

HCoV-229E Mpro Suppresses RLR-Mediated Innate Immune Signalling Through Cleavage of NEMO and Through Other Mechanisms

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. The Mpro of HCoV-229E Dampens the IFN-β and NF-κB Responses
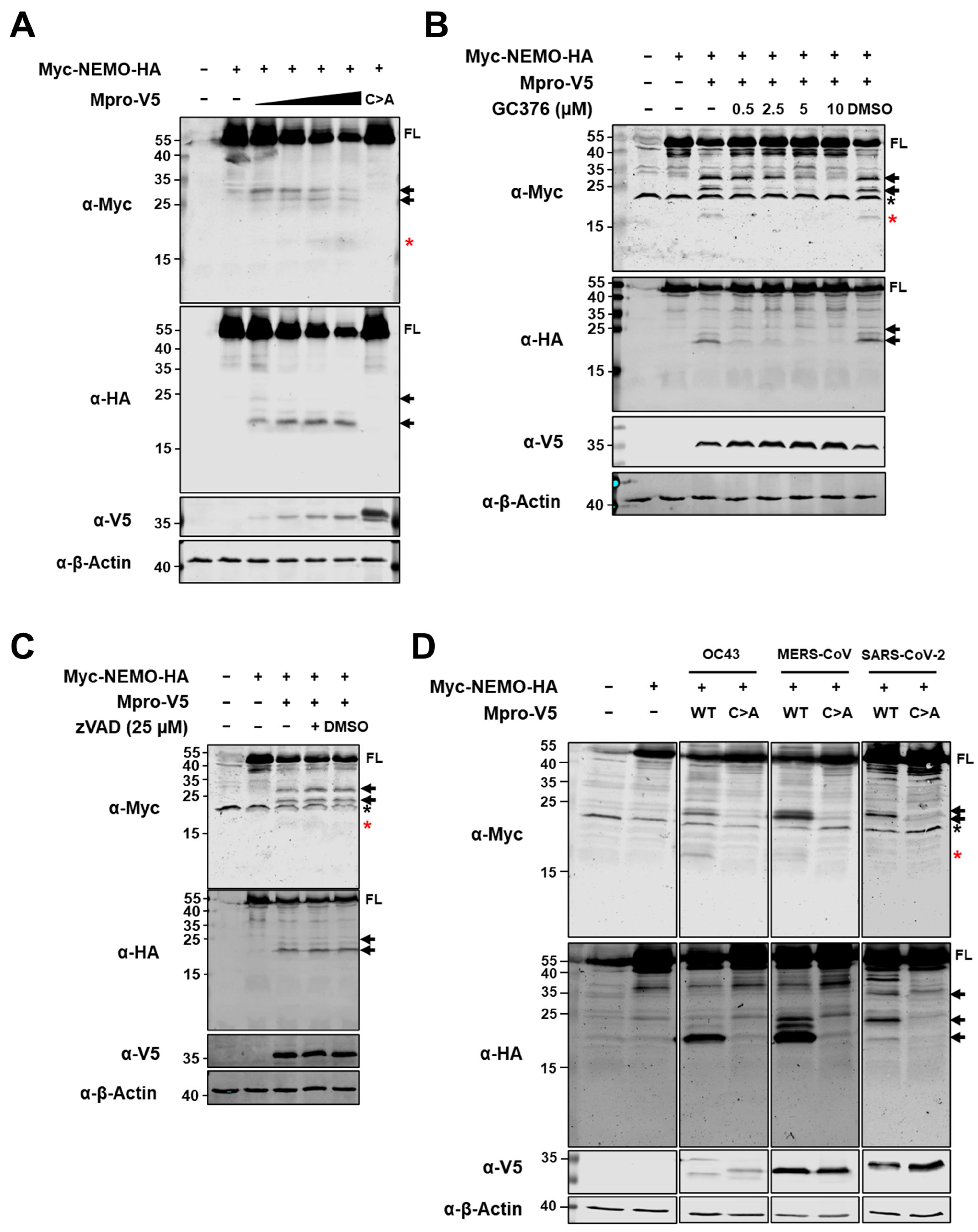
2.2. Ectopic Expression of HCoV-229E Mpro Induces NEMO Cleavage by Means of Its Catalytic Activity
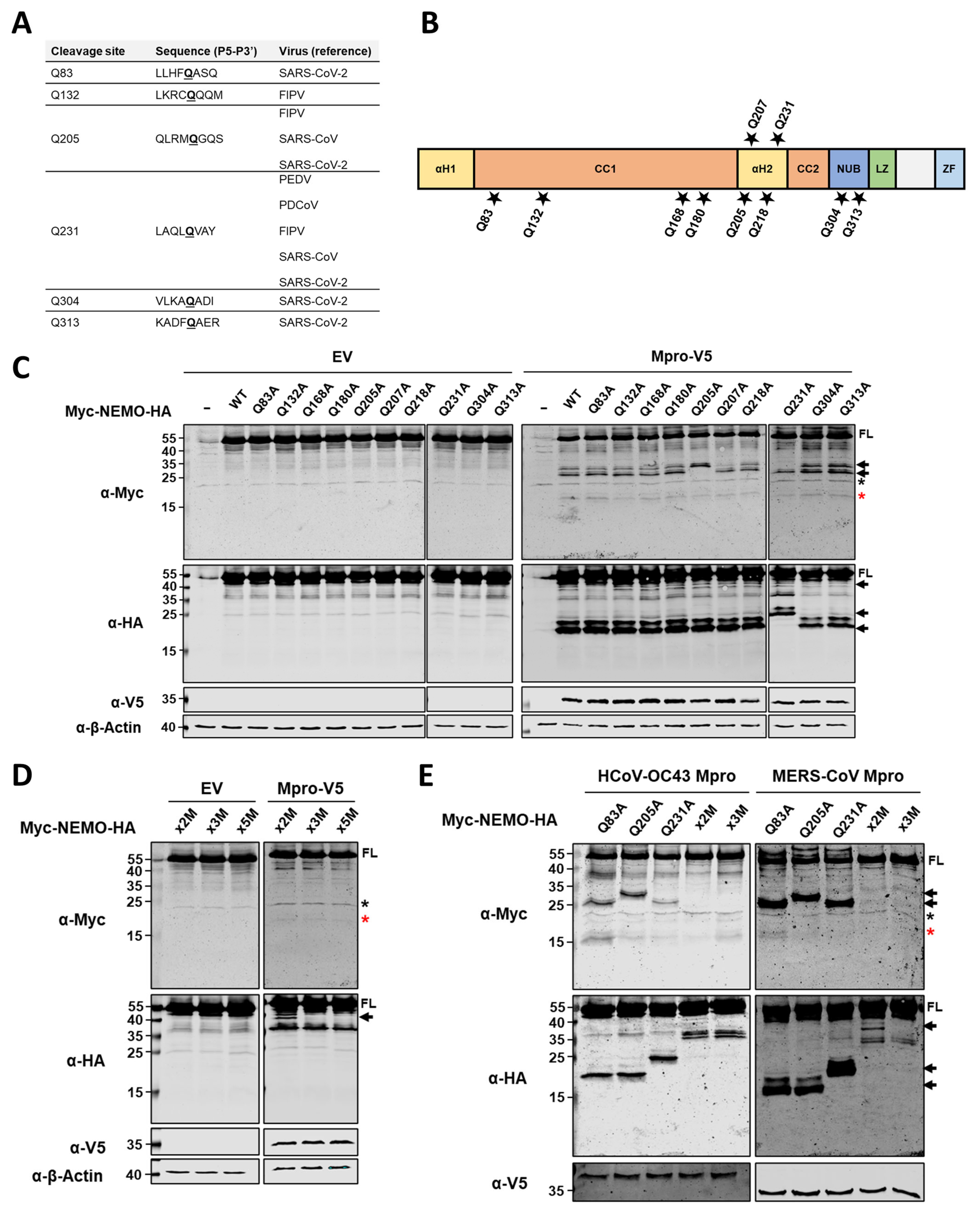
2.3. HCoV-229E Mpro Cleaves NEMO at Glutamine Residues 83, 205 and 231
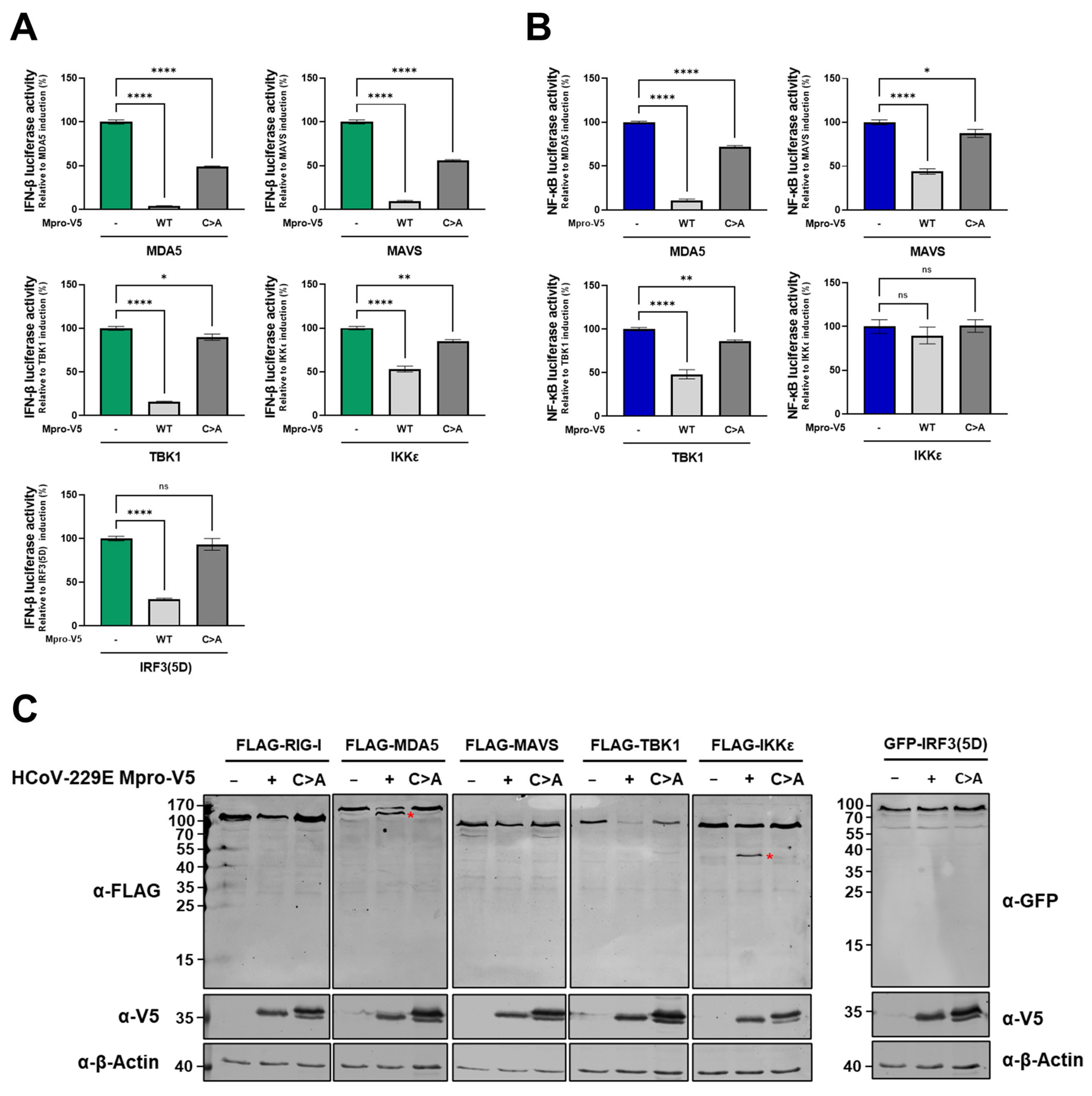
2.4. Besides by Cleavage of NEMO, HCoV-229E Mpro Likely Abrogates NF-κB Induction Through Other Mechanisms
2.5. HCoV-229E Mpro Disrupts the RLR Signalling Pathway Downstream of NEMO
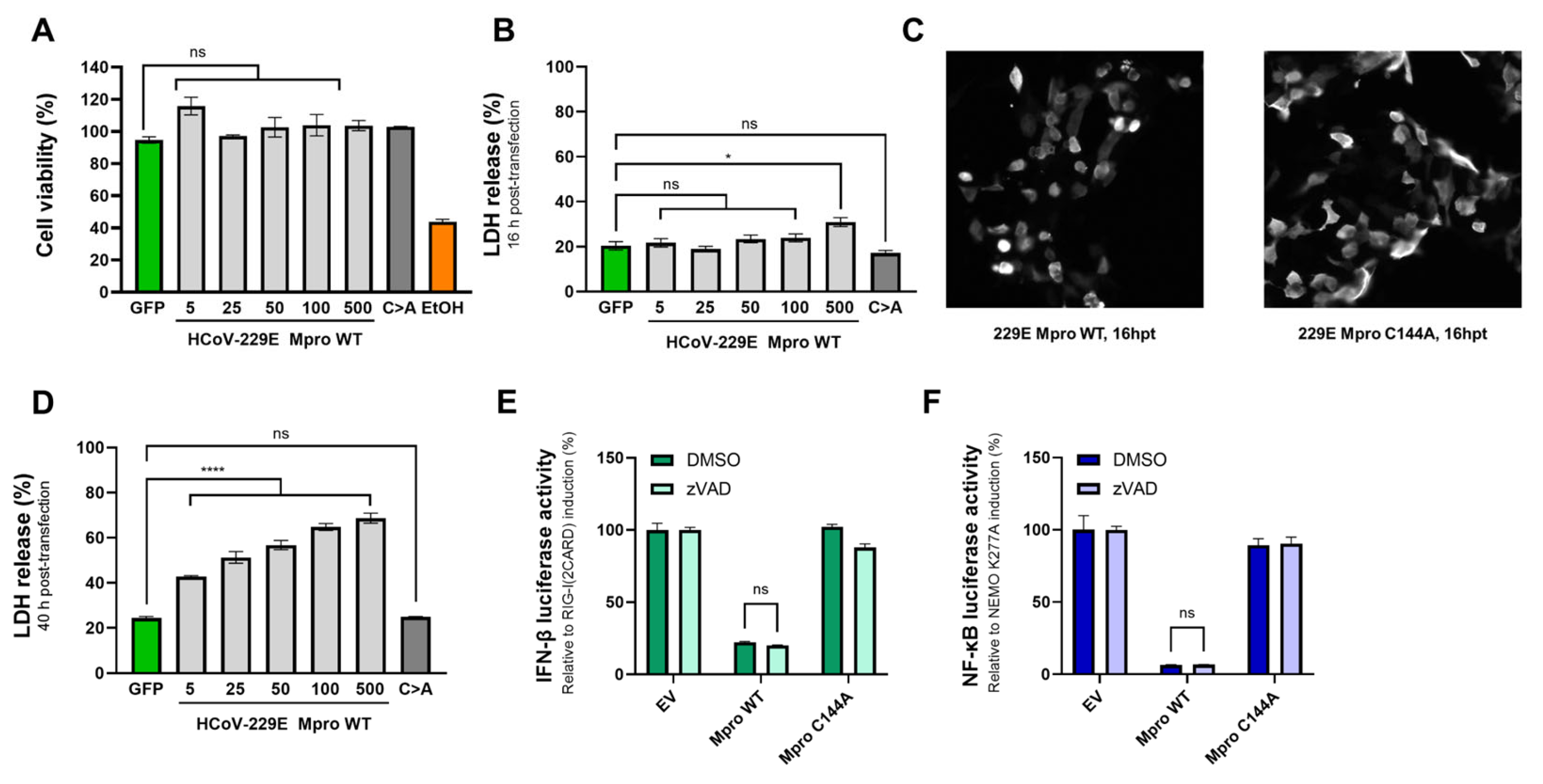
2.6. The Effect of Mpro on IFN-β and NF-κB Induction Is Independent of Mpro-Mediated Toxicity
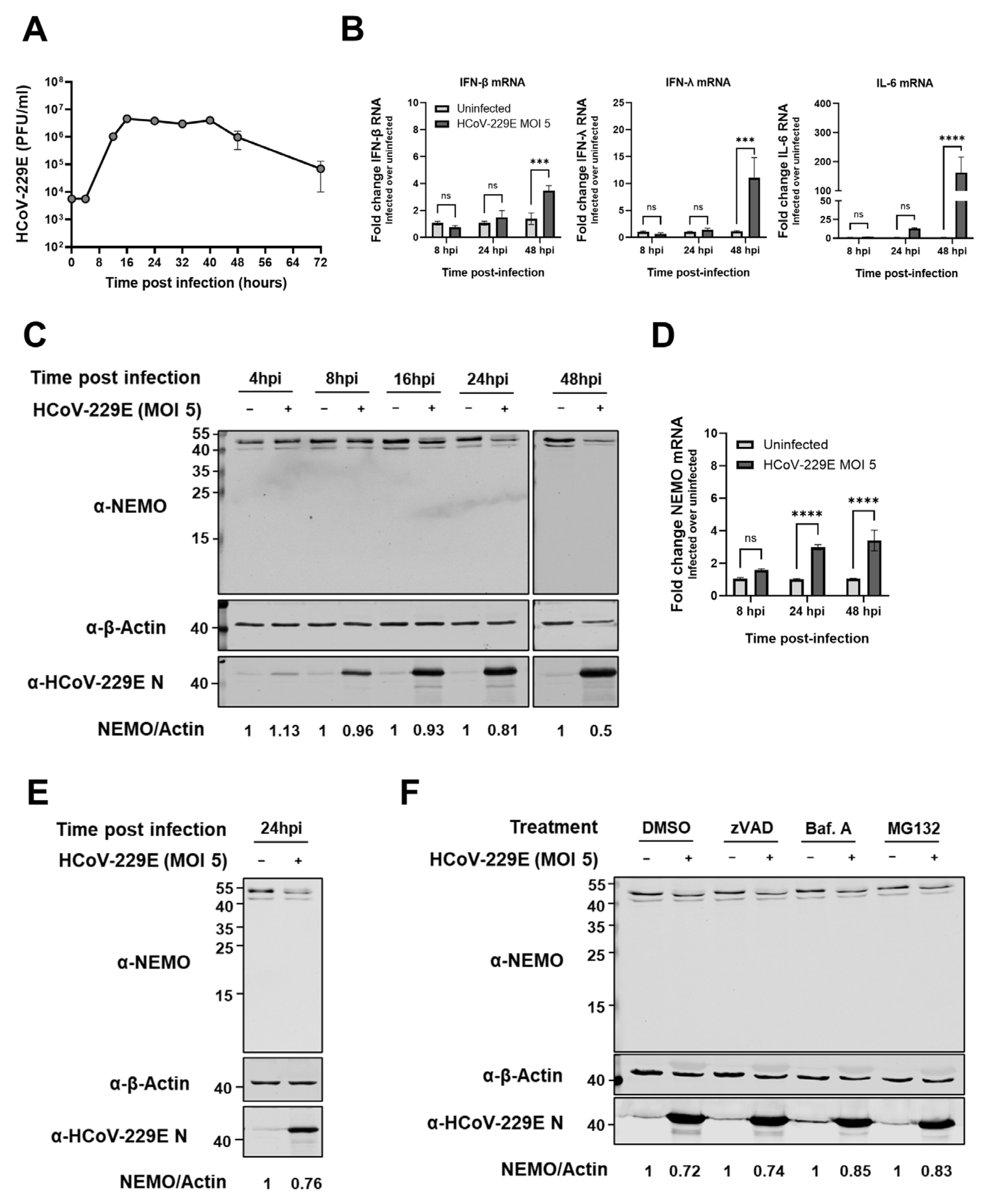
2.7. HCoV-229E Infection Leads to a Delayed Antiviral Innate Immune Response and Reduced NEMO Protein Expression Levels
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Virus Infections
4.2. Virus Infections
4.3. Plaque Assays
4.4. Plasmids Used for Transfections
4.5. Immunoblotting Analysis
4.6. Dual-Luciferase Reporter Assay
4.7. Immunofluorescence Microscopy
4.8. RNA Isolation and RT-qPCR
4.9. Cell Death Assays
4.10. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Minkoff, J.M.; tenOever, B. Innate immune evasion strategies of SARS-CoV-2. Nat. Rev. Microbiol. 2023, 21, 178–194. [Google Scholar] [CrossRef]
- Kasuga, Y.; Zhu, B.; Jang, K.J.; Yoo, J.S. Innate immune sensing of coronavirus and viral evasion strategies. Exp. Mol. Med. 2021, 53, 723–736. [Google Scholar] [CrossRef]
- Yoo, J.S.; Kato, H.; Fujita, T. Sensing viral invasion by RIG-I like receptors. Curr. Opin. Microbiol. 2014, 20, 131–138. [Google Scholar] [CrossRef]
- Loo, Y.M.; Gale, M., Jr. Immune signaling by RIG-I-like receptors. Immunity 2011, 34, 680–692. [Google Scholar] [CrossRef]
- Luan, X.; Wang, L.; Song, G.; Zhou, W. Innate immune responses to RNA: Sensing and signaling. Front. Immunol. 2024, 15, 1287940. [Google Scholar] [CrossRef]
- Rehwinkel, J.; Gack, M.U. RIG-I-like receptors: Their regulation and roles in RNA sensing. Nat. Rev. Immunol. 2020, 20, 537–551. [Google Scholar] [CrossRef]
- Lin, R.; Heylbroeck, C.; Pitha, P.M.; Hiscott, J. Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol. Cell. Biol. 1998, 18, 2986–2996. [Google Scholar] [CrossRef]
- Sato, M.; Tanaka, N.; Hata, N.; Oda, E.; Taniguchi, T. Involvement of the IRF family transcription factor IRF-3 in virus-induced activation of the IFN-beta gene. FEBS Lett. 1998, 425, 112–116. [Google Scholar] [CrossRef]
- Paz, S.; Sun, Q.; Nakhaei, P.; Romieu-Mourez, R.; Goubau, D.; Julkunen, I.; Lin, R.; Hiscott, J. Induction of IRF-3 and IRF-7 phosphorylation following activation of the RIG-I pathway. Cell. Mol. Biol. 2006, 52, 17–28. [Google Scholar]
