
Signature Construction Associated with Tumor-Infiltrating Macrophages Identifies IRF8 as a Novel Biomarker for Immunotherapy in Advanced Gastric Cancer

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
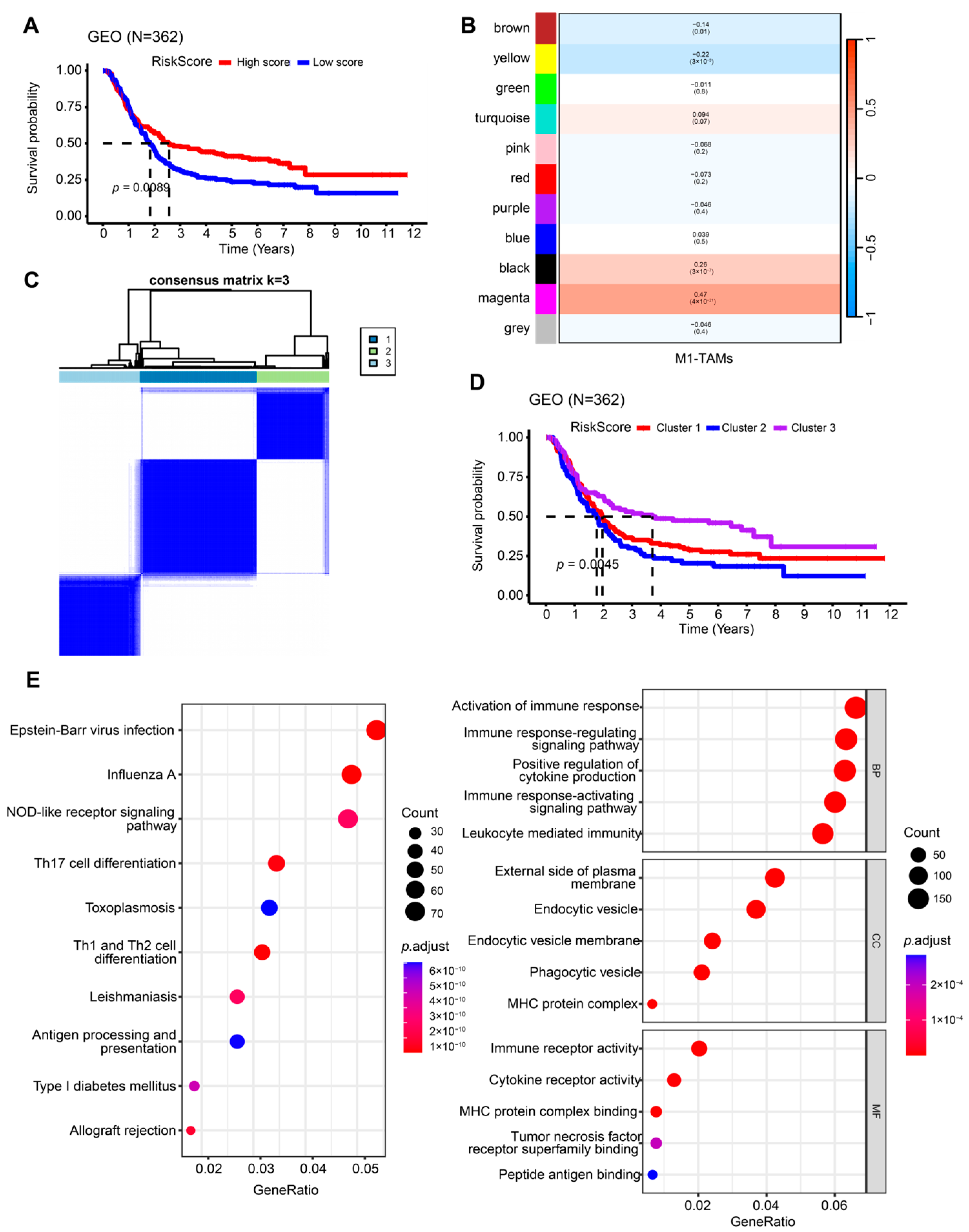
2.1. Identification of M1-like TAM-Related Subgroups
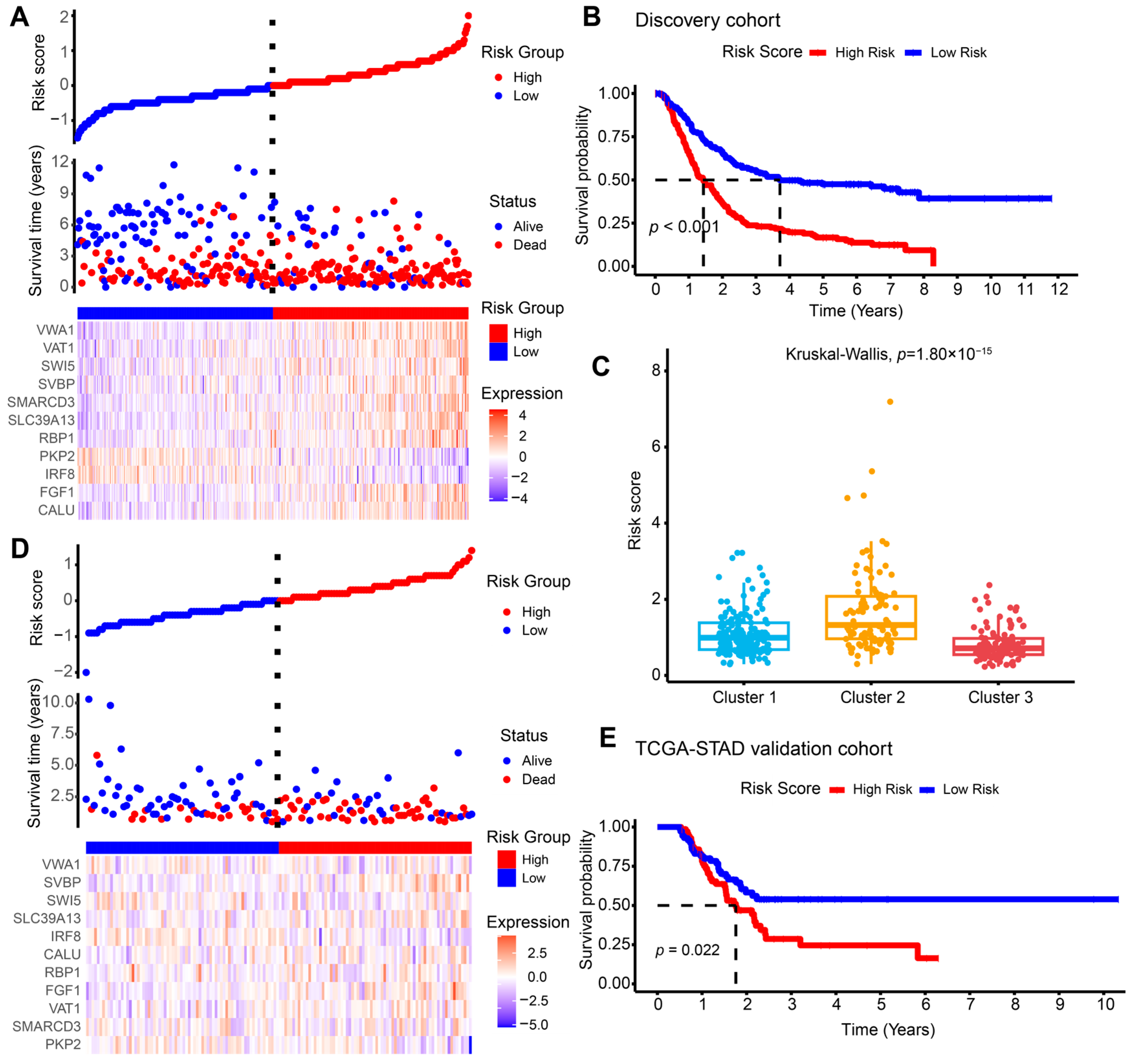
2.2. Construction and Validation of the M1-like TAM-Related Risk Signature for Independent Prognostic Prediction
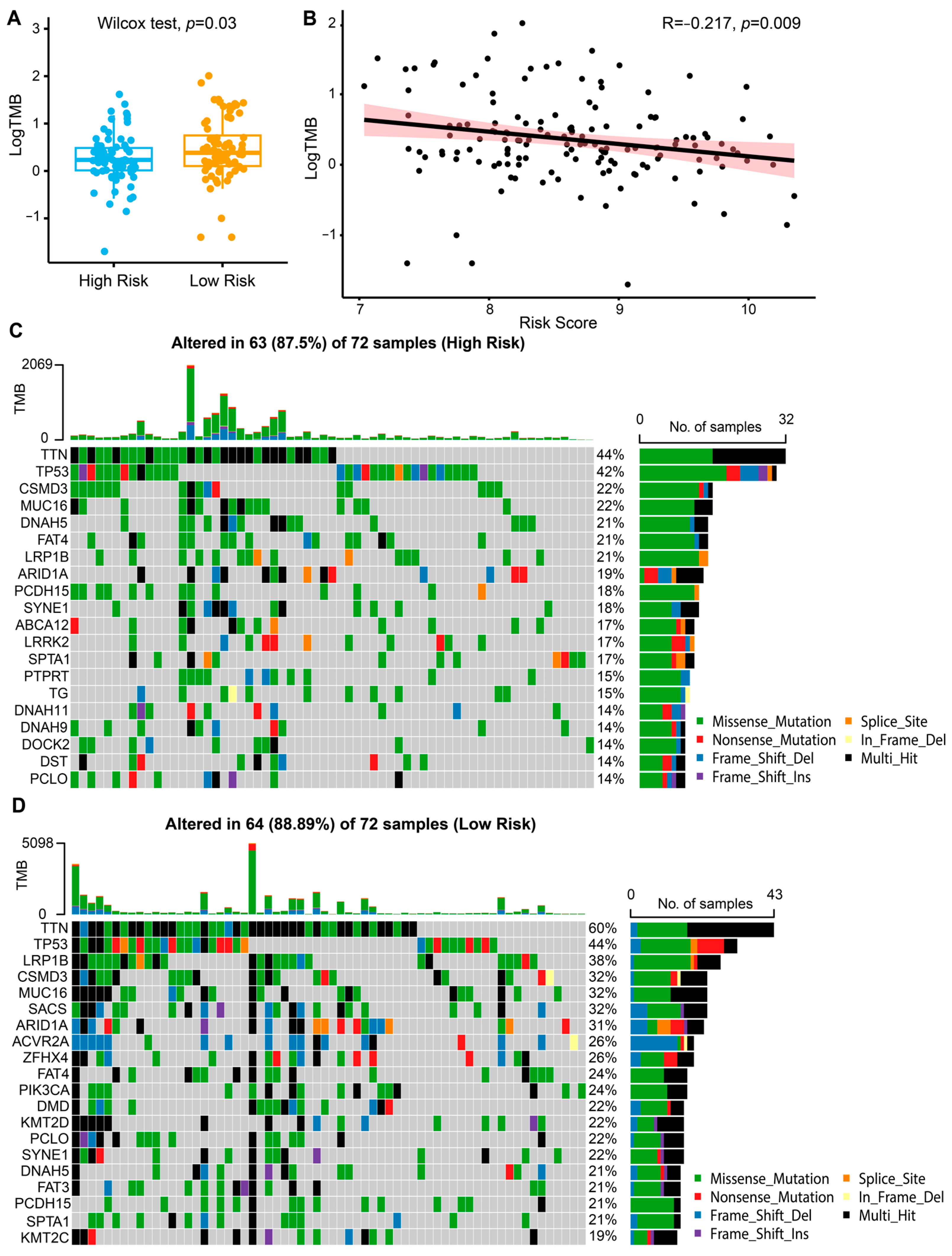
2.3. Immune Landscape and Predictive Value of the Risk Model for Immunotherapy Response
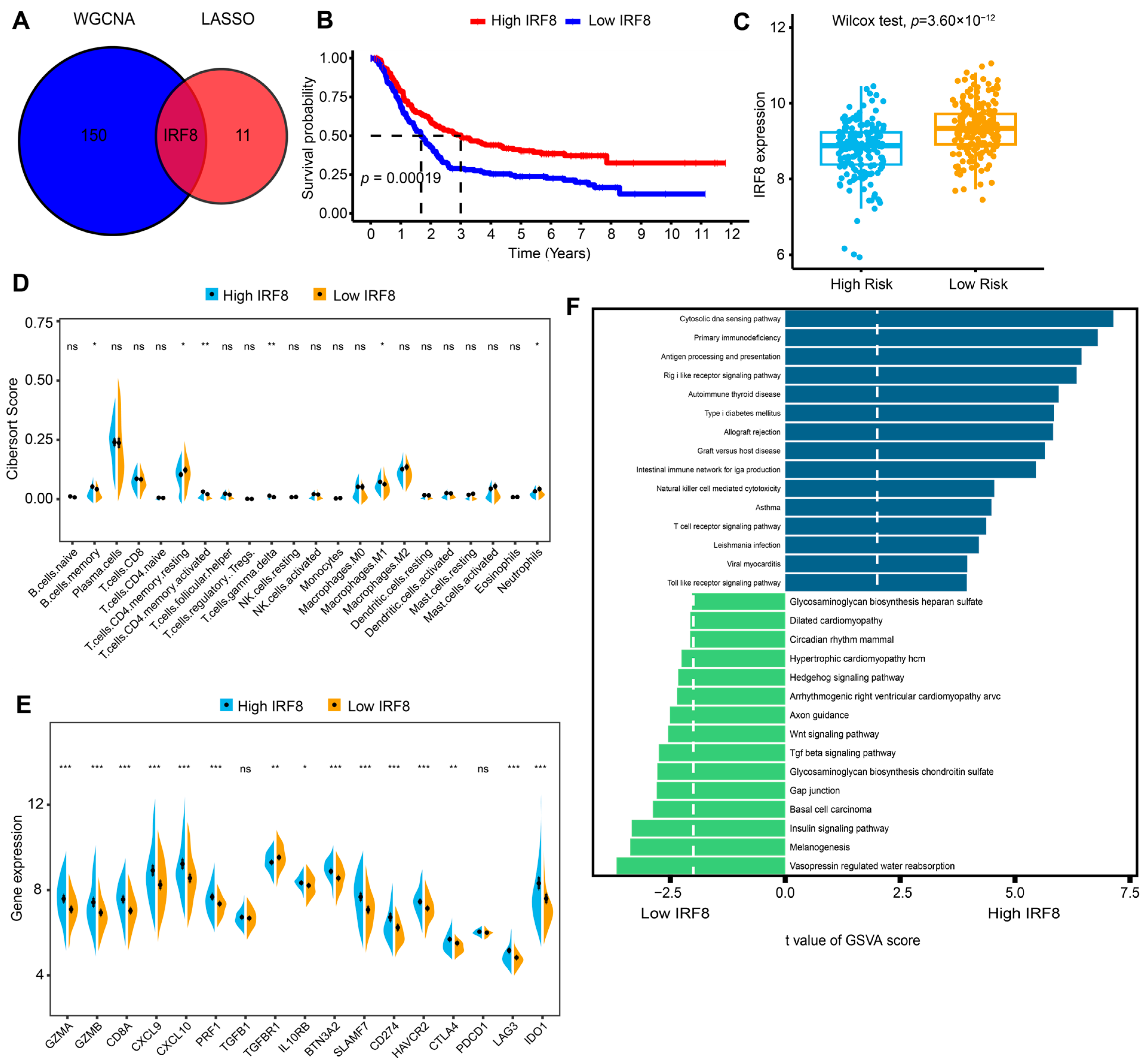
2.4. Identifying IRF8 as a Hub Gene in the Risk Signature and Further Investigating Its Role in Immune Activation
2.5. IRF8 Overexpression Suppresses GC Progression In Vivo
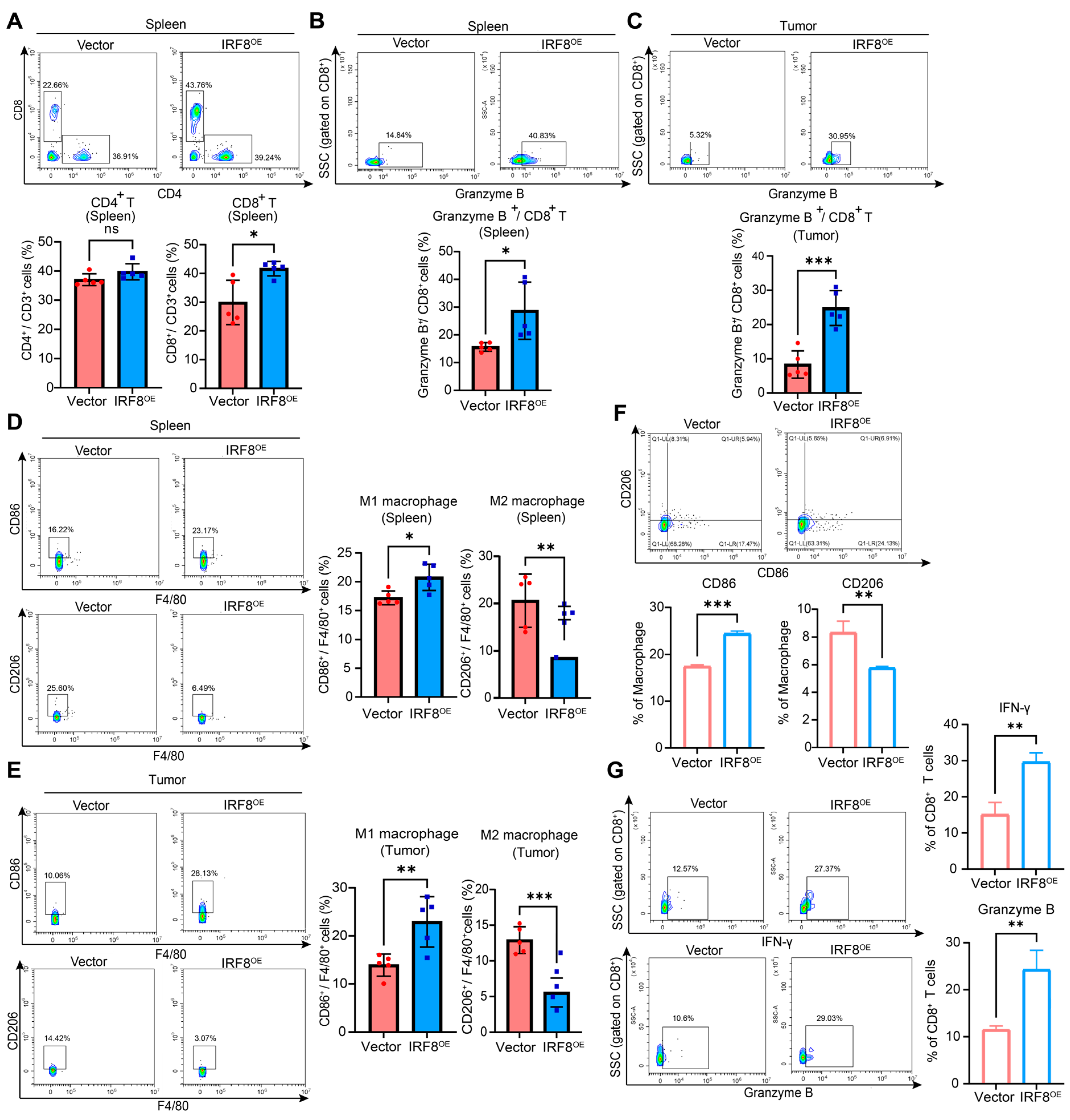
2.6. IRF8 Reshapes the Suppressive TIME in GC
2.7. IRF8 Overexpression Augments PD-1 Blockade in Syngeneic GC Models
2.8. High IRF8 Expression Exhibits a Favorable Response to Anti-PD-1 Therapy
3. Discussion
4. Materials and Methods
4.1. Data Collection and Preprocessing
4.2. Analysis of Tumor-Infiltrating Immune Cells
4.3. WGCNA
4.4. Identification of M1-like TAM-Related Subgroups via Consensus Clustering
4.5. Construction and Validation of M1-like TAM-Related Prognostic Risk Model
4.6. Functional Enrichment Analysis
4.7. Somatic Mutation Analysis
4.8. Immunotherapy Response Prediction
4.9. Cell Culture and Stable Transfection
4.10. Preparation of CM
4.11. Generation of Human T Cells
4.12. Western Blotting
4.13. Animal Models
4.14. Multicolor Flow Cytometry Analysis
4.15. IHC Staining
4.16. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer Statistics, 2024. CA Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef] [PubMed]
- Guan, W.-L.; He, Y.; Xu, R.-H. Gastric Cancer Treatment: Recent Progress and Future Perspectives. J. Hematol. Oncol. 2023, 16, 57. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, I.; Ramachandran, S.; Zabel, C.; Gaikwad, S.; Srivastava, S.K. The Evolutionary Legacy of Immune Checkpoint Inhibitors. Semin. Cancer Biol. 2022, 86, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, T.; Wang, Y.A. Gastric Cancer Immunosuppressive Microenvironment Heterogeneity: Implications for Therapy Development. Trends Cancer 2024, 10, 627–642. