
Immune Cell Interactions and Immune Checkpoints in the Tumor Microenvironment of Gastric Cancer


 , , , , and
, , , , and 
Abstract
1. Introduction
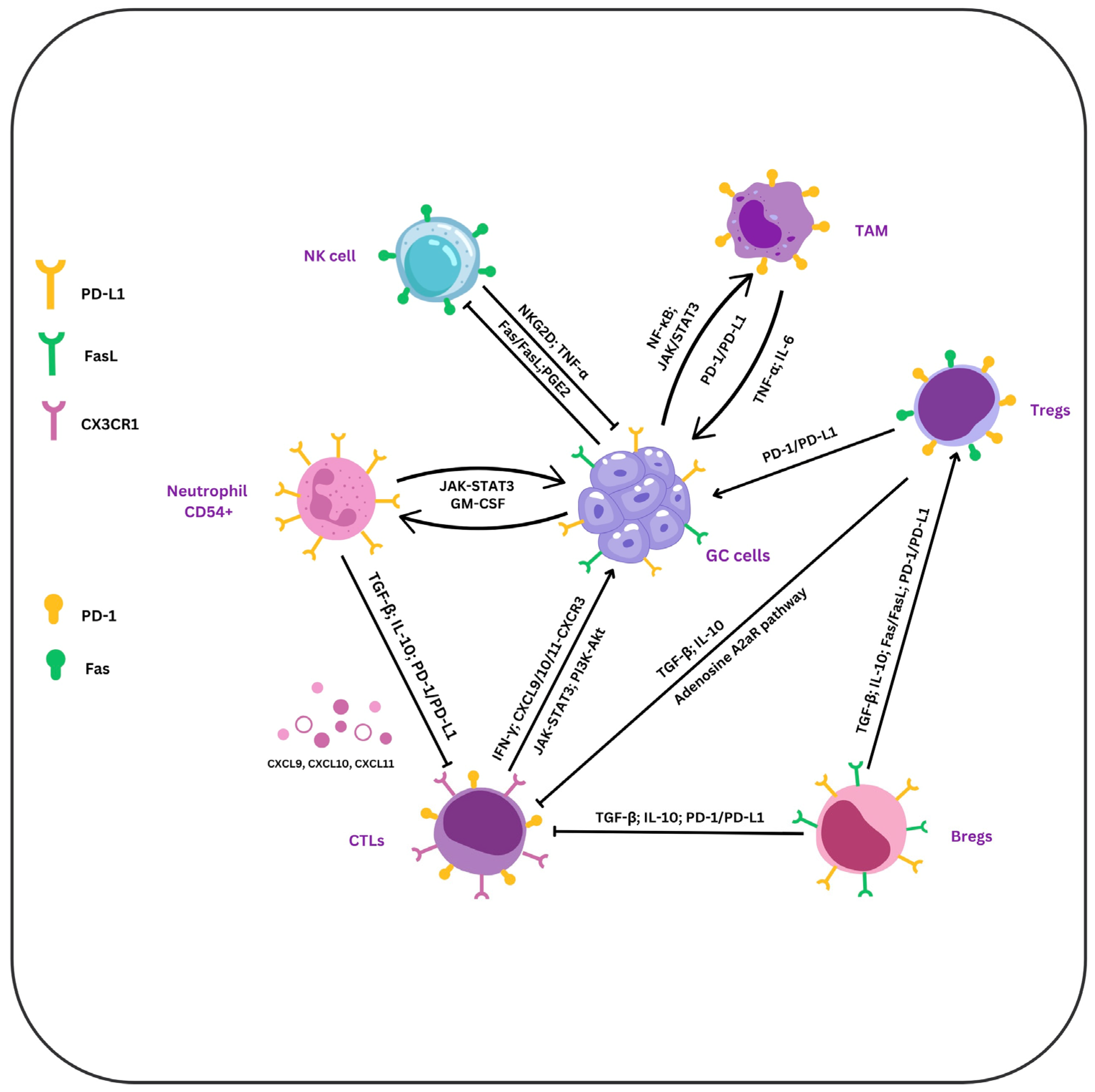
2. TILs
2.1. T Lymphocytes
2.1.1. CD8+ T Cells
The Link Between Tumor-Elicited Immunosuppression and CTLs
2.1.2. CD4+ T Cells
2.2. B Lymphocytes
2.2.1. Regulatory B Cells (Bregs)
2.2.2. Tertiary Lymphoid Structures (TLSs)
2.3. NK Cells
3. TAMs
4. PD-1, PD-L1, and PD-L2
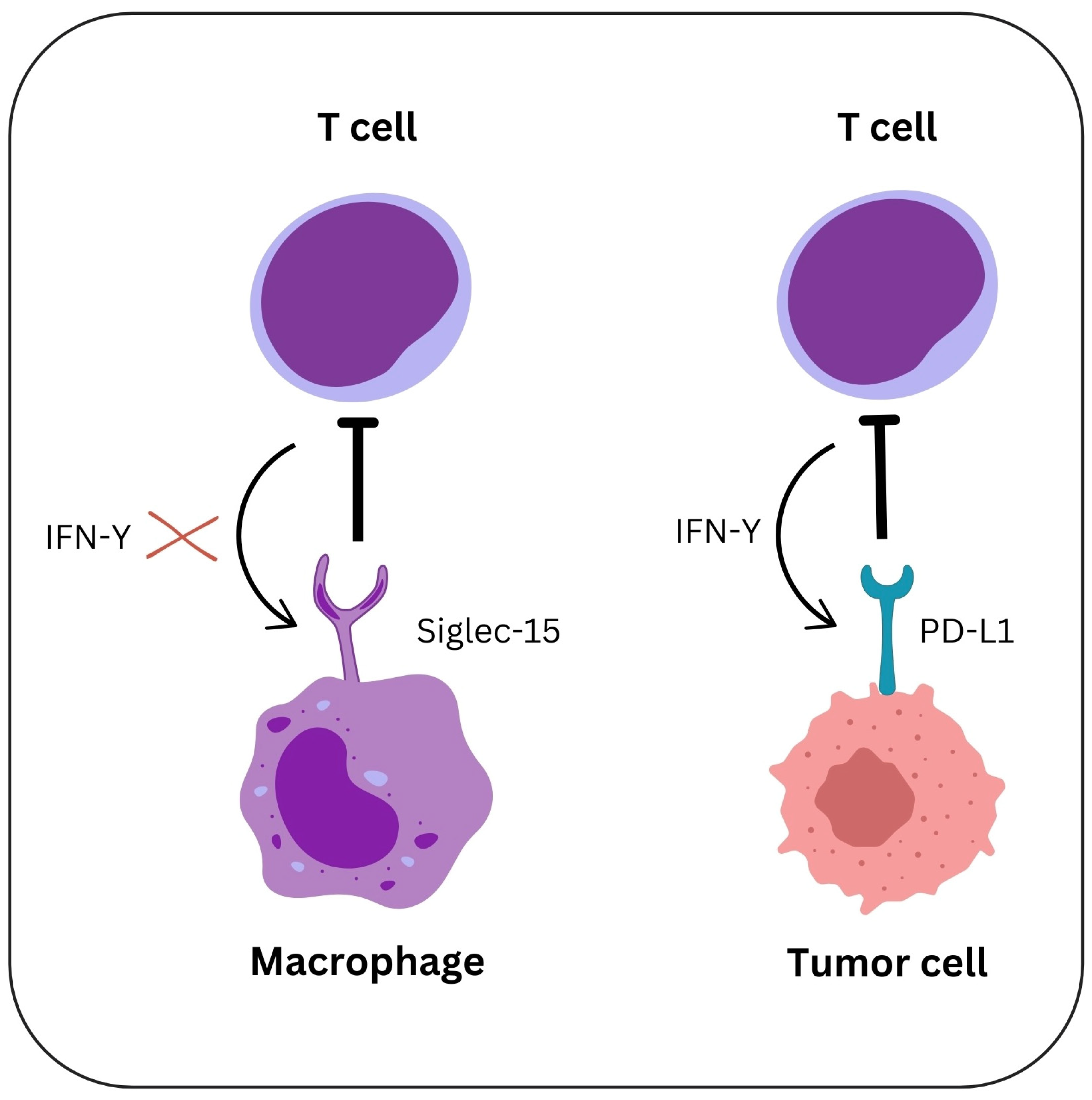
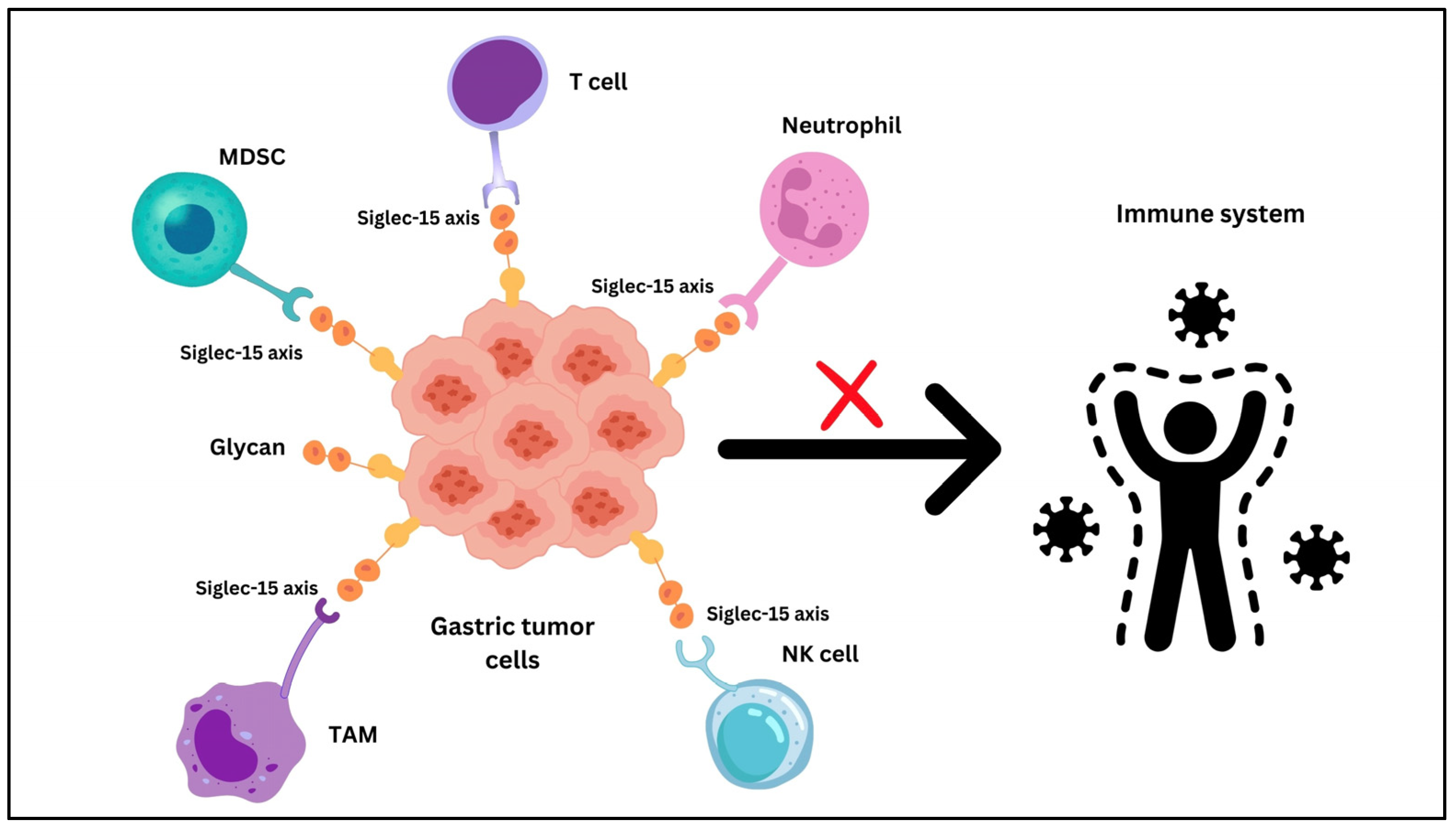
5. Siglec-15
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| A2AR—Adenosine A2A Receptor |
| APC—Antigen Presenting Cell |
| ARID1A—AT-rich Interactive Domain-containing protein 1A |
| Bregs—B Regulatory Cells |
| CAR NK—Chimeric Antigen Receptor Natural killer cells |
| CIN—Chromosomal Instability |
| CTLs—Cytotoxic T Lymphocytes |
| CXCL9, CXCL10, and CXCL11—C-X-C Motif Chemokine Ligand 9, 10, and 11 |
| CXCR3—C-X-C Motif Chemokine Receptor 3 |
| DAP12—DNAX-Activation Protein of 12 kDa |
| DCs—Dendritic Cells |
| DNA—Deoxyribonucleic Acid |
| EBV—Epstein–Barr Virus |
| EGFR—epidermal growth factor receptor |
| Fas/FasL—Fas/Fas Ligand |
| FDC—follicular dendritic cell |
| FOXP3—Forkhead Box Protein P3 |
| GC—Gastric Cancer |
| GS—Genomic Stable |
| H. pylori—Helicobacter pylori |
| HER-2—Human Epidermal Growth Factor Receptor 2 |
| HLA—Human Leucocyte Antigen |
| IFN-γ—Interferon-gamma |
| Ig—Immunoglobulin |
| IL-10—Interleukin-10 |
| IL-17—interleukin-17 |
| IL-21—interleukin-21 |
| IL-6—Interleukin-6 |
| IL-8—Interleukin-8 |
| JAK/STAT3—Janus Kinase/Signal Transducer and Activator of Transcription 3 |
| mAbs—monoclonal antibodies |
| MDSCs—Myeloid-Derived Suppressor Cells |
| MHC—major histocompatibility complex |
| MMR—Mismatch Repair |
| MSI—Microsatellite Instability |
| NF-κB/NFAT—Nuclear Factor kappa-light-chain-enhancer of activated B cells/Nuclear Factor of Activated T-cells |
| NK—Natural Killer |
| NKG2D—Natural Killer Group 2 Member D |
| NSCLC—non-small cell lung cancer |
| PD-1—Programmed Cell Death-Protein-1 receptor |
| PD-L1—Programmed Cell Death Ligand-1 |
| PD-L2—Programmed Cell Death Ligand-2 |
| PGE2—Prostaglandin E2 |
| PI3K-AKT—Phosphoinositide 3-Kinase-Protein Kinase B |
| RA—Rheumatoid Arthritis |
| Sialyl-Tn—Sialylated Tn Antigen |
| Siglec-15—Sialic Acid-Binding Immunoglobulin-Like Lectin 15 |
| TAMs—Tumor-Associated Macrophages |
| TCR—T Cell Receptor |
| TGF-β—Transforming Growth Factor-beta |
| Th—T helper |
| TILs—Tumor-Infiltrating Lymphocytes |
| TLSs—Tertiary lymphoid structures |
| TME—Tumor Microenvironment |
| TNF-α—Tumor Necrosis Factor-alpha |
| Tregs—Regulatory T Cells |
| TRM—Tissue-Resident Memory |
| VEGF—Vascular Endothelial Growth Factor |
References
- Filho, A.M.; Laversanne, M.; Ferlay, J.; Colombet, M.; Piñeros, M.; Znaor, A.; Parkin, D.M.; Soerjomataram, I.; Bray, F. The GLOBOCAN 2022 cancer estimates: Data sources, methods, and a snapshot of the cancer burden worldwide. Int. J. Cancer 2024. [Google Scholar] [CrossRef] [PubMed]
- Cisło, M.; Filip, A.A.; Offerhaus, G.J.A.; Ciseł, B.; Rawicz-Pruszyński, K.; Skierucha, M.; Polkowski, W.P. Piotr Distinct molecular subtypes of gastric cancer: From Laurén to molecular pathology. Oncotarget 2018, 9, 19427–19442. Available online: https://www.oncotarget.com/article/24827/text/ (accessed on 27 December 2024). [CrossRef]
- Kim, M.; Seo, A.N. Molecular Pathology of Gastric Cancer. J. Gastric Cancer 2022, 22, 273–305. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Kim, W.G.; Kwon, C.H.; Park, D.Y. Differences in immune contextures among different molecular subtypes of gastric cancer and their prognostic impact. Gastric Cancer 2019, 22, 1164–1175. [Google Scholar] [CrossRef]
- Wang, G.; Yang, L.; Wang, Y.; Hu, R.; Zhang, K.; Guo, T.; Chen, B.; Jiang, X.; Cui, R. Characterization of Immune-Related Molecular Subtypes and a Prognostic Signature Correlating with the Response to Immunotherapy in Patients with Gastric Cancer. Front. Immunol. 2022, 13, 939836. [Google Scholar] [CrossRef] [PubMed]
- Ma, E.S.; Wang, Z.X.; Zhu, M.Q.; Zhao, J. Immune evasion mechanisms and therapeutic strategies in gastric cancer. World J. Gastrointest. Oncol. 2022, 14, 216–229. [Google Scholar] [CrossRef]
- Wang, J.; Liu, T.; Huang, T.; Shang, M.; Wang, X. The mechanisms on evasion of anti-tumor immune responses in gastric cancer. Front. Oncol. 2022, 12, 943806. [Google Scholar] [CrossRef]