- Zhao, T.; Yang, L.; Sun, Q.; Arguello, M.; Ballard, D.W.; Hiscott, J.; Lin, R. The NEMO adaptor bridges the nuclear factor-kappaB and interferon regulatory factor signaling pathways. Nat. Immunol. 2007, 8, 592–600. [Google Scholar] [CrossRef]
- Lei, J.; Hilgenfeld, R. RNA-virus proteases counteracting host innate immunity. FEBS Lett. 2017, 591, 3190–3210. [Google Scholar] [CrossRef]
- Schulz, K.S.; Mossman, K.L. Viral Evasion Strategies in Type I IFN Signaling—A Summary of Recent Developments. Front. Immunol. 2016, 7, 498. [Google Scholar] [CrossRef]
- Versteeg, G.A.; García-Sastre, A. Viral tricks to grid-lock the type I interferon system. Curr. Opin. Microbiol. 2010, 13, 508–516. [Google Scholar] [CrossRef]
- Lee, H.J.; Shieh, C.K.; Gorbalenya, A.E.; Koonin, E.V.; La Monica, N.; Tuler, J.; Bagdzhadzhyan, A.; Lai, M.M. The complete sequence (22 kilobases) of murine coronavirus gene 1 encoding the putative proteases and RNA polymerase. Virology 1991, 180, 567–582. [Google Scholar] [CrossRef]
- Ziebuhr, J.; Siddell, S.G. Processing of the human coronavirus 229E replicase polyproteins by the virus-encoded 3C-like proteinase: Identification of proteolytic products and cleavage sites common to pp1a and pp1ab. J. Virol. 1999, 73, 177–185. [Google Scholar] [CrossRef]
- Barretto, N.; Jukneliene, D.; Ratia, K.; Chen, Z.; Mesecar, A.D.; Baker, S.C. The papain-like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. J. Virol. 2005, 79, 15189–15198. [Google Scholar] [CrossRef]
- Lindner, H.A.; Fotouhi-Ardakani, N.; Lytvyn, V.; Lachance, P.; Sulea, T.; Ménard, R. The papain-like protease from the severe acute respiratory syndrome coronavirus is a deubiquitinating enzyme. J. Virol. 2005, 79, 15199–15208. [Google Scholar] [CrossRef]
- Mielech, A.M.; Kilianski, A.; Baez-Santos, Y.M.; Mesecar, A.D.; Baker, S.C. MERS-CoV papain-like protease has deISGylating and deubiquitinating activities. Virology 2014, 450–451, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Chen, G.; Guo, B.; Cheng, G.; Tang, H. PLP2, a potent deubiquitinase from murine hepatitis virus, strongly inhibits cellular type I interferon production. Cell Res. 2008, 18, 1105–1113. [Google Scholar] [CrossRef]
- Chen, X.; Yang, X.; Zheng, Y.; Yang, Y.; Xing, Y.; Chen, Z. SARS coronavirus papain-like protease inhibits the type I interferon signaling pathway through interaction with the STING-TRAF3-TBK1 complex. Protein Cell 2014, 5, 369–381. [Google Scholar] [CrossRef]