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Xie, J.; Huang, W.; Chen, H.; Xi, S.; Han, Z.; Huang, L.; Lin, T.; Zhao, L.-Y.; Hu, Y.-F.; et al. Tumor Immune Microenvironment and Chemosensitivity Signature for Predicting Response to Chemotherapy in Gastric Cancer. Cancer Immunol. Res. 2019, 7, 2065–2073. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Yu, B.; Wang, F.; Yang, J. Single-Cell RNA Sequencing to Map Tumor Heterogeneity in Gastric Carcinogenesis Paving Roads to Individualized Therapy. Cancer Immunol. Immunother. CII 2024, 73, 233. [Google Scholar] [CrossRef] [PubMed]
- Locati, M.; Curtale, G.; Mantovani, A. Diversity, Mechanisms, and Significance of Macrophage Plasticity. Annu. Rev. Pathol. 2020, 15, 123–147. [Google Scholar] [CrossRef] [PubMed]
- Najafi, M.; Hashemi Goradel, N.; Farhood, B.; Salehi, E.; Nashtaei, M.S.; Khanlarkhani, N.; Khezri, Z.; Majidpoor, J.; Abouzaripour, M.; Habibi, M.; et al. Macrophage Polarity in Cancer: A Review. J. Cell. Biochem. 2019, 120, 2756–2765. [Google Scholar] [CrossRef]
- Lin, Y.; Xu, J.; Lan, H. Tumor-Associated Macrophages in Tumor Metastasis: Biological Roles and Clinical Therapeutic Applications. J. Hematol. Oncol. 2019, 12, 76. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Ruffell, B. Macrophages as Regulators of Tumour Immunity and Immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef]
- Lf, T.; Pf, F.; Dm, S. Clinical Evaluation of Macrophages in Cancer: Role in Treatment, Modulation and Challenges. Cancer Immunol. Immunother. CII 2017, 66, 1509–1527. [Google Scholar] [CrossRef]
- Mills, C.D.; Lenz, L.L.; Harris, R.A. A Breakthrough: Macrophage-Directed Cancer Immunotherapy. Cancer Res. 2016, 76, 513–516. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Yan, L.; Jiu, J.; Liu, H.; Li, D.; Li, X.; Zhang, J.; Li, S.; Fan, Z.; Lv, Z.; et al. PSME2 Offers Value as a Biomarker of M1 Macrophage Infiltration in Pan-Cancer and Inhibits Osteosarcoma Malignant Phenotypes. Int. J. Biol. Sci. 2024, 20, 1452–1470. [Google Scholar] [CrossRef]