- Tian, C.; Jing, H.; Wang, C.; Wang, W.; Cui, Y.; Chen, J.; Sha, D. Prognostic role of tumour-infiltrating lymphocytes assessed by H&E-stained section in gastric cancer: A systematic review and meta-analysis. Br. Med. J. Open 2021, 11, e044163. [Google Scholar] [CrossRef]
- Cao, X.; Kang, Y.; Tai, P.; Zhang, P.; Lin, X.; Xu, F.; Nie, Z.; He, B. Prognostic role of tumor-infiltrating lymphocytes in gastric cancer: A systematic review and meta-analysis. Clin. Res. Hepatol. Gastroenterol. 2024, 49, 102510. [Google Scholar] [CrossRef]
- Chiu, Y.M.; Tsai, C.L.; Kao, J.T.; Hsieh, C.T.; Shieh, D.C.; Lee, Y.J.; Tsay, G.J.; Cheng, K.S.; Wu, Y.Y. PD-1 and PD-L1 Up-regulation Promotes T-cell Apoptosis in Gastric Adenocarcinoma. Anticancer Res. 2018, 38, 2069–2078. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Lu, Q.; Sanmamed, M.F.; Wang, J. Siglec-15 as an Emerging Target for Next-generation Cancer Immunotherapy. Clin. Cancer Res. 2021, 27, 680–688. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Guo, Y.; Li, B.; Shen, M.; Yi, Y.; Li, L.; Zhao, X.; Yang, L. Siglec-15 on macrophages suppress the immune microenvironment in patients with PD-L1 negative non-metastasis lung adenocarcinoma. Cancer Gene Ther. 2024, 31, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Ren, X. Immunosuppressive checkpoint Siglec-15: A vital new piece of the cancer immunotherapy jigsaw puzzle. Cancer Biol. Med. 2019, 16, 205–210. [Google Scholar] [CrossRef]
- Chen, X.; Mo, S.; Zhang, Y.; Ma, H.; Lu, Z.; Yu, S.; Chen, J. Analysis of a novel immune checkpoint, Siglec-15, in pancreatic ductal adenocarcinoma. J. Pathol. Clin. Res. 2022, 8, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Paijens, S.T.; Vledder, A.; de Bruyn, M.; Nijman, H.W. Tumor-infiltrating lymphocytes in the immunotherapy era. Cell. Mol. Immunol. 2021, 18, 842–859. [Google Scholar] [CrossRef] [PubMed]
- Frankowska, K.; Zarobkiewicz, M.; Dąbrowska, I.; Bojarska-Junak, A. Tumor infiltrating lymphocytes and radiological picture of the tumor. Med. Oncol. 2023, 40, 176. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.P.; Chen, J.N.; Xiao, L.; He, Q.; Feng, Z.Y.; Zhang, Z.G.; Liu, J.P.; Wei, H.B.; Shao, C.K. The implication of tumor-infiltrating lymphocytes in Epstein-Barr virus-associated gastric carcinoma. Hum. Pathol. 2019, 85, 82–91. [Google Scholar] [CrossRef]
- Ma, J.; Li, J.; Hao, Y.; Nie, Y.; Li, Z.; Qian, M.; Liang, Q.; Yu, J.; Zeng, M.; Wu, K. Differentiated tumor immune micro-environment of Epstein–Barr virus-associated and negative gastric cancer: Implication in prognosis and immunotherapy. Oncotarget 2017, 8, 67094–67103. [Google Scholar] [CrossRef] [PubMed]
- Knochelmann, H.M.; Dwyer, C.J.; Bailey, S.R.; Amaya, S.M.; Elston, D.M.; Mazza-McCrann, J.M.; Paulos, C.M. When worlds collide: Th17 and Treg cells in cancer and autoimmunity. Cell. Mol. Immunol. 2018, 15, 458–469. [Google Scholar] [CrossRef] [PubMed]
- Wegrzyn, A.S.; Kedzierska, A.E.; Obojski, A. Identification and classification of distinct surface markers of T regulatory cells. Front. Immunol. 2023, 13, 1055805. [Google Scholar] [CrossRef] [PubMed]
- Alcover, A.; Alarcón, B.; Di Bartolo, V. Cell Biology of T Cell Receptor Expression and Regulation. Annu. Rev. Immunol. 2018, 36, 103–125. [Google Scholar] [CrossRef]
- Li, F.; Li, C.; Cai, X.; Xie, Z.; Zhou, L.; Cheng, B.; Zhong, R.; Xiong, S.; Li, J.; Chen, Z.; et al. The association between CD8+ tumor-infiltrating lymphocytes and the clinical outcome of cancer immunotherapy: A systematic review and meta-analysis. EClinicalMedicine 2021, 41, 101134. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, J. Anatomy of a murder: How cytotoxic T cells and NK cells are activated, develop, and eliminate their targets. Immunol. Rev. 2010, 235, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Jameson, S.C.; Masopust, D. Understanding Subset Diversity in T Cell Memory. Immunity 2018, 48, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Golubovskaya, V.; Wu, L. Different Subsets of T Cells, Memory, Effector Functions, and CAR-T Immunotherapy. Cancers 2016, 8, 36. [Google Scholar] [CrossRef]