- Li, S.W.; Wang, C.Y.; Jou, Y.J.; Huang, S.H.; Hsiao, L.H.; Wan, L.; Lin, Y.J.; Kung, S.H.; Lin, C.W. SARS Coronavirus Papain-Like Protease Inhibits the TLR7 Signaling Pathway through Removing Lys63-Linked Polyubiquitination of TRAF3 and TRAF6. Int. J. Mol. Sci. 2016, 17, 678. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Wang, D.; Zhou, J.; Pan, T.; Chen, J.; Yang, Y.; Lv, M.; Ye, X.; Peng, G.; Fang, L.; et al. Porcine Deltacoronavirus nsp5 Antagonizes Type I Interferon Signaling by Cleaving STAT2. J. Virol. 2017, 91, e00003-17. [Google Scholar] [CrossRef]
- Wang, D.; Fang, L.; Shi, Y.; Zhang, H.; Gao, L.; Peng, G.; Chen, H.; Li, K.; Xiao, S. Porcine Epidemic Diarrhea Virus 3C-Like Protease Regulates Its Interferon Antagonism by Cleaving NEMO. J. Virol. 2016, 90, 2090–2101. [Google Scholar] [CrossRef]
- Zhu, X.; Fang, L.; Wang, D.; Yang, Y.; Chen, J.; Ye, X.; Foda, M.F.; Xiao, S. Porcine deltacoronavirus nsp5 inhibits interferon-β production through the cleavage of NEMO. Virology 2017, 502, 33–38. [Google Scholar] [CrossRef]
- Chen, S.; Tian, J.; Li, Z.; Kang, H.; Zhang, J.; Huang, J.; Yin, H.; Hu, X.; Qu, L. Feline Infectious Peritonitis Virus Nsp5 Inhibits Type I Interferon Production by Cleaving NEMO at Multiple Sites. Viruses 2019, 12, 43. [Google Scholar] [CrossRef]
- Wenzel, J.; Lampe, J.; Müller-Fielitz, H.; Schuster, R.; Zille, M.; Müller, K.; Krohn, M.; Körbelin, J.; Zhang, L.; Özorhan, Ü.; et al. The SARS-CoV-2 main protease M(pro) causes microvascular brain pathology by cleaving NEMO in brain endothelial cells. Nat. Neurosci. 2021, 24, 1522–1533. [Google Scholar] [CrossRef]
- Chen, J.; Li, Z.; Guo, J.; Xu, S.; Zhou, J.; Chen, Q.; Tong, X.; Wang, D.; Peng, G.; Fang, L.; et al. SARS-CoV-2 nsp5 Exhibits Stronger Catalytic Activity and Interferon Antagonism than Its SARS-CoV Ortholog. J. Virol. 2022, 96, e0003722. [Google Scholar] [CrossRef]
- Hameedi, M.A.; Prates, E.T.; Garvin, M.R.; Mathews, I.I.; Amos, B.K.; Demerdash, O.; Bechthold, M.; Iyer, M.; Rahighi, S.; Kneller, D.W.; et al. Structural and functional characterization of NEMO cleavage by SARS-CoV-2 3CLpro. Nat. Commun. 2022, 13, 5285. [Google Scholar] [CrossRef]
- van Huizen, M.; Vendrell, X.M.; de Gruyter, H.L.M.; Boomaars-van der Zanden, A.L.; van der Meer, Y.; Snijder, E.J.; Kikkert, M.; Myeni, S.K. The Main Protease of Middle East Respiratory Syndrome Coronavirus Induces Cleavage of Mitochondrial Antiviral Signaling Protein to Antagonize the Innate Immune Response. Viruses 2024, 16, 256. [Google Scholar] [CrossRef]
- Zhang, Y.; Kandwal, S.; Fayne, D.; Stevenson, N.J. MERS-CoV-nsp5 expression in human epithelial BEAS 2b cells attenuates type I interferon production by inhibiting IRF3 nuclear translocation. Cell. Mol. Life Sci. 2024, 81, 433. [Google Scholar] [CrossRef]
- Channappanavar, R.; Perlman, S. Pathogenic human coronavirus infections: Causes and consequences of cytokine storm and immunopathology. Semin. Immunopathol. 2017, 39, 529–539. [Google Scholar] [CrossRef]