- Luo, F.; Li, H.; Ma, W.; Cao, J.; Chen, Q.; Lu, F.; Qiu, M.; Zhou, P.; Xia, Z.; Zeng, K.; et al. The BCL-2 Inhibitor APG-2575 Resets Tumor-Associated Macrophages toward the M1 Phenotype, Promoting a Favorable Response to Anti-PD-1 Therapy via NLRP3 Activation. Cell. Mol. Immunol. 2024, 21, 60–79. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Marchesi, F.; Garlanda, C. Macrophages as Tools and Targets in Cancer Therapy. Nat. Rev. Drug Discov. 2022, 21, 799–820. [Google Scholar] [CrossRef]
- Xia, Y.; Rao, L.; Yao, H.; Wang, Z.; Ning, P.; Chen, X. Engineering Macrophages for Cancer Immunotherapy and Drug Delivery. Adv. Mater. 2020, 32, 2002054. [Google Scholar] [CrossRef]
- Li, J.; Sun, J.; Zeng, Z.; Liu, Z.; Ma, M.; Zheng, Z.; He, Y.; Kang, W. Tumour-Associated Macrophages in Gastric Cancer: From Function and Mechanism to Application. Clin. Transl. Med. 2023, 13, e1386. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.A.; Yarchoan, M.; Jaffee, E.; Swanton, C.; Quezada, S.A.; Stenzinger, A.; Peters, S. Development of Tumor Mutation Burden as an Immunotherapy Biomarker: Utility for the Oncology Clinic. Ann. Oncol. 2019, 30, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.-F.; Liu, C.-J.; Liu, L.-L.; Zhang, Q.; Guo, A.-Y. Expression Profile of Immune Checkpoint Genes and Their Roles in Predicting Immunotherapy Response. Brief. Bioinform. 2021, 22, bbaa176. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The Tumor Microenvironment at a Glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef]
- Lubiński, J.; Lener, M.R.; Marciniak, W.; Pietrzak, S.; Derkacz, R.; Cybulski, C.; Gronwald, J.; Dębniak, T.; Jakubowska, A.; Huzarski, T.; et al. Serum Essential Elements and Survival after Cancer Diagnosis. Nutrients 2023, 15, 2611. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, B.; Zhao, Y.; Shao, X.; Wang, M.; Ma, F.; Yang, L.; Nie, M.; Jin, P.; Yao, K.; et al. Metabolomic Machine Learning Predictor for Diagnosis and Prognosis of Gastric Cancer. Nat. Commun. 2024, 15, 1657. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Qin, D.; Tao, Z.; Wang, B.; Xie, Y.; Wang, Y.; Li, B.; Cao, J.; Qiao, X.; Zhong, S.; et al. Identification of Cuproptosis-Related Subtypes, Construction of a Prognosis Model, and Tumor Microenvironment Landscape in Gastric Cancer. Front. Immunol. 2022, 13, 1056932. [Google Scholar] [CrossRef]