- Kaech, S.M.; Wherry, E.J.; Ahmed, R. Effector and memory T-cell differentiation: Implications for vaccine development. Nat. Rev. Immunol. 2002, 2, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Damei, I.; Trickovic, T.; Mami-Chouaib, F.; Corgnac, S. Tumor-resident memory T cells as a biomarker of the response to cancer immunotherapy. Front. Immunol. 2023, 14, 1205984. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Bergsbaken, T.; Edelblum, K.L. The multifunctional nature of CD103 (αEβ7 integrin) signaling in tissue-resident lymphocytes. Am. J. Physiol. Cell Physiol. 2022, 323, C1161–C1167. [Google Scholar] [CrossRef]
- Sullivan, P.M.; Reed, S.J.; Kalia, V.; Sarkar, S. Solid Tumor Microenvironment Can Harbor and Support Functional Properties of Memory T Cells. Front. Immunol. 2021, 12, 706150. [Google Scholar] [CrossRef] [PubMed]
- Borràs, D.M.; Verbandt, S.; Ausserhofer, M.; Sturm, G.; Lim, J.; Verge, G.A.; Vanmeerbeek, I.; Laureano, R.S.; Govaerts, J.; Sprooten, J.; et al. Single cell dynamics of tumor specificity vs bystander activity in CD8+ T cells define the diverse immune landscapes in colorectal cancer. Cell Discov. 2023, 9, 114. [Google Scholar] [CrossRef]
- Beumer-Chuwonpad, A.; Taggenbrock, R.L.R.E.; Ngo, T.A.; van Gisbergen, K.P.J.M. The Potential of Tissue-Resident Memory T Cells for Adoptive Immunotherapy against Cancer. Cells 2021, 10, 2234. [Google Scholar] [CrossRef]
- Simoni, Y.; Becht, E.; Fehlings, M.; Loh, C.Y.; Koo, S.L.; Teng, K.W.W.; Yeong, J.P.S.; Nahar, R.; Zhang, T.; Kared, H.; et al. Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 2018, 557, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Hu, J.; Ping, Y.; Xu, L.; Liao, G.; Jiang, Z.; Pang, B.; Sun, S.; Zhang, Y.; Xiao, Y.; et al. Single-Cell Transcriptomic Analysis Reveals a Tumor-Reactive T Cell Signature Associated with Clinical Outcome and Immunotherapy Response in Melanoma. Front. Immunol. 2021, 12, 758288. [Google Scholar] [CrossRef] [PubMed]
- Martín-Sierra, C.; Martins, R.; Laranjeira, P.; Coucelo, M.; Abrantes, A.M.; Oliveira, R.C.; Tralhão, J.G.; Botelho, M.F.; Furtado, E.; Domingues, M.R.; et al. Functional and Phenotypic Characterization of Tumor-Infiltrating Leukocyte Subsets and Their Contribution to the Pathogenesis of Hepatocellular Carcinoma and Cholangiocarcinoma. Transl. Oncol. 2019, 12, 1468–1479. [Google Scholar] [CrossRef]
- Philip, M.; Schietinger, A. CD8+ T cell differentiation and dysfunction in cancer. Nat. Rev. Immunol. 2022, 22, 209–223. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Jiang, A. Dendritic Cells and CD8 T Cell Immunity in Tumor Microenvironment. Front. Immunol. 2018, 9, 3059. [Google Scholar] [CrossRef] [PubMed]
- Svensson-Arvelund, J.; Cuadrado-Castano, S.; Pantsulaia, G.; Kim, K.; Aleynick, M.; Hammerich, L.; Upadhyay, R.; Yellin, M.; Marsh, H.; Oreper, D.; et al. Expanding cross-presenting dendritic cells enhances oncolytic virotherapy and is critical for long-term anti-tumor immunity. Nat. Commun. 2022, 13, 7149. [Google Scholar] [CrossRef] [PubMed]
- Baessler, A.; Vignali, D.A.A. T Cell Exhaustion. Annu. Rev. Immunol. 2024, 42, 179–206. [Google Scholar] [CrossRef] [PubMed]
- Blank, C.U.; Haining, W.N.; Held, W.; Hogan, P.G.; Kallies, A.; Lugli, E.; Lynn, R.C.; Philip, M.; Rao, A.; Restifo, N.P.; et al. Defining ‘T cell exhaustion’. Nat. Rev. Immunol. 2019, 19, 665–674. [Google Scholar] [CrossRef]
- Zhang, J.; Lei, F.; Tan, H. The development of CD8 T-cell exhaustion heterogeneity and the therapeutic potentials in cancer. Front. Immunol. 2023, 14, 1166128. [Google Scholar] [CrossRef]
- Han, S.H.; Kim, M.; Chung, Y.R.; Woo, J.W.; Choi, H.Y.; Park, S.Y. Expression of HLA class I is associated with immune cell infiltration and patient outcome in breast cancer. Sci. Rep. 2022, 12, 20367. [Google Scholar] [CrossRef] [PubMed]
- Rodems, T.S.; Heninger, E.; Stahlfeld, C.N.; Gilsdorf, C.S.; Carlson, K.N.; Kircher, M.R.; Singh, A.; Krueger, T.E.; Beebe, D.J.; Jarrard, D.F.; et al. Reversible epigenetic alterations regulate class I HLA loss in prostate cancer. Commun. Biol. 2022, 5, 897. [Google Scholar] [CrossRef] [PubMed]
- Palma, M.B.; Tronik-Le Roux, D.; Amín, G.; Castañeda, S.; Möbbs, A.M.; Scarafia, M.A.; La Greca, A.; Daouya, M.; Poras, I.; Inda, A.M.; et al. HLA-G gene editing in tumor cell lines as a novel alternative in cancer immunotherapy. Sci. Rep. 2021, 11, 22158. [Google Scholar] [CrossRef]
- Thelen, M.; Wennhold, K.; Lehmann, J.; Garcia-Marquez, M.; Klein, S.; Kochen, E.; Lohneis, P.; Lechner, A.; Wagener-Ryczek, S.; Plum, P.S.; et al. Cancer-specific immune evasion and substantial heterogeneity within cancer types provide evidence for personalized immunotherapy. NPJ Precis. Oncol. 2021, 5, 52. [Google Scholar] [CrossRef]
