
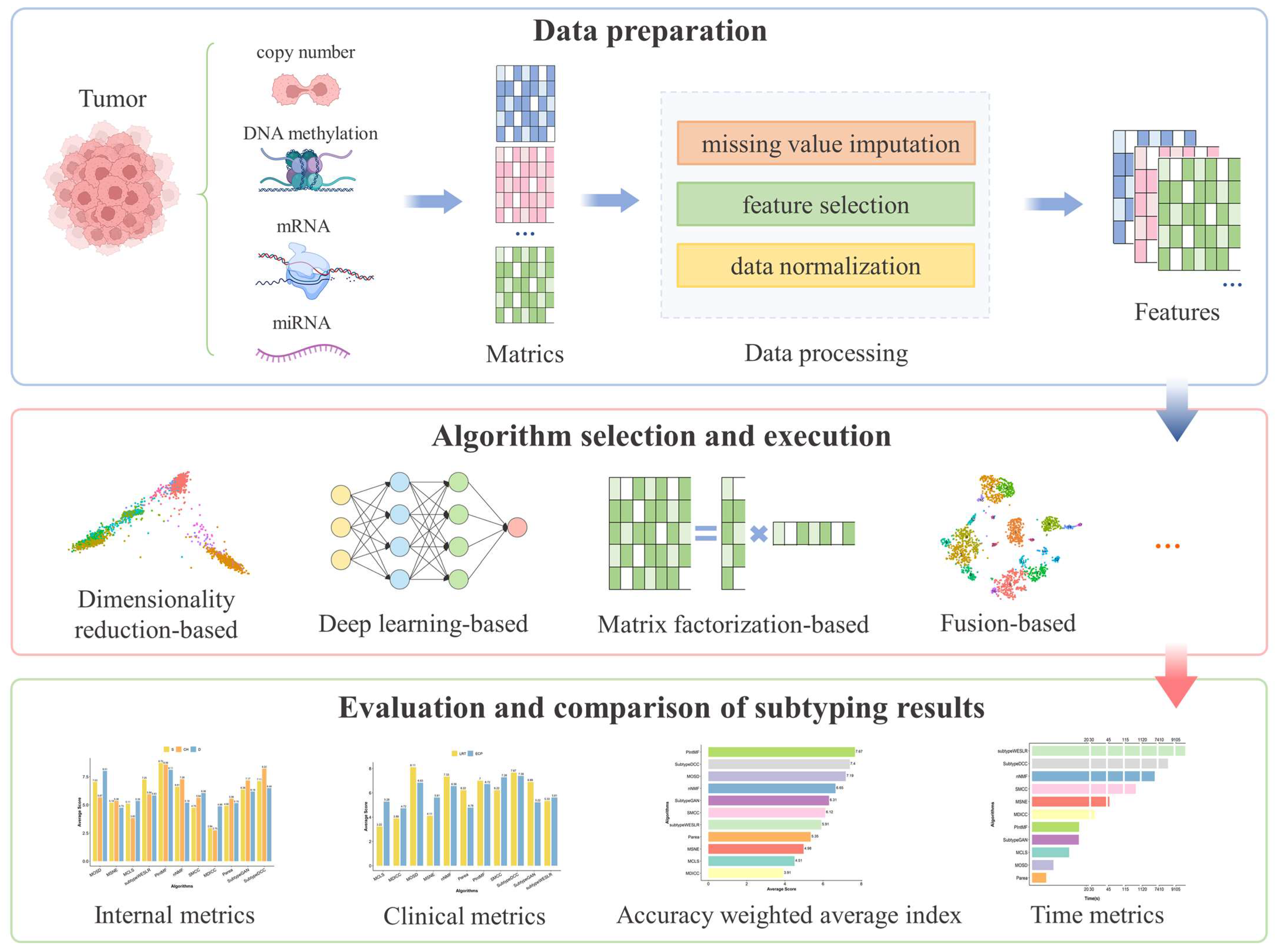
Comprehensive Evaluation of Multi-Omics Clustering Algorithms for Cancer Molecular Subtyping

 ,
,
Abstract
1. Introduction
2. Results
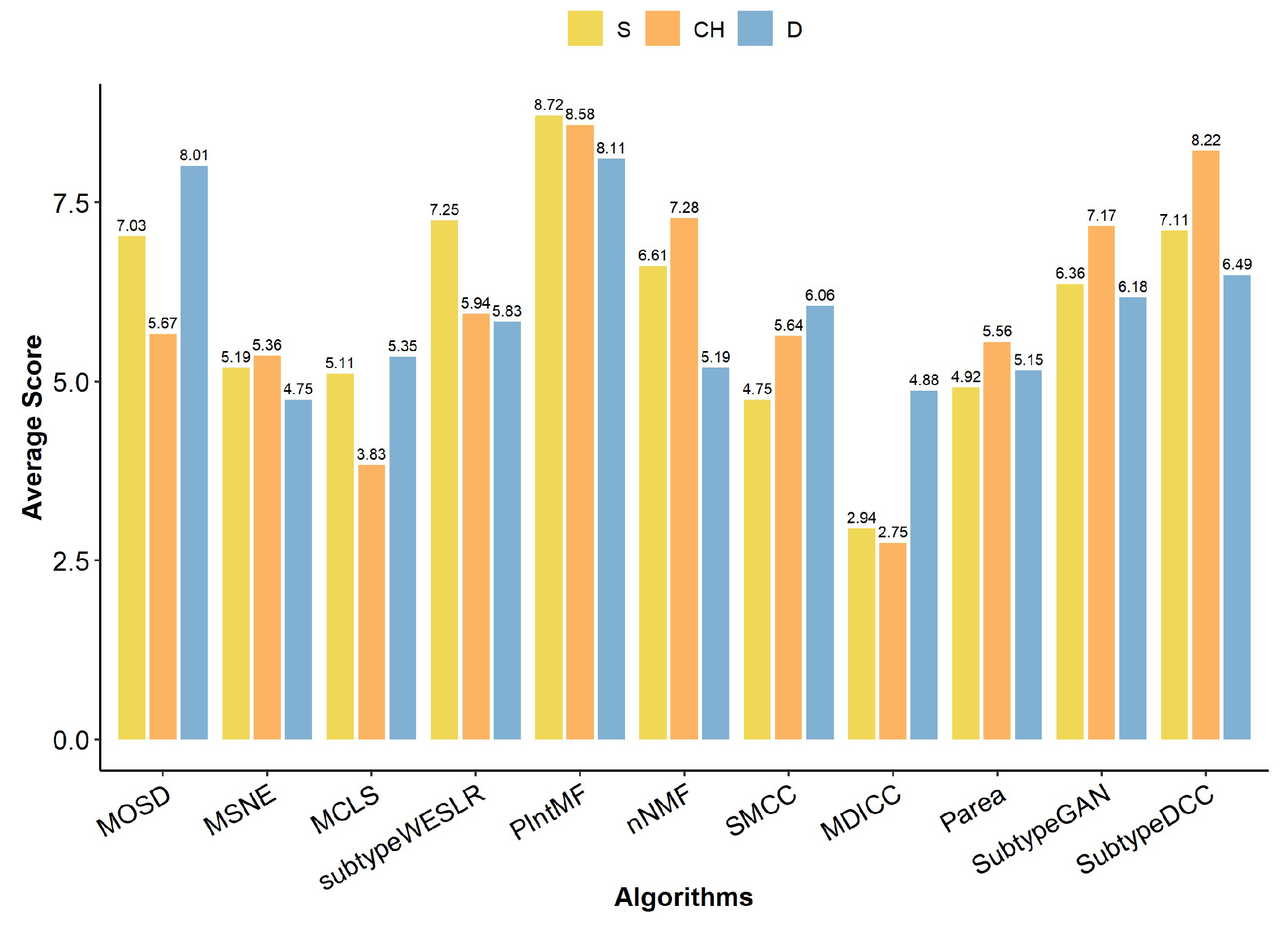
