
In Vitro Infection of Human Macrophages with Porphyromonas gingivalis W83


Abstract
:1. Introduction
2. Results
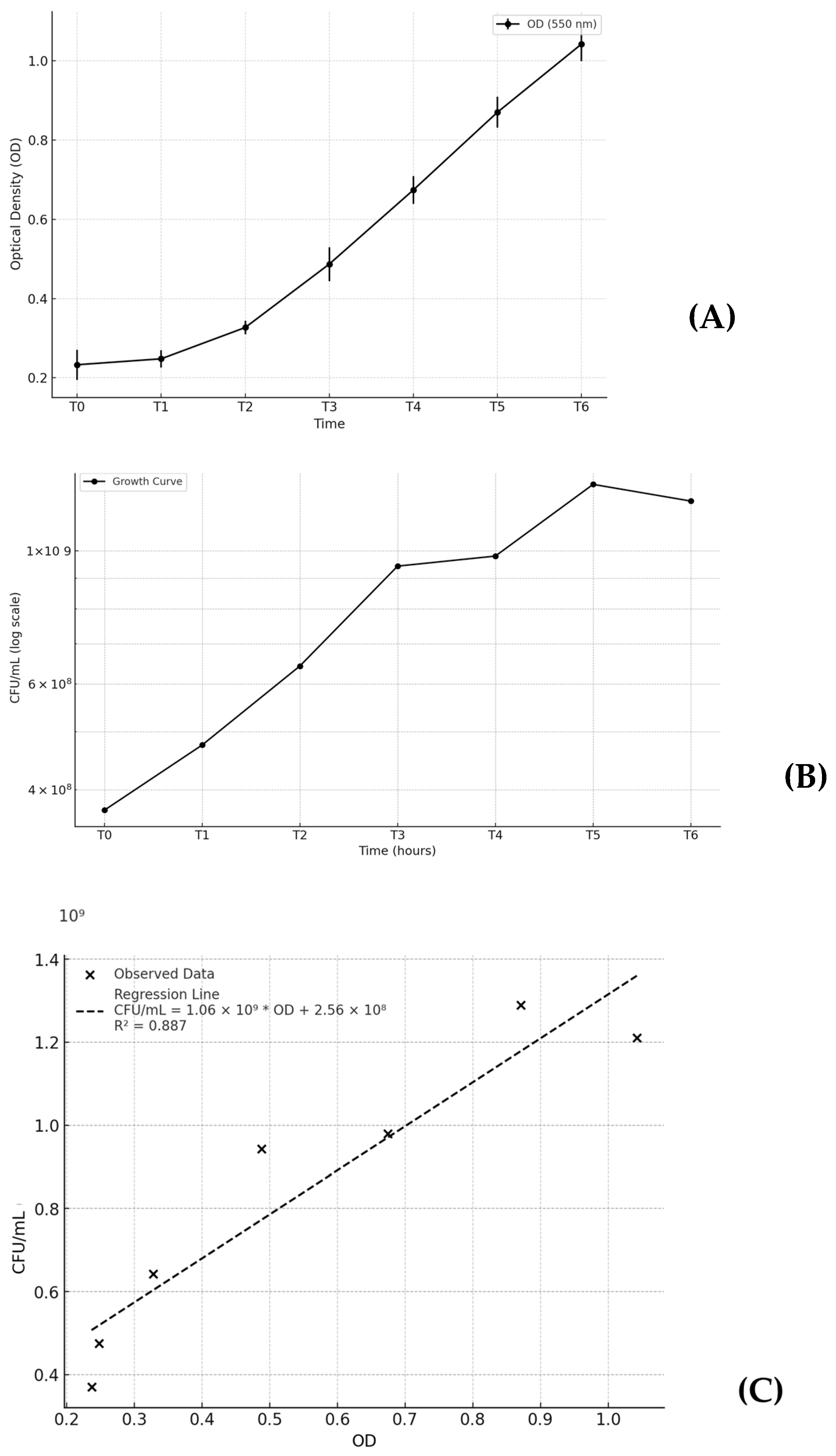
2.1. Growth Curve Optimization
2.2. Quantitative PCR (qPCR) Standard Curve:
2.3. Macrophage Activation and Polarization
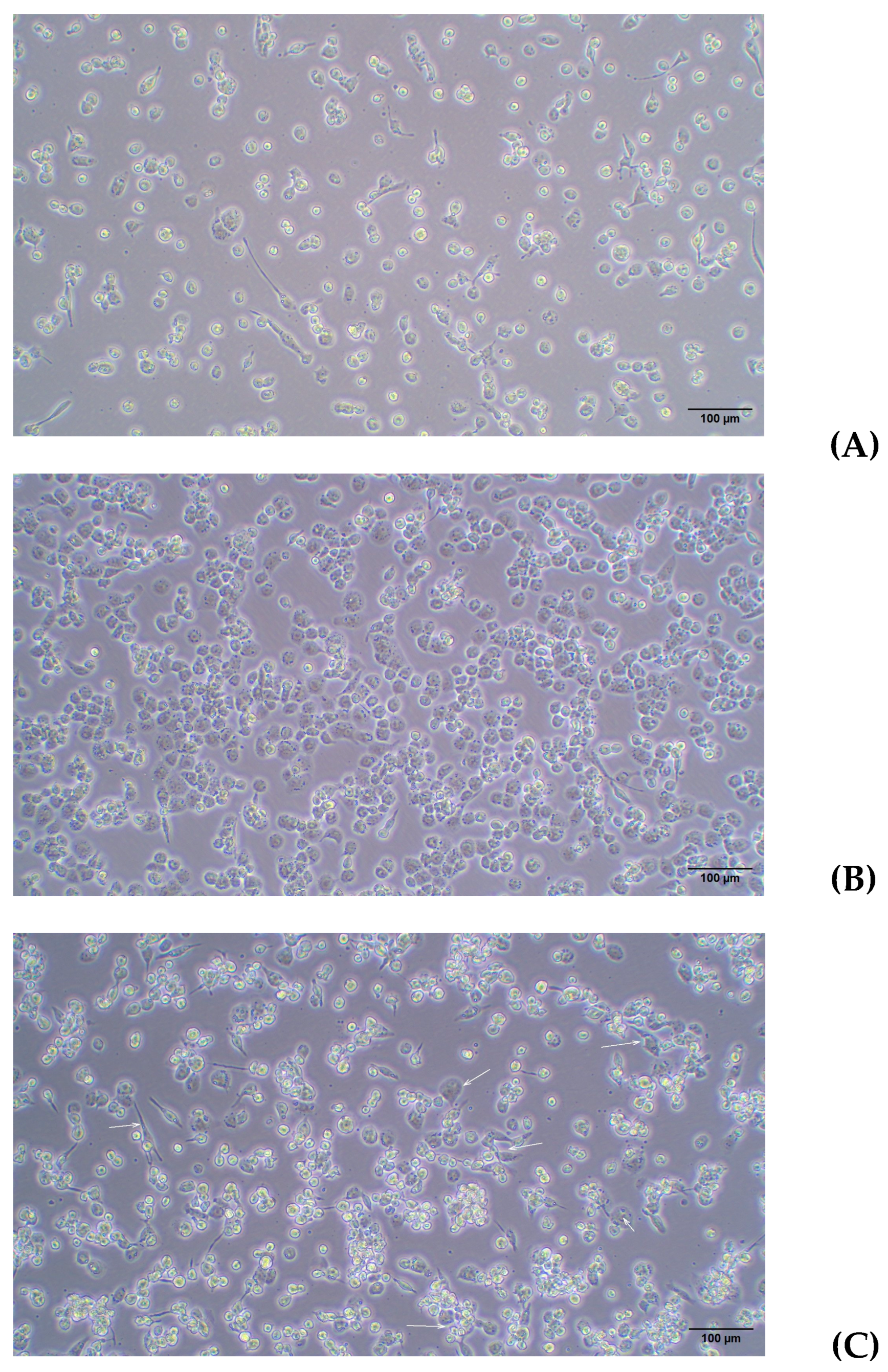
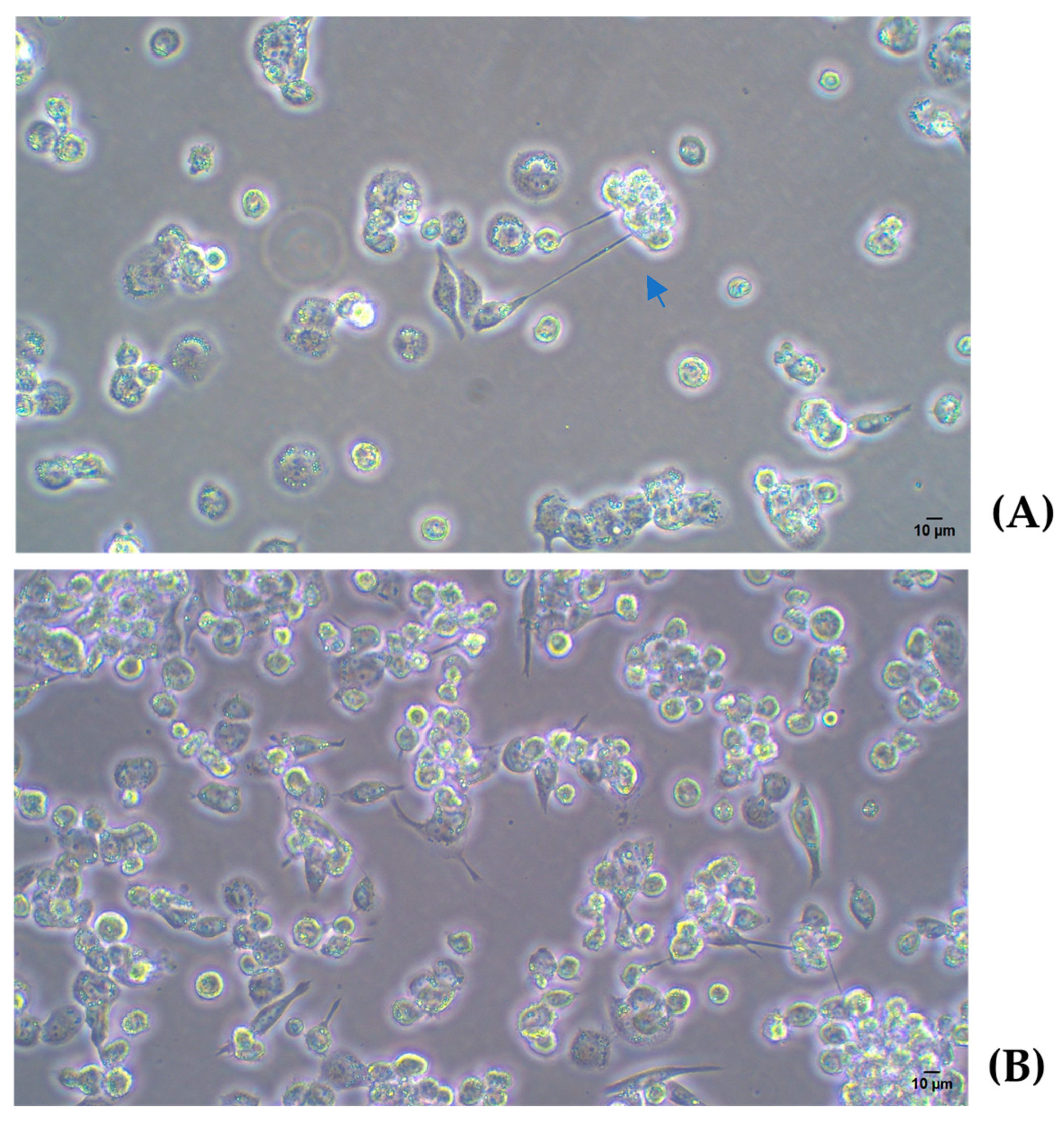