- Prendergast, G.C.; Mondal, A.; Dey, S.; Laury-Kleintop, L.D.; Muller, A.J. Inflammatory Reprogramming with IDO1 Inhibitors: Turning Immunologically Unresponsive ‘Cold’ Tumors ‘Hot’. Trends Cancer 2018, 4, 38–58. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Li, Q.; Chen, J.; Liu, Y.; Zhao, X.; Tan, B.; Ai, J.; Zhang, Z.; Song, J.; Shan, B. Prevalence of Th17 and Treg cells in gastric cancer patients and its correlation with clinical parameters. Oncol. Rep. 2013, 30, 1215–1222. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Zhao, J.; Xu, X.; Zhang, D.; Shen, H.; Wang, S. Role of adenosine A2a receptor in cancers and autoimmune diseases. Immun. Inflamm. Dis. 2023, 11, e826. [Google Scholar] [CrossRef]
- Meng, X.; Zhu, S.; Dong, Q.; Zhang, S.; Ma, J.; Zhou, C. Expression of Th17/Treg related molecules in gastric cancer tissues. Turk. J. Gastroenterol. 2018, 29, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Kindlund, B.; Sjöling, Å.; Yakkala, C.; Adamsson, J.; Janzon, A.; Hansson, L.E.; Hermansson, M.; Janson, P.; Winqvist, O.; Lundin, S.B. CD4+ regulatory T cells in gastric cancer mucosa are proliferating and express high levels of IL-10 but little TGF-β. Gastric Cancer 2017, 20, 116–125. [Google Scholar] [CrossRef]
- Ng, T.H.; Britton, G.J.; Hill, E.V.; Verhagen, J.; Burton, B.R.; Wraith, D.C. Regulation of adaptive immunity; the role of interleukin-10. Front. Immunol. 2013, 4, 129. [Google Scholar] [CrossRef]
- Wang, Y.A.; Li, X.L.; Mo, Y.Z.; Fan, C.M.; Tang, L.; Xiong, F.; Guo, C.; Xiang, B.; Zhou, M.; Ma, J.; et al. Effects of tumor metabolic microenvironment on regulatory T cells. Mol. Cancer 2018, 17, 168. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; He, Y.; Huang, J.; Tao, Y.; Liu, S. Metabolism of Dendritic Cells in Tumor Microenvironment: For Immuno-therapy. Front. Immunol. 2021, 12, 613492. [Google Scholar] [CrossRef]
- Wu, M.S.; Lin, J.T.; Hsu, P.N.; Lin, C.Y.; Hsieh, Y.T.; Chiu, Y.H.; Hsueh, P.R.; Liao, K.W. Preferential induction of transforming growth factor-beta production in gastric epithelial cells and monocytes by Helicobacter pylori soluble proteins. J. Infect. Dis. 2007, 196, 1386–1393. [Google Scholar] [CrossRef] [PubMed]
- Kao, J.Y.; Zhang, M.; Miller, M.J.; Mills, J.C.; Wang, B.; Liu, M.; Eaton, K.A.; Zou, W.; Berndt, B.E.; Cole, T.S.; et al. Helicobacter pylori immune escape is mediated by dendritic cell-induced Treg skewing and Th17 suppression in mice. Gastroenterology 2010, 138, 1046–1054. [Google Scholar] [CrossRef]
- Larussa, T.; Leone, I.; Suraci, E.; Imeneo, M.; Luzza, F. Helicobacter pylori and T Helper Cells: Mechanisms of Immune Escape and Tolerance. J. Immunol. Res. 2015, 2015, 981328. [Google Scholar] [CrossRef]
- Rezalotfi, A.; Ahmadian, E.; Aazami, H.; Solgi, G.; Ebrahimi, M. Gastric Cancer Stem Cells Effect on Th17/Treg Balance; A Bench to Beside Perspective. Front. Oncol. 2019, 9, 226. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Huang, X.; Han, X.; Zhang, J.; Gao, L.; Chen, H. IL-17A in gastric carcinogenesis: Good or bad? Front. Immunol. 2024, 15, 1501293. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, Q.; Cheng, X.; Zhang, B.; Lin, J.; Tang, Y.; Li, F.; Yang, C.S.; Wang, T.C.; Tu, S. Bone Marrow-Derived Myofibroblasts Promote Gastric Cancer Metastasis by Activating TGF-β1 and IL-6/STAT3 Signalling Loop. OncoTargets Ther. 2020, 13, 10567–10580. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Chen, X.; Herjan, T.; Li, X. The role of interleukin-17 in tumor development and progression. J. Exp. Med. 2020, 217, e20190297. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, T.; Kono, K.; Mizukami, Y.; Kawaguchi, Y.; Mimura, K.; Watanabe, M.; Izawa, S.; Fujii, H. Distribution of Th17 cells and FoxP3(+) regulatory T cells in tumor-infiltrating lymphocytes, tumor-draining lymph nodes and peripheral blood lymphocytes in patients with gastric cancer. Cancer Sci. 2010, 101, 1947–1954. [Google Scholar] [CrossRef] [PubMed]
- Downs-Canner, S.M.; Meier, J.; Vincent, B.G.; Serody, J.S. B Cell Function in the Tumor Microenvironment. Annu. Rev. Immunol. 2022, 40, 169–193. [Google Scholar] [CrossRef] [PubMed]
- Zhang, E.; Ding, C.; Li, S.; Zhou, X.; Aikemu, B.; Fan, X.; Sun, J.; Zheng, M.; Yang, X. Roles and mechanisms of tumour-infiltrating B cells in human cancer: A new force in immunotherapy. Biomark. Res. 2023, 11, 28. [Google Scholar] [CrossRef]