2.1. Internal Metrics of Clustering Algorithm
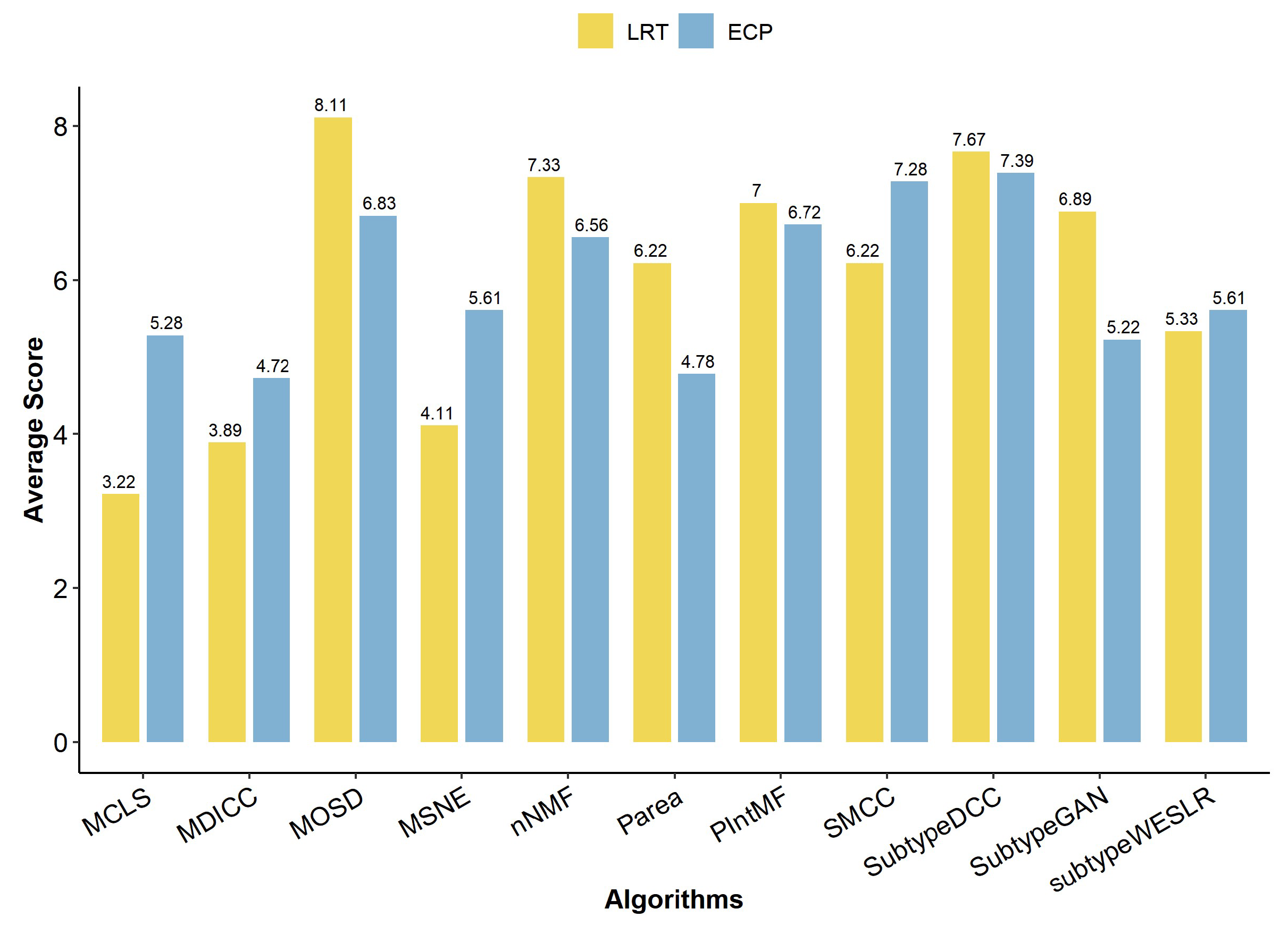
2.2. Clinical Metrics of Clustering Algorithm
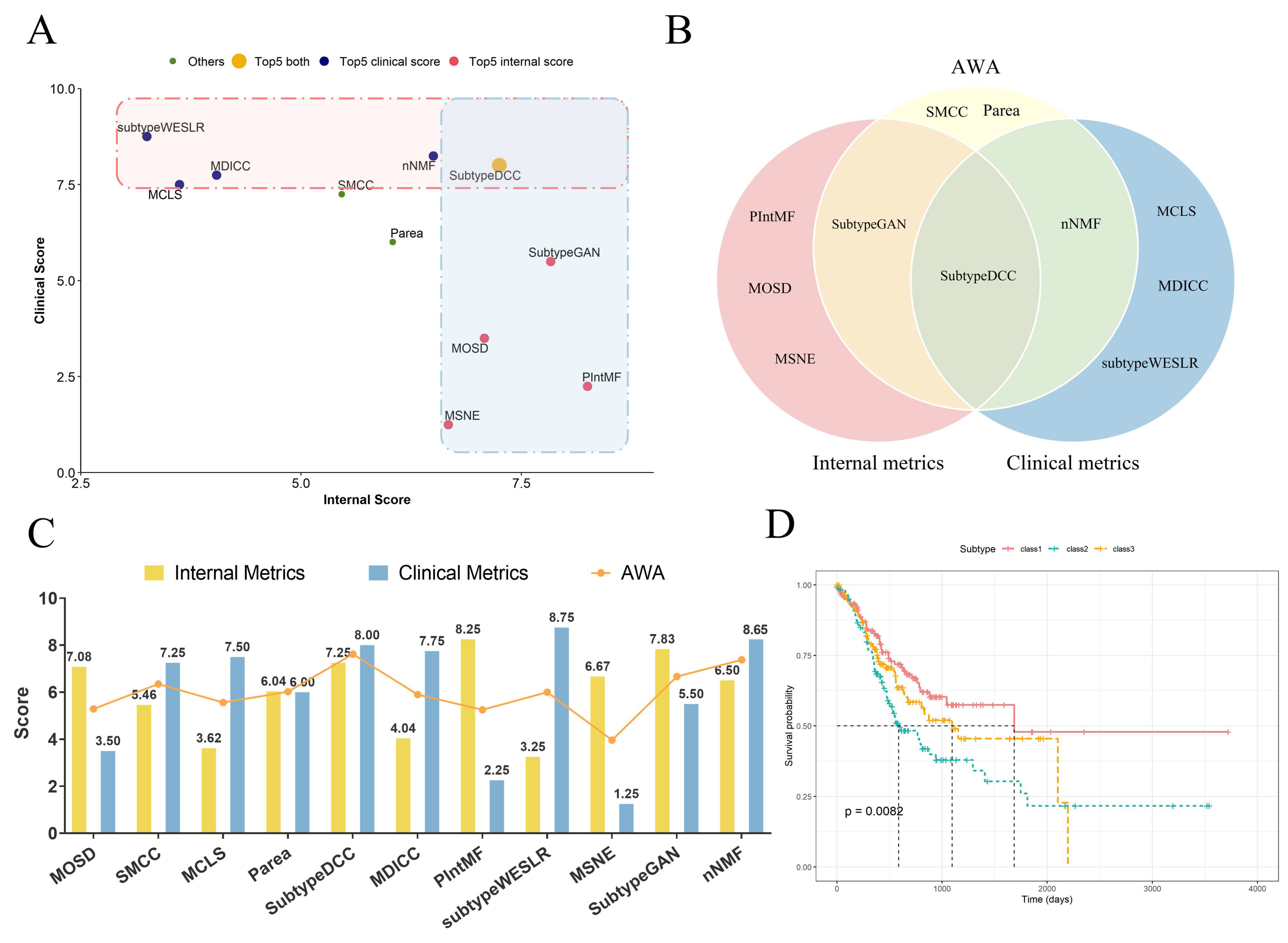
2.3. The Accuracy-Weighted Average Index of Clustering Algorithm
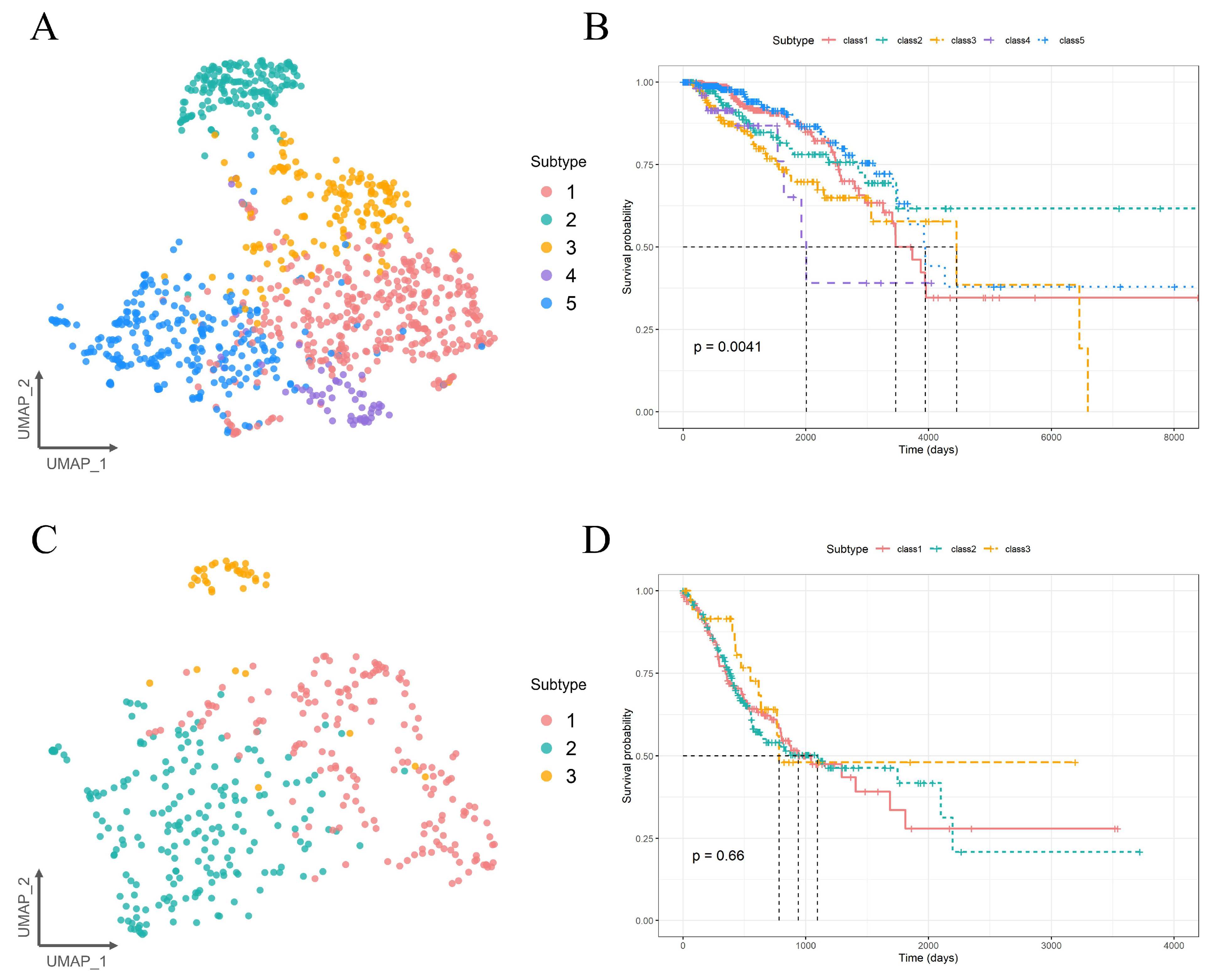
2.4. Molecular Subtyping Performance of PIntMF
3. Discussion
4. Materials and Methods
4.1. Data Sources and Preprocessing
4.2. Multi-Omics Clustering Algorithm Selection
- MOSD [8] first creates affinities for each data type using local scaling. The affinities are then linearly combined into a network by assigning weights to each omics. Finally, spectral clustering is applied to the self-diffusion-enhanced similarity network to identify cancer subtypes.