- Fung, S.Y.; Siu, K.L.; Lin, H.; Yeung, M.L.; Jin, D.Y. SARS-CoV-2 main protease suppresses type I interferon production by preventing nuclear translocation of phosphorylated IRF3. Int. J. Biol. Sci. 2021, 17, 1547–1554. [Google Scholar] [CrossRef]
- Zheng, Y.; Deng, J.; Han, L.; Zhuang, M.W.; Xu, Y.; Zhang, J.; Nan, M.L.; Xiao, Y.; Zhan, P.; Liu, X.; et al. SARS-CoV-2 NSP5 and N protein counteract the RIG-I signaling pathway by suppressing the formation of stress granules. Signal Transduct. Target. Ther. 2022, 7, 22. [Google Scholar] [CrossRef]
- Wathelet, M.G.; Lin, C.H.; Parekh, B.S.; Ronco, L.V.; Howley, P.M.; Maniatis, T. Virus infection induces the assembly of coordinately activated transcription factors on the IFN-beta enhancer in vivo. Mol. Cell 1998, 1, 507–518. [Google Scholar] [CrossRef]
- Weaver, B.K.; Kumar, K.P.; Reich, N.C. Interferon regulatory factor 3 and CREB-binding protein/p300 are subunits of double-stranded RNA-activated transcription factor DRAF1. Mol. Cell Biol. 1998, 18, 1359–1368. [Google Scholar] [CrossRef]
- Brady, G.; Haas, D.A.; Farrell, P.J.; Pichlmair, A.; Bowie, A.G. Molluscum Contagiosum Virus Protein MC005 Inhibits NF-κB Activation by Targeting NEMO-Regulated IκB Kinase Activation. J. Virol. 2017, 91, e00545-17. [Google Scholar] [CrossRef]
- Chen, J.; Wang, D.; Sun, Z.; Gao, L.; Zhu, X.; Guo, J.; Xu, S.; Fang, L.; Li, K.; Xiao, S. Arterivirus nsp4 Antagonizes Interferon Beta Production by Proteolytically Cleaving NEMO at Multiple Sites. J. Virol. 2019, 93, e00385-19. [Google Scholar] [CrossRef]
- Wang, D.; Fang, L.; Li, K.; Zhong, H.; Fan, J.; Ouyang, C.; Zhang, H.; Duan, E.; Luo, R.; Zhang, Z.; et al. Foot-and-mouth disease virus 3C protease cleaves NEMO to impair innate immune signaling. J. Virol. 2012, 86, 9311–9322. [Google Scholar] [CrossRef]
- Zhang, H.; Jiang, P.; Chen, Z.; Wang, D.; Zhou, Y.; Zhu, X.; Xiao, S.; Fang, L. Norovirus 3C-Like protease antagonizes interferon-β production by cleaving NEMO. Virology 2022, 571, 12–20. [Google Scholar] [CrossRef]
- Frelin, C.; Imbert, V.; Bottero, V.; Gonthier, N.; Samraj, A.K.; Schulze-Osthoff, K.; Auberger, P.; Courtois, G.; Peyron, J.F. Inhibition of the NF-kappaB survival pathway via caspase-dependent cleavage of the IKK complex scaffold protein and NF-kappaB essential modulator NEMO. Cell Death Differ. 2008, 15, 152–160. [Google Scholar] [CrossRef]
- Chuck, C.P.; Chong, L.T.; Chen, C.; Chow, H.F.; Wan, D.C.; Wong, K.B. Profiling of substrate specificity of SARS-CoV 3CL. PLoS ONE 2010, 5, e13197. [Google Scholar] [CrossRef] [PubMed]
- Chuck, C.P.; Chow, H.F.; Wan, D.C.; Wong, K.B. Profiling of substrate specificities of 3C-like proteases from group 1, 2a, 2b, and 3 coronaviruses. PLoS ONE 2011, 6, e27228. [Google Scholar] [CrossRef] [PubMed]