- Michaud, D.; Steward, C.R.; Mirlekar, B.; Pylayeva-Gupta, Y. Regulatory B cells in cancer. Immunol. Rev. 2021, 299, 74–92. [Google Scholar] [CrossRef] [PubMed]
- Catalán, D.; Mansilla, M.A.; Ferrier, A.; Soto, L.; Oleinika, K.; Aguillón, J.C.; Aravena, O. Immunosuppressive Mechanisms of Regulatory B Cells. Front. Immunol. 2021, 12, 611795. [Google Scholar] [CrossRef] [PubMed]
- Sarvaria, A.; Madrigal, J.A.; Saudemont, A. B cell regulation in cancer and anti-tumor immunity. Cell. Mol. Immunol. 2017, 14, 662–674. [Google Scholar] [CrossRef]
- Moreira, H.; Dobosz, A.; Cwynar-Zając, Ł.; Nowak, P.; Czyżewski, M.; Barg, M.; Reichert, P.; Królikowska, A.; Barg, E. Unraveling the role of Breg cells in digestive tract cancer and infectious immunity. Front. Immunol. 2022, 13, 981847. [Google Scholar] [CrossRef] [PubMed]
- Sautès-Fridman, C.; Lawand, M.; Giraldo, N.A.; Kaplon, H.; Germain, C.; Fridman, W.H.; Dieu-Nosjean, M.C. Tertiary Lymphoid Structures in Cancers: Prognostic Value, Regulation, and Manipulation for Therapeutic Intervention. Front. Immunol. 2016, 7, 407. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wu, S. Tertiary lymphoid structures are critical for cancer prognosis and therapeutic response. Front. Immunol. 2023, 13, 1063711. [Google Scholar] [CrossRef]
- Yamakoshi, Y.; Tanaka, H.; Sakimura, C.; Deguchi, S.; Mori, T.; Tamura, T.; Toyokawa, T.; Muguruma, K.; Hirakawa, K.; Ohira, M. Immunological potential of tertiary lymphoid structures surrounding the primary tumor in gastric cancer. Int. J. Oncol. 2020, 57, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Groen-van Schooten, T.S.; Franco Fernandez, R.; van Grieken, N.C.T.; Bos, E.N.; Seidel, J.; Saris, J.; Martínez-Ciarpaglini, C.; Fleitas, T.C.; Thommen, D.S.; de Gruijl, T.D.; et al. Mapping the complexity and diversity of tertiary lymphoid structures in primary and peritoneal metastatic gastric cancer. J. Immunother. Cancer 2024, 12, e009243. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Wu, X.; Zhang, C.; Liang, Y.; Cheng, S.; Zhang, H.; Shen, L.; Chen, Y. Analyzing the associations between tertiary lymphoid structures and postoperative prognosis, along with immunotherapy response in gastric cancer: Findings from pooled cohort studies. J. Cancer Res. Clin. Oncol. 2024, 150, 153. [Google Scholar] [CrossRef] [PubMed]
- Tan, R.; Nie, M.; Long, W. The role of B cells in cancer development. Front. Oncol. 2022, 12, 958756. [Google Scholar] [CrossRef]
- Yang, Y.; Chen, X.; Pan, J.; Ning, H.; Zhang, Y.; Bo, Y.; Ren, X.; Li, J.; Qin, S.; Wang, D.; et al. Pan-cancer single-cell dissection reveals phenotypically distinct B cell subtypes. Cell 2024, 187, 4790–4811.e22. [Google Scholar] [CrossRef]
- Wolf, N.K.; Kissiov, D.U.; Raulet, D.H. Roles of natural killer cells in immunity to cancer, and applications to immunotherapy. Nature reviews. Immunology 2023, 23, 90–105. [Google Scholar] [CrossRef]
- Sanseviero, E. NK Cell-Fc Receptors Advance Tumor Immunotherapy. J. Clin. Med. 2019, 8, 1667. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Wei, Y. Therapeutic Potential of Natural Killer Cells in Gastric Cancer. Front. Immunol. 2019, 9, 3095. [Google Scholar] [CrossRef] [PubMed]
- Yajima, T.; Hoshino, K.; Muranushi, R.; Mogi, A.; Onozato, R.; Yamaki, E.; Kosaka, T.; Tanaka, S.; Shirabe, K.; Yoshikai, Y.; et al. Fas/FasL signaling is critical for the survival of exhausted antigen-specific CD8+ T cells during tumor immune response. Mol. Immunol. 2019, 107, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.Y.; Feng, Y.; Mao, Q.S.; Ma, P.; Liu, J.Z.; Lu, W.; Liu, Y.F.; Chen, X.; Hu, Y.L.; Xue, W.J. Diagnostic and prognostic value of the peripheral natural killer cell levels in gastric cancer. Exp. Ther. Med. 2020, 20, 3816–3822. [Google Scholar] [CrossRef] [PubMed]
- Saito, H.; Osaki, T.; Ikeguchi, M. Decreased NKG2D expression on NK cells correlates with impaired NK cell function in patients with gastric cancer. Gastric Cancer 2012, 15, 27–33. [Google Scholar] [CrossRef]
- Li, T.; Zhang, Q.; Jiang, Y.; Yu, J.; Hu, Y.; Mou, T.; Chen, G.; Li, G. Gastric cancer cells inhibit natural killer cell proliferation and induce apoptosis via prostaglandin E2. Oncoimmunology 2015, 5, e1069936. [Google Scholar] [CrossRef] [PubMed]
- Rafei, H.; Daher, M.; Rezvani, K. Chimeric antigen receptor (CAR) natural killer (NK)-cell therapy: Leveraging the power of innate immunity. Br. J. Haematol. 2021, 193, 216–230. [Google Scholar] [CrossRef] [PubMed]