- MSNE [9] first constructs similarity networks of samples for complete or partial multi-omics data. Then, the integrated similarity of samples is captured through random walk on multiple similarity networks. Finally, the cancer subtypes are obtained with the Skip-gram method by projecting the samples into a low-dimensional space for k-mean clustering.
- MCLS [10] first utilizes complete multi-omics data to construct a latent subspace using principal component analysis (PCA)-based feature extraction and singular value decomposition (SVD). Then, a projection matrix of each omics is learned to project the incomplete multi-omics data to the latent subspace. Finally, the samples are clustered using spectral clustering in the latent subspace.
- Subtype-WESLR [11] is based on a sparse subspace learning framework. First, it employs a weighted ensemble strategy to fuse base clustering obtained from different methods as prior knowledge. Then, the sample feature profiles of each data type are projected to a common latent subspace corresponding to the subspace consistency. Finally, the common subspace is optimized by an iterative method to identify cancer subtypes.
- PIntMF [12] is a matrix factorization model with non-negativity and sparsity constraints. First, the original matrix is decomposed into a product of a common basis matrix (W) and a specific coefficient matrix (Hk). Then, sparsity is added to W and Hk via lasso penalization and equality constraints are applied to improve interpretability. Finally, W and Hk are iteratively updated until the similarity of W is stable, and hierarchical clustering is performed on W to obtain the sample subtypes.
- nNMF [13] combines the strengths of intNMF [34] and SNF [35]. It first uses intNMF to construct a stable consensus matrix for each data type. Then, theses consensus matrices are integrated into a single consensus matrix by SNF. Finally, spectral clustering is performed on this single consensus matrix to determine cancer subtypes.
- SMCC [14] first constructs sample-sample similarity networks based on Euclidean distance. Then, it integrates weighted least squares, low-rank subspace representation, and entropy to fuse networks. Finally, co-regularization is used to measure and minimize the distribution difference between the similarity networks and the fused network, and cancer subtypes are obtained through clustering.
- MDICC [15] first constructs affinity matrices for different omics data based on Gaussian kernel functions. After fusing them using low-rank subspace representation and entropy, the integrated matrix is clustered using K-means++ to obtain cancer subtypes.
- Parea [16] is based on multi-view hierarchical clustering and data fusion. It first selects hierarchical clustering methods to represent each omics data into separate views. Then, hierarchical clustering is used again to identify cancer subtypes by creating a fusion object.
- Subtype-GAN [17] is a deep adversarial learning method based on a multi-input multi-output neural network. First, the features of each omics data are extracted from relatively independent layers. Then, after inputting these features into the same shared layer, the subtypes of the samples are obtained by consensus GMM clustering through the hidden factors of the shared layer.
- Subtype-DCC [18] combines deep clustering and decoupled comparison learning. First, data pairs are constructed from the pair construction backbone (PCB) through data augmentation, and features are extracted from the data augmentation using a shared deep neural network. Then, the instance-level contrastive head (ICH) and the cluster-level contrastive head (CCH) are used for contrastive learning in the row and column spaces of the feature matrix, respectively. After training, the CCH is used to predict cancer subtypes.
