of Evans Oxazolidinones with LiOH/H2O2. Org. Process Res. Dev. 2019, 23, 1378–1385. [Google Scholar] [CrossRef]
- Xu, X.; Peng, L.; Chang, X.; Guo, C. Ni/Chiral Sodium Carboxylate Dual Catalyzed Asymmetric O-Propargylation. J. Am. Chem. Soc. 2021, 143, 21048–21055. [Google Scholar] [CrossRef]
- Romanski, J.; Nowak, P.; Chapuis, C.; Jurczak, J. Total synthesis of (5S)-dihydroyashabushiketol. Tetrahedron Asymmetry 2011, 22, 787–790. [Google Scholar] [CrossRef]
- Fang, Z.; Wills, M. Asymmetric Transfer Hydrogenation of Functionalized Acetylenic Ketones. J. Org. Chem. 2013, 78, 8594–8605. [Google Scholar] [CrossRef]
- Hayashi, Y.; Saitoh, T.; Arase, H.; Kawauchi, G.; Takeda, N.; Shimasaki, Y.; Sato, I. Two-Pot Synthesis of Chiral 1,3-syn-Diols through Asymmetric Organocatalytic Aldol and Wittig Reactions Followed by Domino Hemiacetal/Oxy-Michael Reactions. Chem. Eur. J. 2018, 24, 4909–4915. [Google Scholar] [CrossRef] [PubMed]
- Gadakh, S.K.; Sudalai, S. Co-catalyzed two-stereocentered hydrolytic kinetic resolution: Application to the synthesis of yashabushidiols A and B and the lactone unit of compactin and mevinolin. Tetrahedron Asymmetry 2015, 26, 118–123. [Google Scholar] [CrossRef]
- Hiroya, K.; Jouka, R.; Kameda, M.; Yasuhara, A.; Sakamoto, T. Cyclization Reactions of 2-Alkynylbenzyl Alcohol and 2-Alkynylbenzylamine Derivatives Promoted by Tetrabutylammonium Fluoride. Tetrahedron 2001, 57, 9697–9710. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nam, S.-Y.; Cho, J.; Jung, S.M.; Lee, H.-J.; Ryu, H.W.; Oh, S.-R.; Lee, K.-I. Practical Preparation of (3S)-Hydroxy-5-Phenylpentanoic Acid: Asymmetric Synthesis of (S)-Daphneolone and (S)-Dihydroyashabushiketol, and Formal Synthesis of (3S,5S)-Yashabushidiol B. Int. J. Mol. Sci. 2025, 26, 1476. https://doi.org/10.3390/ijms26041476
Nam S-Y, Cho J, Jung SM, Lee H-J, Ryu HW, Oh S-R, Lee K-I. Practical Preparation of (3S)-Hydroxy-5-Phenylpentanoic Acid: Asymmetric Synthesis of (S)-Daphneolone and (S)-Dihydroyashabushiketol, and Formal Synthesis of (3S,5S)-Yashabushidiol B. International Journal of Molecular Sciences. 2025; 26(4):1476. https://doi.org/10.3390/ijms26041476
Chicago/Turabian StyleNam, So-Yeon, Joungmo Cho, Simon MoonGeun Jung, Hyun-Jun Lee, Hyung Won Ryu, Sei-Ryang Oh, and Kee-In Lee. 2025. "Practical Preparation of (3S)-Hydroxy-5-Phenylpentanoic Acid: Asymmetric Synthesis of (S)-Daphneolone and (S)-Dihydroyashabushiketol, and Formal Synthesis of (3S,5S)-Yashabushidiol B" International Journal of Molecular Sciences 26, no. 4: 1476. https://doi.org/10.3390/ijms26041476
APA StyleNam, S.-Y., Cho, J., Jung, S. M., Lee, H.-J., Ryu, H. W., Oh, S.-R., & Lee, K.-I. (2025). Practical Preparation of (3S)-Hydroxy-5-Phenylpentanoic Acid: Asymmetric Synthesis of (S)-Daphneolone and (S)-Dihydroyashabushiketol, and Formal Synthesis of (3S,5S)-Yashabushidiol B. International Journal of Molecular Sciences, 26(4), 1476. https://doi.org/10.3390/ijms26041476