- Scheck, M.K.; Hofheinz, R.D.; Lorenzen, S. HER2-Positive Gastric Cancer and Antibody Treatment: State of the Art and Future Developments. Cancers 2024, 16, 1336. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Xu, X.; Wei, S.; Jiang, P.; Xue, L.; Wang, J. Tumor-associated macrophages: Potential therapeutic strategies and future prospects in cancer. J. Immunother. Cancer 2021, 9, e001341. [Google Scholar] [CrossRef] [PubMed]
- Yerolatsite, M.; Torounidou, N.; Gogadis, A.; Kapoulitsa, F.; Ntellas, P.; Lampri, E.; Tolia, M.; Batistatou, A.; Katsanos, K.; Mauri, D. TAMs and PD-1 Networking in Gastric Cancer: A Review of the Literature. Cancers 2023, 16, 196. [Google Scholar] [CrossRef] [PubMed]
- Takamiya, R.; Ohtsubo, K.; Takamatsu, S.; Taniguchi, N.; Angata, T. The interaction between Siglec-15 and tumor-associated sialyl-Tn antigen enhances TGF-β secretion from monocytes/macrophages through the DAP12-Syk pathway. Glycobiology 2013, 23, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Bai, R.; Li, Y.; Jian, L.; Yang, Y.; Zhao, L.; Wei, M. The hypoxia-driven crosstalk between tumor and tumor-associated macrophages: Mechanisms and clinical treatment strategies. Mol. Cancer 2022, 21, 177. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sun, J.; Liu, L.N.; Flies, D.B.; Nie, X.; Toki, M.; Zhang, J.; Song, C.; Zarr, M.; Zhou, X.; et al. Siglec-15 as an immune suppressor and potential target for normalization cancer immunotherapy. Nat. Med. 2019, 25, 656–666. [Google Scholar] [CrossRef]
- Kang, F.B.; Chen, W.; Wang, L.; Zhang, Y.Z. The diverse functions of Siglec-15 in bone remodeling and antitumor responses. Pharmacol. Res. 2020, 155, 104728. [Google Scholar] [CrossRef]
- Jiang, K.Y.; Qi, L.L.; Kang, F.B.; Wang, L. The intriguing roles of Siglec family members in the tumor microenvironment. Biomark. Res. 2022, 10, 22. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Yang, Z.; Wang, J.; Wang, Q.; Zhang, H.; Ju, X. Research progress on tumour associated macrophages in gastric cancer (Review). Oncol. Rep. 2021, 45, 35. [Google Scholar] [CrossRef]
- Kim, B.G.; Malek, E.; Choi, S.H.; Ignatz-Hoover, J.J.; Driscoll, J.J. Novel therapies emerging in oncology to target the TGF-β pathway. J. Hematol. Oncol. 2021, 14, 55. [Google Scholar] [CrossRef] [PubMed]
- Farshidpour, M.; Ahmed, M.; Junna, S.; Merchant, J.L. Myeloid-derived suppressor cells in gastrointestinal cancers: A systemic review. World J. Gastrointest. Oncol. 2021, 13, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Beury, D.W.; Parker, K.H.; Nyandjo, M.; Sinha, P.; Carter, K.A.; Ostrand-Rosenberg, S. Cross-talk among myeloid-derived suppressor cells, macrophages, and tumor cells impacts the inflammatory milieu of solid tumors. J. Leukoc. Biol. 2014, 96, 1109–1118. [Google Scholar] [CrossRef] [PubMed]
- Parvez, A.; Choudhary, F.; Mudgal, P.; Khan, R.; Qureshi, K.A.; Farooqi, H.; Aspatwar, A. PD-1 and PD-L1: Architects of immune symphony and immunotherapy breakthroughs in cancer treatment. Front. Immunol. 2023, 14, 1296341. [Google Scholar] [CrossRef] [PubMed]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.W.; Li, Y.; Yang, Y.; Yang, H.K.; Dong, J.M.; Xiao, Z.H.; He, X.; Guo, J.H.; Wang, R.Q.; Dai, B.; et al. Tumor immunotherapy resistance: Revealing the mechanism of PD-1/PD-L1-mediated tumor immune escape. Biomed. Pharmacother. 2024, 171, 116203. [Google Scholar] [CrossRef]
- Chen, J.; Jiang, C.C.; Jin, L.; Zhang, X.D. Regulation of PD-L1: A novel role of pro-survival signalling in cancer. Ann. Oncol. 2016, 27, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Song, T.L.; Nairismägi, M.L.; Laurensia, Y.; Lim, J.Q.; Tan, J.; Li, Z.M.; Pang, W.L.; Kizhakeyil, A.; Wijaya, G.C.; Huang, D.C.; et al. Oncogenic activation of the STAT3 pathway drives PD-L1 expression in natural killer/T-cell lymphoma. Blood 2018, 132, 1146–1158. [Google Scholar] [CrossRef] [PubMed]
- Mandai, M.; Hamanishi, J.; Abiko, K.; Matsumura, N.; Baba, T.; Konishi, I. Dual Faces of IFNγ in Cancer Progression: A Role of PD-L1 Induction in the Determination of Pro- and Antitumor Immunity. Clin. Cancer Res. 2016, 22, 2329–2334. [Google Scholar] [CrossRef]
- Mimura, K.; Teh, J.L.; Okayama, H.; Shiraishi, K.; Kua, L.F.; Koh, V.; Smoot, D.T.; Ashktorab, H.; Oike, T.; Suzuki, Y.; et al. PD-L1 expression is mainly regulated by interferon gamma associated with JAK-STAT pathway in gastric cancer. Cancer Sci. 2018, 109, 43–53. [Google Scholar] [CrossRef]