4.3. Metrics for Evaluating Subtyping Performance
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhao, L.; Lee, V.H.F.; Ng, M.K.; Yan, H.; Bijlsma, M.F. Molecular Subtyping of Cancer: Current Status and Moving toward Clinical Applications. Brief. Bioinform. 2019, 20, 572–584. [Google Scholar] [CrossRef] [PubMed]
- Ivanisevic, T.; Sewduth, R.N. Multi-Omics Integration for the Design of Novel Therapies and the Identification of Novel Biomarkers. Proteomes 2023, 11, 34. [Google Scholar] [CrossRef]
- Subramanian, I.; Verma, S.; Kumar, S.; Jere, A.; Anamika, K. Multi-Omics Data Integration, Interpretation, and Its Application. Bioinform. Biol. Insig. 2020, 14, 117793221989905. [Google Scholar] [CrossRef] [PubMed]
- Rappoport, N.; Shamir, R. Multi-Omic and Multi-View Clustering Algorithms: Review and Cancer Benchmark. Nucleic Acids Res. 2018, 46, 10546–10562. [Google Scholar] [CrossRef]
- Athieniti, E.; Spyrou, G.M. A Guide to Multi-Omics Data Collection and Integration for Translational Medicine. Comput. Struct. Biotec 2023, 21, 134–149. [Google Scholar] [CrossRef]
- Tini, G.; Marchetti, L.; Priami, C.; Scott-Boyer, M.-P. Multi-Omics Integration—A Comparison of Unsupervised Clustering Methodologies. Brief. Bioinform. 2019, 20, 1269–1279. [Google Scholar] [CrossRef]
- Chauvel, C.; Novoloaca, A.; Veyre, P.; Reynier, F.; Becker, J. Evaluation of Integrative Clustering Methods for the Analysis of Multi-Omics Data. Brief. Bioinform. 2020, 21, 541–552. [Google Scholar] [CrossRef]
- Duan, X.; Ding, X.; Zhao, Z. Multi-Omics Integration with Weighted Affinity and Self-Diffusion Applied for Cancer Subtypes Identification. J. Transl. Med. 2024, 22, 79. [Google Scholar] [CrossRef]
- Xu, H.; Gao, L.; Huang, M.; Duan, R. A Network Embedding Based Method for Partial Multi-Omics Integration in Cancer Subtyping. Methods 2021, 192, 67–76. [Google Scholar] [CrossRef]
- Ye, X. Multi-Omics Clustering for Cancer Subtyping Based on Latent Subspace Learning. Comput. Biol. Med. 2023, 164, 107223. [Google Scholar] [CrossRef]
- Song, W.; Wang, W.; Dai, D.-Q. Subtype-WESLR: Identifying Cancer Subtype with Weighted Ensemble Sparse Latent Representation of Multi-View Data. Brief. Bioinform. 2022, 23, bbab398. [Google Scholar] [CrossRef] [PubMed]
- Pierre-Jean, M.; Mauger, F.; Deleuze, J.-F.; Le Floch, E. PIntMF: Penalized Integrative Matrix Factorization Method for Multi-Omics Data. Bioinformatics 2022, 38, 900–907. [Google Scholar] [CrossRef] [PubMed]
- Chalise, P.; Ni, Y.; Fridley, B.L. Network-Based Integrative Clustering of Multiple Types of Genomic Data Using Non-Negative Matrix Factorization. Comput. Biol. Med. 2020, 118, 103625. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.; Yang, Y.; Qiu, Y.; Zou, Q. SMCC: A Novel Clustering Method for Single- and Multi-Omics Data Based on Co-Regularized Network Fusion. IEEE/ACM Trans. Comput. Biol. Bioinform. 2024, 14, 1–9. [Google Scholar] [CrossRef]
- Yang, Y.; Tian, S.; Qiu, Y.; Zhao, P.; Zou, Q. MDICC: Novel Method for Multi-Omics Data Integration and Cancer Subtype Identification. Brief. Bioinform. 2022, 23, bbac132. [Google Scholar] [CrossRef]
- Pfeifer, B.; Bloice, M.D.; Schimek, M.G. Parea: Multi-View Ensemble Clustering for Cancer Subtype Discovery. J. Biomed. Inform. 2023, 143, 104406. [Google Scholar] [CrossRef]
- Yang, H.; Chen, R.; Li, D.; Wang, Z. Subtype-GAN: A Deep Learning Approach for Integrative Cancer Subtyping of Multi-Omics Data. Bioinformatics 2021, 37, 2231–2237. [Google Scholar] [CrossRef]
- Zhao, J.; Zhao, B.; Song, X.; Lyu, C.; Chen, W.; Xiong, Y.; Wei, D.-Q. Subtype-DCC: Decoupled Contrastive Clustering Method for Cancer Subtype Identification Based on Multi-Omics Data. Brief. Bioinform. 2023, 24, bbad025. [Google Scholar] [CrossRef]
- Vamathevan, J.; Clark, D.; Czodrowski, P.; Dunham, I.; Ferran, E.; Lee, G.; Li, B.; Madabhushi, A.; Shah, P.; Spitzer, M.; et al. Applications of Machine Learning in Drug Discovery and Development. Nat. Rev. Drug Discov. 2019, 18, 463–477. [Google Scholar] [CrossRef]