- Chia, N.-Y.; Tan, P. Molecular Classification of Gastric Cancer. Ann. Oncol. 2016, 27, 763–769. [Google Scholar] [CrossRef]
- Fuchs, C.S.; Doi, T.; Jang, R.W.; Muro, K.; Satoh, T.; Machado, M.; Sun, W.; Jalal, S.I.; Shah, M.A.; Metges, J.-P.; et al. Safety and Efficacy of Pembrolizumab Monotherapy in Patients With Previously Treated Advanced Gastric and Gastroesophageal Junction Cancer: Phase 2 Clinical KEYNOTE-059 Trial. JAMA Oncol. 2018, 4, e180013. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.-K.; Boku, N.; Satoh, T.; Ryu, M.-H.; Chao, Y.; Kato, K.; Chung, H.C.; Chen, J.-S.; Muro, K.; Kang, W.K.; et al. Nivolumab in Patients with Advanced Gastric or Gastro-Oesophageal Junction Cancer Refractory to, or Intolerant of, at Least Two Previous Chemotherapy Regimens (ONO-4538-12, ATTRACTION-2): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet 2017, 390, 2461–2471. [Google Scholar] [CrossRef]
- Chen, S.; Saeed, A.F.U.H.; Liu, Q.; Jiang, Q.; Xu, H.; Xiao, G.G.; Rao, L.; Duo, Y. Macrophages in Immunoregulation and Therapeutics. Signal Transduct. Target. Ther. 2023, 8, 207. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, Y. Tumor-Associated Macrophages: From Basic Research to Clinical Application. J. Hematol. Oncol. 2017, 10, 58. [Google Scholar] [CrossRef]
- Gambardella, V.; Castillo, J.; Tarazona, N.; Gimeno-Valiente, F.; Martínez-Ciarpaglini, C.; Cabeza-Segura, M.; Roselló, S.; Roda, D.; Huerta, M.; Cervantes, A.; et al. The Role of Tumor-Associated Macrophages in Gastric Cancer Development and Their Potential as a Therapeutic Target. Cancer Treat. Rev. 2020, 86, 102015. [Google Scholar] [CrossRef] [PubMed]
- You, T.; Tang, H.; Wu, W.; Gao, J.; Li, X.; Li, N.; Xu, X.; Xing, J.; Ge, H.; Xiao, Y.; et al. POSTN Secretion by Extracellular Matrix Cancer-Associated Fibroblasts (eCAFs) Correlates with Poor ICB Response via Macrophage Chemotaxis Activation of Akt Signaling Pathway in Gastric Cancer. Aging Dis. 2023, 14, 2177–2192. [Google Scholar] [CrossRef]