- Ju, X.; Zhang, H.; Zhou, Z.; Chen, M.; Wang, Q. Tumor-associated macrophages induce PD-L1 expression in gastric cancer cells through IL-6 and TNF-α signaling. Exp. Cell Res. 2020, 396, 112315. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, R.; Zhang, W.; Naseem, M.; Puccini, A.; Berger, M.D.; Soni, S.; McSkane, M.; Baba, H.; Lenz, H.J. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation—A target for novel cancer therapy. Cancer Treat. Rev. 2018, 63, 40–47. [Google Scholar] [CrossRef]
- Zhang, C.; Li, Z.; Xu, L.; Che, X.; Wen, T.; Fan, Y.; Li, C.; Wang, S.; Cheng, Y.; Wang, X.; et al. CXCL9/10/11, a regulator of PD-L1 expression in gastric cancer. BMC Cancer 2018, 18, 462. [Google Scholar] [CrossRef]
- Wang, T.T.; Zhao, Y.L.; Peng, L.S.; Chen, N.; Chen, W.; Lv, Y.P.; Mao, F.Y.; Zhang, J.Y.; Cheng, P.; Teng, Y.S.; et al. Tumour-activated neutrophils in gastric cancer foster immune suppression and disease progression through GM-CSF-PD-L1 pathway. Gut 2017, 66, 1900–1911. [Google Scholar] [CrossRef]
- Shan, Z.G.; Yan, Z.B.; Peng, L.S.; Cheng, P.; Teng, Y.S.; Mao, F.Y.; Fan, K.; Zhuang, Y.; Zhao, Y.L. Granulocyte-Macrophage Colony-Stimulating Factor-Activated Neutrophils Express B7-H4 That Correlates with Gastric Cancer Progression and Poor Patient Survival. J. Immunol. Res. 2021, 2021, 6613247. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J.; Lee, C.S. The Role of the AT-Rich Interaction Domain 1A Gene (ARID1A) in Human Carcinogenesis. Genes 2023, 15, 5. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.B.; Ahn, J.M.; Bae, W.J.; Sung, C.O.; Lee, D. Functional loss of ARID1A is tightly associated with high PD-L1 expression in gastric cancer. Int. J. Cancer 2019, 145, 916–926. [Google Scholar] [CrossRef] [PubMed]
- Nakano, H.; Saito, M.; Nakajima, S.; Saito, K.; Nakayama, Y.; Kase, K.; Yamada, L.; Kanke, Y.; Hanayama, H.; Onozawa, H.; et al. PD-L1 over-expression in EBV-positive gastric cancer is caused by unique genomic or epigenomic mechanisms. Sci. Rep. 2021, 11, 1982. [Google Scholar] [CrossRef] [PubMed]
- Saito, R.; Abe, H.; Kunita, A.; Yamashita, H.; Seto, Y.; Fukayama, M. Overexpression and gene amplification of PD-L1 in cancer cells and PD-L1+ immune cells in Epstein-Barr virus-associated gastric cancer: The prognostic implications. Mod. Pathol. 2017, 30, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Dong, Y.; Liu, H.; Wang, Y.; Zhao, S.; Xuan, Q.; Wang, Y.; Zhang, Q. The clinicopathological and prognostic significance of PD-L1 expression in gastric cancer: A meta-analysis of 10 studies with 1,901 patients. Sci. Rep. 2016, 6, 37933. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Nered, S.; Tryakin, A.; Abgaryan, M.; Artamonova, E.; Stroganova, A.; Stilidi, I. Microsatellite instability (MSI) in patients with gastric cancer (GC) and correlation with PD-L1 expression. J. Clin. Oncol. 2024, 42 (Suppl. S3), 389. [Google Scholar] [CrossRef]
- Mansuri, N.; Birkman, E.M.; Heuser, V.D.; Lintunen, M.; Ålgars, A.; Sundström, J.; Ristamäki, R.; Lehtinen, L.; Carpén, O. Association of tumor-infiltrating T lymphocytes with intestinal-type gastric cancer molecular subtypes and outcome. Virchows Arch. Int. J. Pathol. 2021, 478, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Jiang, K.Y.; Qi, L.L.; Liu, X.B.; Wang, Y.; Wang, L. Prognostic value of Siglec-15 expression in patients with solid tumors: A meta-analysis. Front. Oncol. 2023, 12, 1073932. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Tian, X.; Yan, L.; Guan, X.; Dong, B.; Zhao, M.; Lv, A.; Liu, D.; Wu, J.; Hao, C. Expression and Function of Siglec-15 in RLPS and Its Correlation with PD-L1: Bioinformatics Analysis and Clinicopathological Evidence. Int. J. Med. Sci. 2022, 19, 1977–1988. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Zheng, J.; Shao, Y.; Zhu, L.; Yang, T. Siglec-15 as multifunctional molecule involved in osteoclast differentiation, cancer immunity and microbial infection. Prog. Biophys. Mol. Biol. 2023, 177, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Zhang, B.; Wang, X.; Zeng, Z.; Huang, Z.; Zhang, L.; Wei, F.; Ren, X.; Yang, L. Expression signature, prognosis value, and immune characteristics of Siglec-15 identified by pan-cancer analysis. Oncoimmunology 2020, 9, 1807291. [Google Scholar] [CrossRef]