- Olsen, T.G.; Jackson, B.H.; Feeser, T.A.; Kent, M.N.; Moad, J.C.; Krishnamurthy, S.; Lunsford, D.D.; Soans, R.E. Diagnostic Performance of Deep Learning Algorithms Applied to Three Common Diagnoses in Dermatopathology. J. Pathol. Inform. 2018, 9, 32. [Google Scholar] [CrossRef]
- Esteva, A.; Kuprel, B.; Novoa, R.A.; Ko, J.; Swetter, S.M.; Blau, H.M.; Thrun, S. Dermatologist–Level Classification of Skin Cancer with Deep Neural Networks. Nature 2017, 542, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-S. Exploring Cancer Genomic Data from the Cancer Genome Atlas Project. BMB Rep. 2016, 49, 607–611. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.C.; Korkut, A.; Kanchi, R.S.; Hegde, A.M.; Lenoir, W.; Liu, W.; Liu, Y.; Fan, H.; Shen, H.; Ravikumar, V.; et al. A Comprehensive Pan-Cancer Molecular Study of Gynecologic and Breast Cancers. Cancer Cell 2018, 33, 690–705.e9. [Google Scholar] [CrossRef]
- Robertson, A.G.; Kim, J.; Al-Ahmadie, H.; Bellmunt, J.; Guo, G.; Cherniack, A.D.; Hinoue, T.; Laird, P.W.; Hoadley, K.A.; Akbani, R.; et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell 2017, 171, 540–556.e25. [Google Scholar] [CrossRef]
- Creighton, C.J.; Morgan, M.; Gunaratne, P.H.; Wheeler, D.A.; Gibbs, R.A.; Gordon Robertson, A.; Chu, A.; Beroukhim, R.; Cibulskis, K.; Signoretti, S.; et al. Comprehensive Molecular Characterization of Clear Cell Renal Cell Carcinoma. Nature 2013, 499, 43–49. [Google Scholar] [CrossRef]
- Collisson, E.A.; Campbell, J.D.; Brooks, A.N.; Berger, A.H.; Lee, W.; Chmielecki, J.; Beer, D.G.; Cope, L.; Creighton, C.J.; Danilova, L.; et al. Comprehensive Molecular Profiling of Lung Adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- Raphael, B.J.; Hruban, R.H.; Aguirre, A.J.; Moffitt, R.A.; Yeh, J.J.; Stewart, C.; Robertson, A.G.; Cherniack, A.D.; Gupta, M.; Getz, G.; et al. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203.e13. [Google Scholar] [CrossRef]
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Arachchi, H.; Arora, A.; Auman, J.T.; Ayala, B.; et al. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef]
- Lei, Z.; Tan, I.B.; Das, K.; Deng, N.; Zouridis, H.; Pattison, S.; Chua, C.; Feng, Z.; Guan, Y.K.; Ooi, C.H.; et al. Identification of Molecular Subtypes of Gastric Cancer With Different Responses to PI3-Kinase Inhibitors and 5-Fluorouracil. Gastroenterology 2013, 145, 554–565. [Google Scholar] [CrossRef]
- Levine, D.A. Integrated Genomic Characterization of Endometrial Carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [CrossRef]
- Robertson, A.G.; Shih, J.; Yau, C.; Gibb, E.A.; Oba, J.; Mungall, K.L.; Hess, J.M.; Uzunangelov, V.; Walter, V.; Danilova, L.; et al. Integrative Analysis Identifies Four Molecular and Clinical Subsets in Uveal Melanoma. Cancer Cell 2017, 32, 204–220.e15. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Zhu, H.; Dong, L.; Shi, W.; Chen, R.; Song, Z.; Huang, C.; Li, J.; Dong, X.; Zhou, Y.; et al. Integrated Proteogenomic Characterization of HBV-Related Hepatocellular Carcinoma. Cell 2019, 179, 561–577.e22. [Google Scholar] [CrossRef] [PubMed]
- Petralia, F.; Tignor, N.; Reva, B.; Koptyra, M.; Chowdhury, S.; Rykunov, D.; Krek, A.; Ma, W.; Zhu, Y.; Ji, J.; et al. Integrated Proteogenomic Characterization across Major Histological Types of Pediatric Brain Cancer. Cell 2020, 183, 1962–1985.e31. [Google Scholar] [CrossRef]
- Chalise, P.; Fridley, B.L. Integrative Clustering of Multi-Level ‘omic Data Based on Non-Negative Matrix Factorization Algorithm. PLoS ONE 2017, 12, e0176278. [Google Scholar] [CrossRef]
- Wang, B.; Mezlini, A.M.; Demir, F.; Fiume, M.; Tu, Z.; Brudno, M.; Haibe-Kains, B.; Goldenberg, A. Similarity Network Fusion for Aggregating Data Types on a Genomic Scale. Nat. Methods 2014, 11, 333–337. [Google Scholar] [CrossRef]
- Rousseeuw, P.J. Silhouettes: A Graphical Aid to the Interpretation and Validation of Cluster Analysis. J. Comput. Appl. Math. 1987, 20, 53–65. [Google Scholar] [CrossRef]