- Kiemer, L.; Lund, O.; Brunak, S.; Blom, N. Coronavirus 3CLpro proteinase cleavage sites: Possible relevance to SARS virus pathology. BMC Bioinform. 2004, 5, 72. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Zhang, Q.; Guo, X.K.; Yu, Z.B.; Xu, A.T.; Tang, J.; Feng, W.H. Porcine reproductive and respiratory syndrome virus nonstructural protein 4 antagonizes beta interferon expression by targeting the NF-κB essential modulator. J. Virol. 2014, 88, 10934–10945. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Fang, L.; Wei, D.; Zhang, H.; Luo, R.; Chen, H.; Li, K.; Xiao, S. Hepatitis A virus 3C protease cleaves NEMO to impair induction of beta interferon. J. Virol. 2014, 88, 10252–10258. [Google Scholar] [CrossRef]
- Resnick, S.J.; Iketani, S.; Hong, S.J.; Zask, A.; Liu, H.; Kim, S.; Melore, S.; Lin, F.Y.; Nair, M.S.; Huang, Y.; et al. Inhibitors of Coronavirus 3CL Proteases Protect Cells from Protease-Mediated Cytotoxicity. J. Virol. 2021, 95, e0237420. [Google Scholar] [CrossRef]
- Shibata, Y.; Oyama, M.; Kozuka-Hata, H.; Han, X.; Tanaka, Y.; Gohda, J.; Inoue, J. p47 negatively regulates IKK activation by inducing the lysosomal degradation of polyubiquitinated NEMO. Nat. Commun. 2012, 3, 1061. [Google Scholar] [CrossRef]
- Fliss, P.M.; Jowers, T.P.; Brinkmann, M.M.; Holstermann, B.; Mack, C.; Dickinson, P.; Hohenberg, H.; Ghazal, P.; Brune, W. Viral mediated redirection of NEMO/IKKγ to autophagosomes curtails the inflammatory cascade. PLoS Pathog. 2012, 8, e1002517. [Google Scholar] [CrossRef]
- Tomar, D.; Singh, R. TRIM13 regulates ubiquitination and turnover of NEMO to suppress TNF induced NF-κB activation. Cell. Signal 2014, 26, 2606–2613. [Google Scholar] [CrossRef]
- Xing, J.; Weng, L.; Yuan, B.; Wang, Z.; Jia, L.; Jin, R.; Lu, H.; Li, X.C.; Liu, Y.J.; Zhang, Z. Identification of a role for TRIM29 in the control of innate immunity in the respiratory tract. Nat. Immunol. 2016, 17, 1373–1380. [Google Scholar] [CrossRef]
- Chathuranga, K.; Kim, T.H.; Lee, H.; Park, J.S.; Kim, J.H.; Chathuranga, W.A.G.; Ekanayaka, P.; Choi, Y.J.; Lee, C.H.; Kim, C.J.; et al. Negative regulation of NEMO signaling by the ubiquitin E3 ligase MARCH2. Embo. J. 2020, 39, e105139. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Ma, Z.; Wu, Y.; Shi, X.; Zhang, Y.; Zhang, M.; Zhang, M.; Wang, L.; Liu, W. SARS-CoV-2 3C-like protease antagonizes interferon-beta production by facilitating the degradation of IRF3. Cytokine 2021, 148, 155697. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Qin, C.; Rao, Y.; Ngo, C.; Feng, J.J.; Zhao, J.; Zhang, S.; Wang, T.Y.; Carriere, J.; Savas, A.C.; et al. SARS-CoV-2 Nsp5 Demonstrates Two Distinct Mechanisms Targeting RIG-I and MAVS To Evade the Innate Immune Response. mBio 2021, 12, e0233521. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.Z.; Wang, S.Y.; Zheng, Z.Q.; Yi, H.; Li, W.W.; Xu, Z.S.; Wang, Y.Y. SARS-CoV-2 membrane glycoprotein M antagonizes the MAVS-mediated innate antiviral response. Cell. Mol. Immunol. 2021, 18, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Cao, Z.; Xie, X.; Zhang, X.; Chen, J.Y.; Wang, H.; Menachery, V.D.; Rajsbaum, R.; Shi, P.Y. Evasion of Type I Interferon by SARS-CoV-2. Cell Rep. 2020, 33, 108234. [Google Scholar] [CrossRef]