- Shafi, S.; Aung, T.N.; Robbins, C.; Zugazagoitia, J.; Vathiotis, I.; Gavrielatou, N.; Yaghoobi, V.; Fernandez, A.; Niu, S.; Liu, L.N.; et al. Development of an immunohistochemical assay for Siglec-15. Lab. Investig. 2022, 102, 771–778. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.Q.; Nong, J.Y.; Zhao, D.; Li, H.Y.; Su, D.; Zhou, L.J.; Dong, Y.J.; Zhang, C.; Che, N.Y.; Zhang, S.C.; et al. The significance of Siglec-15 expression in resectable non-small cell lung cancer. Neoplasma 2020, 67, 1214–1222. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, M.S.; Braoudaki, M.; Patel, H.; Ahmad, I.; Shagufta; Siddiqui, S.S. Novel Siglec-15-Sia axis inhibitor leads to colorectal cancer cell death by targeting miR-6715b-3p and oncogenes. Front. Immunol. 2023, 14, 1254911. [Google Scholar] [CrossRef] [PubMed]
- Korver, W.; Wong, A.; Gebremeskel, S.; Negri, G.L.; Schanin, J.; Chang, K.; Leung, J.; Benet, Z.; Luu, T.; Brock, E.C.; et al. The Inhibitory Receptor Siglec-8 Interacts with FcεRI and Globally Inhibits Intracellular Signaling in Primary Mast Cells Upon Activation. Front. Immunol. 2022, 13, 833728. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. The Study of NC318 Alone or in Combination with Pembrolizumab in Patients with Advanced Non-Small Cell Lung Cancer. ClinicalTrials.gov Identifier: NCT04699123. 2025. Available online: https://clinicaltrials.gov/study/NCT04699123?cond=NC318&rank=2 (accessed on 4 January 2025).
- Shum, E.; Myint, H.; Shaik, J.; Zhou, Q.; Barbu, E.; Morawski, A.; Abukharma, H.; Liu, L.; Nelson, M.; Zeidan, S.; et al. 490 Clinical benefit through Siglec-15 targeting with NC318 antibody in subjects with Siglec-15 positive advanced solid tumors. J. ImmunoTherapy Cancer 2021, 9. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Study of PYX-106 in Solid Tumors. ClinicalTrials.gov Identifier: NCT05718557. 2025. Available online: https://clinicaltrials.gov/study/NCT05718557 (accessed on 4 January 2025).
- Quirino, M.W.L.; Pereira, M.C.; Deodato de Souza, M.F.; Pitta, I.D.R.; Da Silva Filho, A.F.; Albuquerque, M.S.S.; Albuquerque, A.P.B.; Martins, M.R.; Pitta, M.G.D.R.; Rêgo, M.J.B.M. Immunopositivity for Siglec-15 in gastric cancer and its association with clinical and pathological parameters. Eur. J. Histochem. 2021, 65, 3174. [Google Scholar] [CrossRef] [PubMed]
- Moreira, R.S.; da Silva, M.M.; de Melo Vasconcelos, C.F.; da Silva, T.D.; Cordeiro, G.G.; Mattos-Jr, L.A.R.; da Rocha Pitta, M.G.; de Melo Rêgo, M.J.B.; Pereira, M.C. Siglec 15 as a biomarker or a druggable molecule for non-small cell lung cancer. J. Cancer Res. Clin. Oncol. 2023, 149, 17651–17661. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | EBV-Positive GC | MSI GC | GS GC | CIN GC |
|---|---|---|---|---|
| Molecular Features |
|
| ||
| Immune Cell Infiltration |
| |||
| Immunotherapy Response |
|
| Characteristic | Observation |
|---|---|
| Expression Levels |
|
| TME | |
| Prognostic Significance | |
| Immune Cell Infiltration | |
| Immunotherapy Response |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cozac-Szőke, A.-R.; Cozac, D.A.; Negovan, A.; Tinca, A.C.; Vilaia, A.; Cocuz, I.-G.; Sabău, A.H.; Niculescu, R.; Chiorean, D.M.; Tomuț, A.N.; et al. Immune Cell Interactions and Immune Checkpoints in the Tumor Microenvironment of Gastric Cancer. Int. J. Mol. Sci. 2025, 26, 1156. https://doi.org/10.3390/ijms26031156
Cozac-Szőke A-R, Cozac DA, Negovan A, Tinca AC, Vilaia A, Cocuz I-G, Sabău AH, Niculescu R, Chiorean DM, Tomuț AN, et al. Immune Cell Interactions and Immune Checkpoints in the Tumor Microenvironment of Gastric Cancer. International Journal of Molecular Sciences. 2025; 26(3):1156. https://doi.org/10.3390/ijms26031156
Chicago/Turabian StyleCozac-Szőke, Andreea-Raluca, Dan Alexandru Cozac, Anca Negovan, Andreea Cătălina Tinca, Alexandra Vilaia, Iuliu-Gabriel Cocuz, Adrian Horațiu Sabău, Raluca Niculescu, Diana Maria Chiorean, Alexandru Nicușor Tomuț, and et al. 2025. "Immune Cell Interactions and Immune Checkpoints in the Tumor Microenvironment of Gastric Cancer" International Journal of Molecular Sciences 26, no. 3: 1156. https://doi.org/10.3390/ijms26031156
APA StyleCozac-Szőke, A.-R., Cozac, D. A., Negovan, A., Tinca, A. C., Vilaia, A., Cocuz, I.-G., Sabău, A. H., Niculescu, R., Chiorean, D. M., Tomuț, A. N., & Cotoi, O. S. (2025). Immune Cell Interactions and Immune Checkpoints in the Tumor Microenvironment of Gastric Cancer. International Journal of Molecular Sciences, 26(3), 1156. https://doi.org/10.3390/ijms26031156