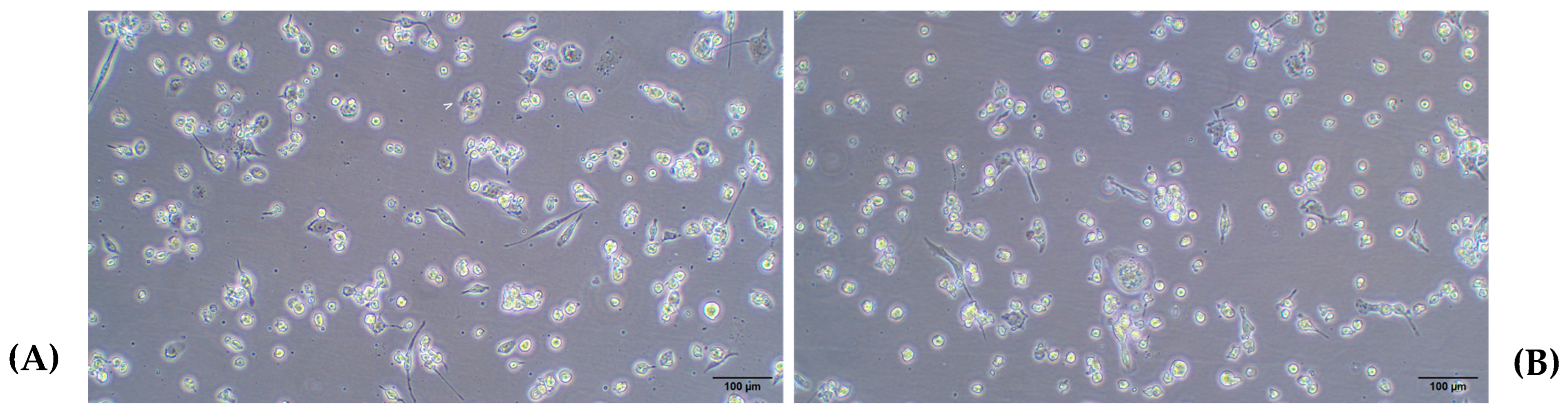
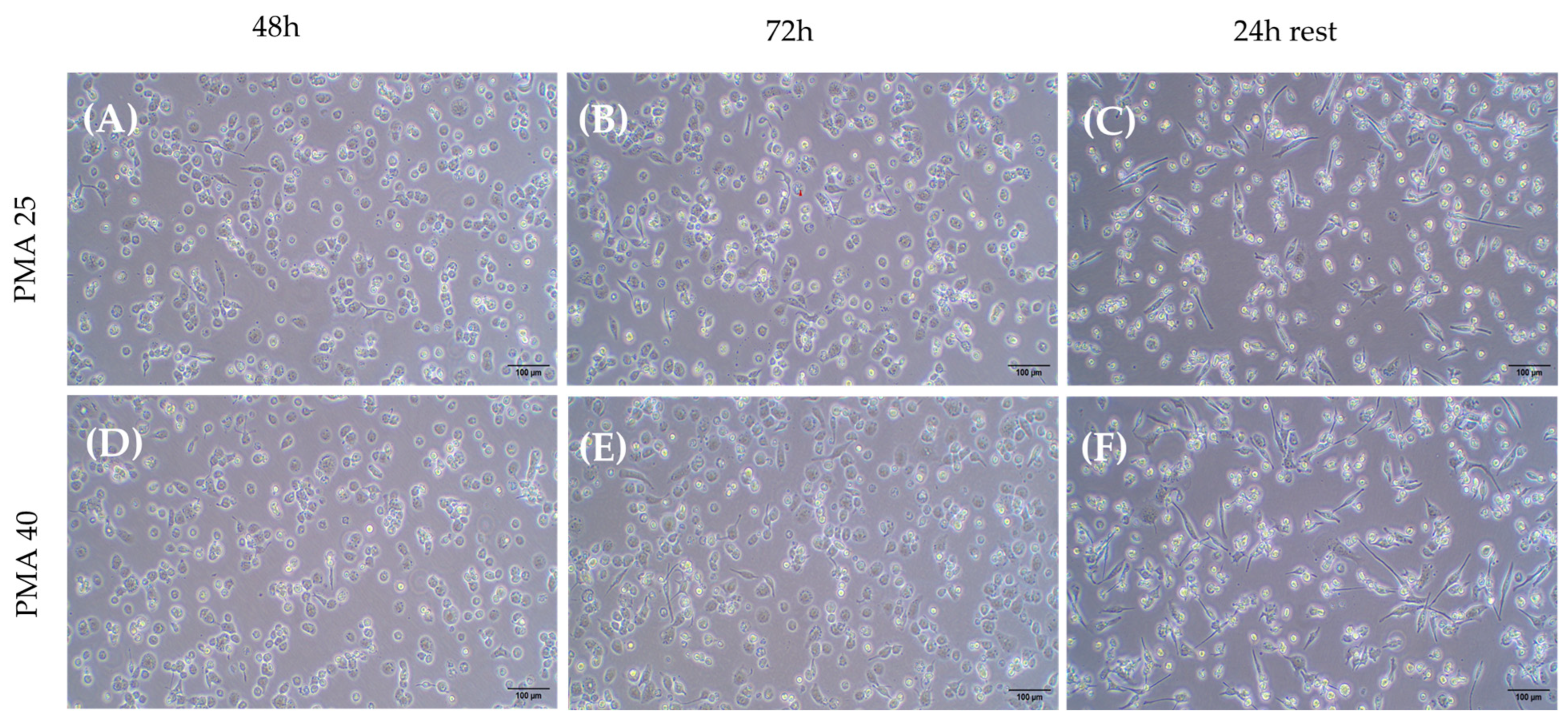