- Lei, X.; Dong, X.; Ma, R.; Wang, W.; Xiao, X.; Tian, Z.; Wang, C.; Wang, Y.; Li, L.; Ren, L.; et al. Activation and evasion of type I interferon responses by SARS-CoV-2. Nat. Commun. 2020, 11, 3810. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, C.; Feng, X.; Nie, L.; Tang, M.; Zhang, H.; Xiong, Y.; Swisher, S.K.; Srivastava, M.; Chen, J. Interactomes of SARS-CoV-2 and human coronaviruses reveal host factors potentially affecting pathogenesis. Embo. J. 2021, 40, e107776. [Google Scholar] [CrossRef]
- Li, W.; Qiao, J.; You, Q.; Zong, S.; Peng, Q.; Liu, Y.; Hu, S.; Liu, W.; Li, S.; Shu, X.; et al. SARS-CoV-2 Nsp5 Activates NF-κB Pathway by Upregulating SUMOylation of MAVS. Front. Immunol. 2021, 12, 750969. [Google Scholar] [CrossRef]
- Lin, C.W.; Lin, K.H.; Hsieh, T.H.; Shiu, S.Y.; Li, J.Y. Severe acute respiratory syndrome coronavirus 3C-like protease-induced apoptosis. FEMS Immunol. Med. Microbiol. 2006, 46, 375–380. [Google Scholar] [CrossRef]
- Song, L.; Wang, D.; Abbas, G.; Li, M.; Cui, M.; Wang, J.; Lin, Z.; Zhang, X.E. The main protease of SARS-CoV-2 cleaves histone deacetylases and DCP1A, attenuating the immune defense of the interferon-stimulated genes. J. Biol. Chem. 2023, 299, 102990. [Google Scholar] [CrossRef]
- Koudelka, T.; Boger, J.; Henkel, A.; Schönherr, R.; Krantz, S.; Fuchs, S.; Rodríguez, E.; Redecke, L.; Tholey, A. N-Terminomics for the Identification of In Vitro Substrates and Cleavage Site Specificity of the SARS-CoV-2 Main Protease. Proteomics 2021, 21, e2000246. [Google Scholar] [CrossRef] [PubMed]
- Bloor, S.; Ryzhakov, G.; Wagner, S.; Butler, P.J.; Smith, D.L.; Krumbach, R.; Dikic, I.; Randow, F. Signal processing by its coil zipper domain activates IKK gamma. Proc. Natl. Acad. Sci. USA 2008, 105, 1279–1284. [Google Scholar] [CrossRef] [PubMed]
- Barral, P.M.; Morrison, J.M.; Drahos, J.; Gupta, P.; Sarkar, D.; Fisher, P.B.; Racaniello, V.R. MDA-5 is cleaved in poliovirus-infected cells. J. Virol. 2007, 81, 3677–3684. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Langereis, M.A.; Lork, M.; Nguyen, M.; Hato, S.V.; Lanke, K.; Emdad, L.; Bhoopathi, P.; Fisher, P.B.; Lloyd, R.E.; et al. Enterovirus 2Apro targets MDA5 and MAVS in infected cells. J. Virol. 2014, 88, 3369–3378. [Google Scholar] [CrossRef] [PubMed]