- Calinski, T.; Harabasz, J. A Dendrite Method for Cluster Analysis. Comm. Stats.—Theory Methods 1974, 3, 1–27. [Google Scholar] [CrossRef]
- Dunn, J.C. Well-Separated Clusters and Optimal Fuzzy Partitions. J. Cybern. 1974, 4, 95–104. [Google Scholar] [CrossRef]
- Mantel, N. Evaluation of Survival Data and Two New Rank Order Statistics Arising in Its Consideration. Cancer Chemother. Rep. 1966, 50, 163–170. [Google Scholar]
- Plackett, R.L. Karl Pearson and the Chi-Squared Test. Int. Stat. Rev. / Rev. Int. De Stat. 1983, 51, 59. [Google Scholar] [CrossRef]
- Mahoney, M.; Magel, R. Estimation of the Power of the Kruskal-Wallis Test. Biom. J. 1996, 38, 613–630. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Datasets | K Value | References |
|---|---|---|
| BRCA | 5 | [23] |
| BLCA | 5 | [24] |
| KIRC | 4 | [25] |
| LUAD | 3 | [26] |
| PAAD | 2 | [27] |
| SKCM | 4 | [28] |
| STAD | 3 | [29] |
| UCEC | 4 | [30] |
| UVM | 4 | [31] |
| PBT | 8 | [32] |
| HCC | 3 | [33] |
| Categories | Algorithms | Year | Implementation | Reference |
|---|---|---|---|---|
| Similarity-based clustering methods | MOSD | 2024 | https://github.com/DXCODEE/MOSD (accessed on 4 March 2024) | [8] |
| MSNE | 2021 | https://github.com/GaoLabXDU/MSNE (accessed on 5 March 2024) | [9] | |
| Dimensionality reduction-based clustering methods | MCLS | 2023 | https://github.com/ShangCS/MCLS (accessed on 9 March 2024) | [10] |
| subtype-WESLR | 2022 | https://github.com/songwenjing123/subtype-WESLR (accessed on 2 April 2024) | [11] | |
| Matrix factorization-based clustering methods | PIntMF | 2022 | https://github.com/mpierrejean/pintmf (accessed on 5 April 2024) | [12] |
| nNMF | 2020 | R v4.3.2 | [13] | |
| Fusion-based clustering methods | SMCC | 2024 | https://github.com/yushanqiu/SMCC (accessed on 5 April 2024) | [14] |
| MDICC | 2022 | https://github.com/yushanqiu/MDICC (accessed on 5 April 2024) | [15] | |
| Parea | 2023 | https://github.com/mdbloice/Pyrea (accessed on 5 April 2024) | [16] | |
| Deep learning-based clustering methods | Subtype-GAN | 2021 | https://github.com/haiyang1986/Subtype-GAN (accessed on 6 April 2024) | [17] |
| Subtype-DCC | 2023 | https://github.com/zhaojingo/Subtype-DCC (accessed on 6 April 2024) | [18] |
| Categories | Metrics | Formulas | Optimum |
|---|---|---|---|
| Internal metrics | S | a: mean distance from the sample point to other points within the same cluster b: mean distance from the sample point to the nearest other cluster | Maximum |
| CH | Bk: inter-cluster covariance matrix Wk: intra-cluster covariance matrix n: sample size k: number of clusters | Maximum | |
| D | d(Ci, Cj): inter-cluster distance diam(Ck): cluster diameter | Maximum | |
| Clinical metrics | LRT | Using log rank test to calculate the p-value | Minimum |
| ECP | χ2 test discrete parameter enrichment Kruskal–Wallis test numerical parameter enrichment | Maximum | |
| Time metrics | RT | Average running time | Minimum |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Wang, L.; Liu, Y.; Li, X.; Ma, J.; Li, M.; Zhu, Y. Comprehensive Evaluation of Multi-Omics Clustering Algorithms for Cancer Molecular Subtyping. Int. J. Mol. Sci. 2025, 26, 963. https://doi.org/10.3390/ijms26030963
Wang J, Wang L, Liu Y, Li X, Ma J, Li M, Zhu Y. Comprehensive Evaluation of Multi-Omics Clustering Algorithms for Cancer Molecular Subtyping. International Journal of Molecular Sciences. 2025; 26(3):963. https://doi.org/10.3390/ijms26030963
Chicago/Turabian StyleWang, Juan, Lingxiao Wang, Yi Liu, Xiao Li, Jie Ma, Mansheng Li, and Yunping Zhu. 2025. "Comprehensive Evaluation of Multi-Omics Clustering Algorithms for Cancer Molecular Subtyping" International Journal of Molecular Sciences 26, no. 3: 963. https://doi.org/10.3390/ijms26030963
APA StyleWang, J., Wang, L., Liu, Y., Li, X., Ma, J., Li, M., & Zhu, Y. (2025). Comprehensive Evaluation of Multi-Omics Clustering Algorithms for Cancer Molecular Subtyping. International Journal of Molecular Sciences, 26(3), 963. https://doi.org/10.3390/ijms26030963