- Shi, T.; Zhang, Y.; Wang, Y.; Song, X.; Wang, H.; Zhou, X.; Liang, K.; Luo, Y.; Che, K.; Wang, X.; et al. DKK1 Promotes Tumor Immune Evasion and Impedes Anti-PD-1 Treatment by Inducing Immunosuppressive Macrophages in Gastric Cancer. Cancer Immunol. Res. 2022, 10, 1506–1524. [Google Scholar] [CrossRef] [PubMed]
- Che, K.; Luo, Y.; Song, X.; Yang, Z.; Wang, H.; Shi, T.; Wang, Y.; Wang, X.; Wu, H.; Yu, L.; et al. Macrophages Reprogramming Improves Immunotherapy of IL-33 in Peritoneal Metastasis of Gastric Cancer. EMBO Mol. Med. 2024, 16, 251–266. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Yu, K.; Zhang, Z.; Gu, Y.; Gu, Y.; Li, W.; Zhang, W.; Shen, Z.; Xu, J.; Qin, J. Blockade of V-Domain Immunoglobulin Suppressor of T-Cell Activation Reprograms Tumour-Associated Macrophages and Improves Efficacy of PD-1 Inhibitor in Gastric Cancer. Clin. Transl. Med. 2024, 14, e1578. [Google Scholar] [CrossRef]
- Gao, J.; Zhao, Z.; Zhang, H.; Huang, S.; Xu, M.; Pan, H. Transcriptomic Characterization and Construction of M2 Macrophage-Related Prognostic and Immunotherapeutic Signature in Ovarian Metastasis of Gastric Cancer. Cancer Immunol. Immunother. CII 2023, 72, 1121–1138. [Google Scholar] [CrossRef]
- Boutilier, A.J.; Elsawa, S.F. Macrophage Polarization States in the Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 6995. [Google Scholar] [CrossRef]
- Takaoka, A.; Tamura, T.; Taniguchi, T. Interferon Regulatory Factor Family of Transcription Factors and Regulation of Oncogenesis. Cancer Sci. 2008, 99, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Wang, H.; Shin, D.-M.; Masiuk, M.; Qi, C.-F.; Morse, H.C. IFN Regulatory Factor 8 Restricts the Size of the Marginal Zone and Follicular B Cell Pools. J. Immunol. 2011, 186, 1458–1466. [Google Scholar] [CrossRef] [PubMed]
- Karki, R.; Lee, E.; Place, D.; Samir, P.; Mavuluri, J.; Sharma, B.R.; Balakrishnan, A.; Malireddi, R.K.S.; Geiger, R.; Zhu, Q.; et al. IRF8 Regulates Transcription of Naips for NLRC4 Inflammasome Activation. Cell 2018, 173, 920–933.e13. [Google Scholar] [CrossRef] [PubMed]