- Pulido, M.R.; Martínez-Salas, E.; Sobrino, F.; Sáiz, M. MDA5 cleavage by the Leader protease of foot-and-mouth disease virus reveals its pleiotropic effect against the host antiviral response. Cell Death Dis. 2020, 11, 718. [Google Scholar] [CrossRef]
- Miyamoto, M.; Himeda, T.; Ishihara, K.; Okuwa, T.; Kobayashi, D.; Nameta, M.; Karasawa, Y.; Chunhaphinyokul, B.; Yoshida, Y.; Tanaka, N.; et al. Theilovirus 3C Protease Cleaves the C-Terminal Domain of the Innate Immune RNA Sensor, Melanoma Differentiation-Associated Gene 5, and Impairs Double-Stranded RNA-Mediated IFN Response. J. Immunol. 2023, 210, 335–347. [Google Scholar] [CrossRef]
- Budroni, V.; Versteeg, G.A. Negative Regulation of the Innate Immune Response through Proteasomal Degradation and Deubiquitination. Viruses 2021, 13, 584. [Google Scholar] [CrossRef]
- Hu, Z.; Xie, Y.; Lu, J.; Yang, J.; Zhang, J.; Jiang, H.; Li, H.; Zhang, Y.; Wu, D.; Zeng, K.; et al. VANGL2 inhibits antiviral IFN-I signaling by targeting TBK1 for autophagic degradation. Sci. Adv. 2023, 9, eadg2339. [Google Scholar] [CrossRef]
- Lin, M.; Zhao, Z.; Yang, Z.; Meng, Q.; Tan, P.; Xie, W.; Qin, Y.; Wang, R.F.; Cui, J. USP38 Inhibits Type I Interferon Signaling by Editing TBK1 Ubiquitination through NLRP4 Signalosome. Mol. Cell 2016, 64, 267–281. [Google Scholar] [CrossRef]
- Liu, D.; Sheng, C.; Gao, S.; Yao, C.; Li, J.; Jiang, W.; Chen, H.; Wu, J.; Pan, C.; Chen, S.; et al. SOCS3 Drives Proteasomal Degradation of TBK1 and Negatively Regulates Antiviral Innate Immunity. Mol. Cell. Biol. 2015, 35, 2400–2413. [Google Scholar] [CrossRef]
- Yang, Y.; Cao, X.; Huang, L.; Yang, A. RNF19a inhibits antiviral immune response to RNA viruses through degradation of TBK1. Mol. Immunol. 2022, 143, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Georgana, I.; Hosmillo, M.; Jahun, A.S.; Emmott, E.; Sorgeloos, F.; Cho, K.O.; Goodfellow, I.G. Porcine Sapovirus Protease Controls the Innate Immune Response and Targets TBK1. Viruses 2024, 16, 247. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shi, H.; Rigolet, P.; Wu, N.; Zhu, L.; Xi, X.G.; Vabret, A.; Wang, X.; Wang, T. Nsp1 proteins of group I and SARS coronaviruses share structural and functional similarities. Infect. Genet. Evol. 2010, 10, 919–924. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Ye, G.; Deng, F.; Wang, G.; Cui, M.; Fang, L.; Xiao, S.; Fu, Z.F.; Peng, G. Structural Basis for the Inhibition of Host Gene Expression by Porcine Epidemic Diarrhea Virus nsp1. J. Virol. 2018, 92, e01896-17. [Google Scholar] [CrossRef]
- Shen, Z.; Wang, G.; Yang, Y.; Shi, J.; Fang, L.; Li, F.; Xiao, S.; Fu, Z.F.; Peng, G. A conserved region of nonstructural protein 1 from alphacoronaviruses inhibits host gene expression and is critical for viral virulence. J. Biol. Chem. 2019, 294, 13606–13618. [Google Scholar] [CrossRef]