- Gatti, G.; Betts, C.; Rocha, D.; Nicola, M.; Grupe, V.; Ditada, C.; Nuñez, N.G.; Roselli, E.; Araya, P.; Dutto, J.; et al. High IRF8 Expression Correlates with CD8 T Cell Infiltration and Is a Predictive Biomarker of Therapy Response in ER-Negative Breast Cancer. Breast Cancer Res. BCR 2021, 23, 40. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Li, Y.; Shi, G.; Du, S.; Wang, X.; Ye, W.; Zhang, Z.; Chu, Y.; Ma, S.; Wang, D.; et al. Hepatic Interferon Regulatory Factor 8 Expression Suppresses Hepatocellular Carcinoma Progression and Enhances the Response to Anti-Programmed Cell Death Protein-1 Therapy. Hepatology 2022, 76, 1602–1616. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Lu, F.; Li, B.; Liu, L.; Zeng, G.; Zhou, Q.; Chen, L. IRF8 Induces Senescence of Lung Cancer Cells to Exert Its Tumor Suppressive Function. Cell Cycle 2019, 18, 3300–3312. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-Intrinsic β-Catenin Signalling Prevents Anti-Tumour Immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef]
- Du, W.; Menjivar, R.E.; Donahue, K.L.; Kadiyala, P.; Velez-Delgado, A.; Brown, K.L.; Watkoske, H.R.; He, X.; Carpenter, E.S.; Angeles, C.V.; et al. WNT Signaling in the Tumor Microenvironment Promotes Immunosuppression in Murine Pancreatic Cancer. J. Exp. Med. 2023, 220, e20220503. [Google Scholar] [CrossRef] [PubMed]
- Gough, N.R.; Xiang, X.; Mishra, L. TGF-β Signaling in Liver, Pancreas, and Gastrointestinal Diseases and Cancer. Gastroenterology 2021, 161, 434–452.e15. [Google Scholar] [CrossRef] [PubMed]
- Larson, C.; Oronsky, B.; Carter, C.A.; Oronsky, A.; Knox, S.J.; Sher, D.; Reid, T.R. TGF-Beta: A Master Immune Regulator. Expert Opin. Ther. Targets 2020, 24, 427–438. [Google Scholar] [CrossRef] [PubMed]
- DeVito, N.C.; Sturdivant, M.; Thievanthiran, B.; Xiao, C.; Plebanek, M.P.; Salama, A.K.S.; Beasley, G.M.; Holtzhausen, A.; Novotny-Diermayr, V.; Strickler, J.H.; et al. Pharmacological Wnt Ligand Inhibition Overcomes Key Tumor-Mediated Resistance Pathways to Anti-PD-1 Immunotherapy. Cell Rep. 2021, 35, 109071. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Bi, Y.; Wei, Y.; Liu, J.; Kuerban, K.; Ye, L. Blocking Wnt/β-Catenin Signal Amplifies Anti-PD-1 Therapeutic Efficacy by Inhibiting Tumor Growth, Migration, and Promoting Immune Infiltration in Glioblastomas. Mol. Cancer Ther. 2021, 20, 1305–1315. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E.; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ Attenuates Tumour Response to PD-L1 Blockade by Contributing to Exclusion of T Cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-J.; Luo, S.-N.; Wu, H.; Zhang, N.-P.; Dong, L.; Liu, T.-T.; Liang, L.; Shen, X.-Z. IRF-2 Inhibits Cancer Proliferation by Promoting AMER-1 Transcription in Human Gastric Cancer. J. Transl. Med. 2022, 20, 68. [Google Scholar] [CrossRef]
- Tian, M.; Wang, X.; Sun, J.; Lin, W.; Chen, L.; Liu, S.; Wu, X.; Shi, L.; Xu, P.; Cai, X.; et al. IRF3 Prevents Colorectal Tumorigenesis via Inhibiting the Nuclear Translocation of β-Catenin. Nat. Commun. 2020, 11, 5762. [Google Scholar] [CrossRef]
- Scheller, M.; Schönheit, J.; Zimmermann, K.; Leser, U.; Rosenbauer, F.; Leutz, A. Cross Talk between Wnt/β-Catenin and Irf8 in Leukemia Progression and Drug Resistance. J. Exp. Med. 2013, 210, 2239–2256. [Google Scholar] [CrossRef]
- Xu, P.; Bailey-Bucktrout, S.; Xi, Y.; Xu, D.; Du, D.; Zhang, Q.; Xiang, W.; Liu, J.; Melton, A.; Sheppard, D.; et al. Innate Antiviral Host Defense Attenuates TGF-β Function through IRF3-Mediated Suppression of Smad Signaling. Mol. Cell 2014, 56, 723–737. [Google Scholar] [CrossRef]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The Sva Package for Removing Batch Effects and Other Unwanted Variation in High-Throughput Experiments. Bioinformatics 2012, 28, 882–883. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust Enumeration of Cell Subsets from Tissue Expression Profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R Package for Weighted Correlation Network Analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Wilkerson, M.D.; Hayes, D.N. ConsensusClusterPlus: A Class Discovery Tool with Confidence Assessments and Item Tracking. Bioinformatics 2010, 26, 1572–1573. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Friedman, J.; Hastie, T.; Tibshirani, R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene Set Variation Analysis for Microarray and RNA-Seq Data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef]