- Maurina, S.F.; O’Sullivan, J.P.; Sharma, G.; Pineda Rodriguez, D.C.; MacFadden, A.; Cendali, F.; Henen, M.A.; Vögeli, B.; Kieft, J.S.; Glasgow, A.; et al. An Evolutionarily Conserved Strategy for Ribosome Binding and Host Translation Inhibition by β-coronavirus Non-structural Protein 1. J. Mol. Biol. 2023, 435, 168259. [Google Scholar] [CrossRef]
- Eriksson, K.K.; Makia, D.; Thiel, V. Generation of recombinant coronaviruses using vaccinia virus as the cloning vector and stable cell lines containing coronaviral replicon RNAs. Methods Mol. Biol. 2008, 454, 237–254. [Google Scholar] [CrossRef]
- Fitzgerald, K.A.; McWhirter, S.M.; Faia, K.L.; Rowe, D.C.; Latz, E.; Golenbock, D.T.; Coyle, A.J.; Liao, S.M.; Maniatis, T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 2003, 4, 491–496. [Google Scholar] [CrossRef]
- Martiáñez-Vendrell, X.; Bloeme-Ter Horst, J.; Hutchinson, R.; Guy, C.; Bowie, A.G.; Kikkert, M. Human Coronavirus 229E Infection Inactivates Pyroptosis Executioner Gasdermin D but Ultimately Leads to Lytic Cell Death Partly Mediated by Gasdermin E. Viruses 2024, 16, 898. [Google Scholar] [CrossRef]
- Gack, M.U.; Shin, Y.C.; Joo, C.H.; Urano, T.; Liang, C.; Sun, L.; Takeuchi, O.; Akira, S.; Chen, Z.; Inoue, S.; et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 2007, 446, 916–920. [Google Scholar] [CrossRef]
- van Kasteren, P.B.; Beugeling, C.; Ninaber, D.K.; Frias-Staheli, N.; van Boheemen, S.; García-Sastre, A.; Snijder, E.J.; Kikkert, M. Arterivirus and nairovirus ovarian tumor domain-containing Deubiquitinases target activated RIG-I to control innate immune signaling. J. Virol. 2012, 86, 773–785. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martiáñez-Vendrell, X.; van Kasteren, P.B.; Myeni, S.K.; Kikkert, M. HCoV-229E Mpro Suppresses RLR-Mediated Innate Immune Signalling Through Cleavage of NEMO and Through Other Mechanisms. Int. J. Mol. Sci. 2025, 26, 1197. https://doi.org/10.3390/ijms26031197
Martiáñez-Vendrell X, van Kasteren PB, Myeni SK, Kikkert M. HCoV-229E Mpro Suppresses RLR-Mediated Innate Immune Signalling Through Cleavage of NEMO and Through Other Mechanisms. International Journal of Molecular Sciences. 2025; 26(3):1197. https://doi.org/10.3390/ijms26031197
Chicago/Turabian StyleMartiáñez-Vendrell, Xavier, Puck B. van Kasteren, Sebenzile K. Myeni, and Marjolein Kikkert. 2025. "HCoV-229E Mpro Suppresses RLR-Mediated Innate Immune Signalling Through Cleavage of NEMO and Through Other Mechanisms" International Journal of Molecular Sciences 26, no. 3: 1197. https://doi.org/10.3390/ijms26031197
APA StyleMartiáñez-Vendrell, X., van Kasteren, P. B., Myeni, S. K., & Kikkert, M. (2025). HCoV-229E Mpro Suppresses RLR-Mediated Innate Immune Signalling Through Cleavage of NEMO and Through Other Mechanisms. International Journal of Molecular Sciences, 26(3), 1197. https://doi.org/10.3390/ijms26031197