- Mayakonda, A.; Lin, D.-C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and Comprehensive Analysis of Somatic Variants in Cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Gu, S.; Pan, D.; Fu, J.; Sahu, A.; Hu, X.; Li, Z.; Traugh, N.; Bu, X.; Li, B.; et al. Signatures of T Cell Dysfunction and Exclusion Predict Cancer Immunotherapy Response. Nat. Med. 2018, 24, 1550–1558. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liao, W.; Wang, Y.; Wang, R.; Fu, B.; Chen, X.; Ouyang, Y.; Bai, B.; Jin, Y.; Lu, Y.; Liu, F.; et al. Signature Construction Associated with Tumor-Infiltrating Macrophages Identifies IRF8 as a Novel Biomarker for Immunotherapy in Advanced Gastric Cancer. Int. J. Mol. Sci. 2025, 26, 1089. https://doi.org/10.3390/ijms26031089
Liao W, Wang Y, Wang R, Fu B, Chen X, Ouyang Y, Bai B, Jin Y, Lu Y, Liu F, et al. Signature Construction Associated with Tumor-Infiltrating Macrophages Identifies IRF8 as a Novel Biomarker for Immunotherapy in Advanced Gastric Cancer. International Journal of Molecular Sciences. 2025; 26(3):1089. https://doi.org/10.3390/ijms26031089
Chicago/Turabian StyleLiao, Wanqian, Yu Wang, Rui Wang, Bibo Fu, Xiangfu Chen, Ying Ouyang, Bing Bai, Ying Jin, Yunxin Lu, Furong Liu, and et al. 2025. "Signature Construction Associated with Tumor-Infiltrating Macrophages Identifies IRF8 as a Novel Biomarker for Immunotherapy in Advanced Gastric Cancer" International Journal of Molecular Sciences 26, no. 3: 1089. https://doi.org/10.3390/ijms26031089
APA StyleLiao, W., Wang, Y., Wang, R., Fu, B., Chen, X., Ouyang, Y., Bai, B., Jin, Y., Lu, Y., Liu, F., Zhang, Y., Shi, D., & Zhang, D. (2025). Signature Construction Associated with Tumor-Infiltrating Macrophages Identifies IRF8 as a Novel Biomarker for Immunotherapy in Advanced Gastric Cancer. International Journal of Molecular Sciences, 26(3), 1089. https://doi.org/10.3390/ijms26031089