2.3.1. Differentiation of THP-1 Cells Into Macrophage Using PMA
2.3.2. M1 Polarization
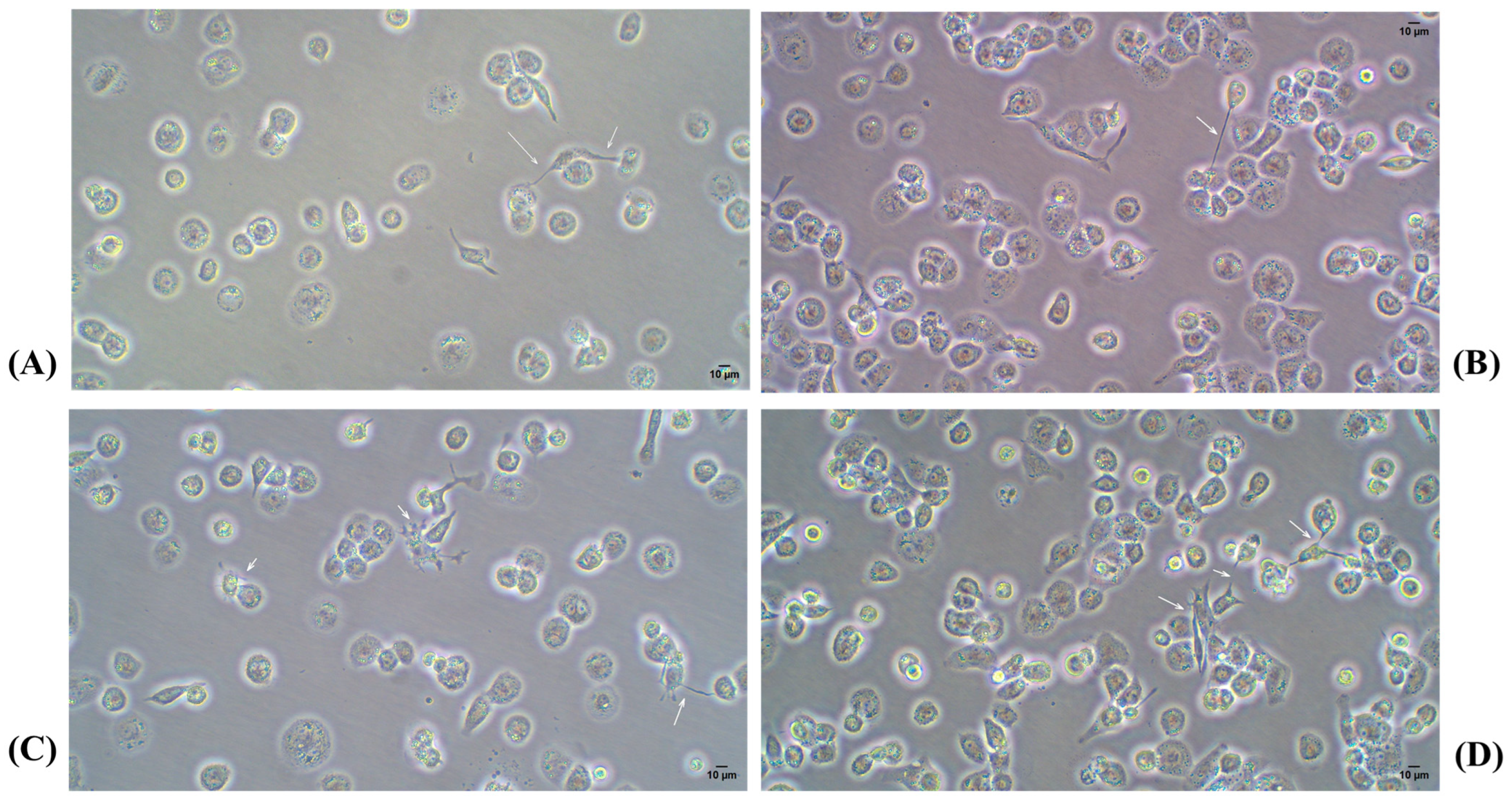
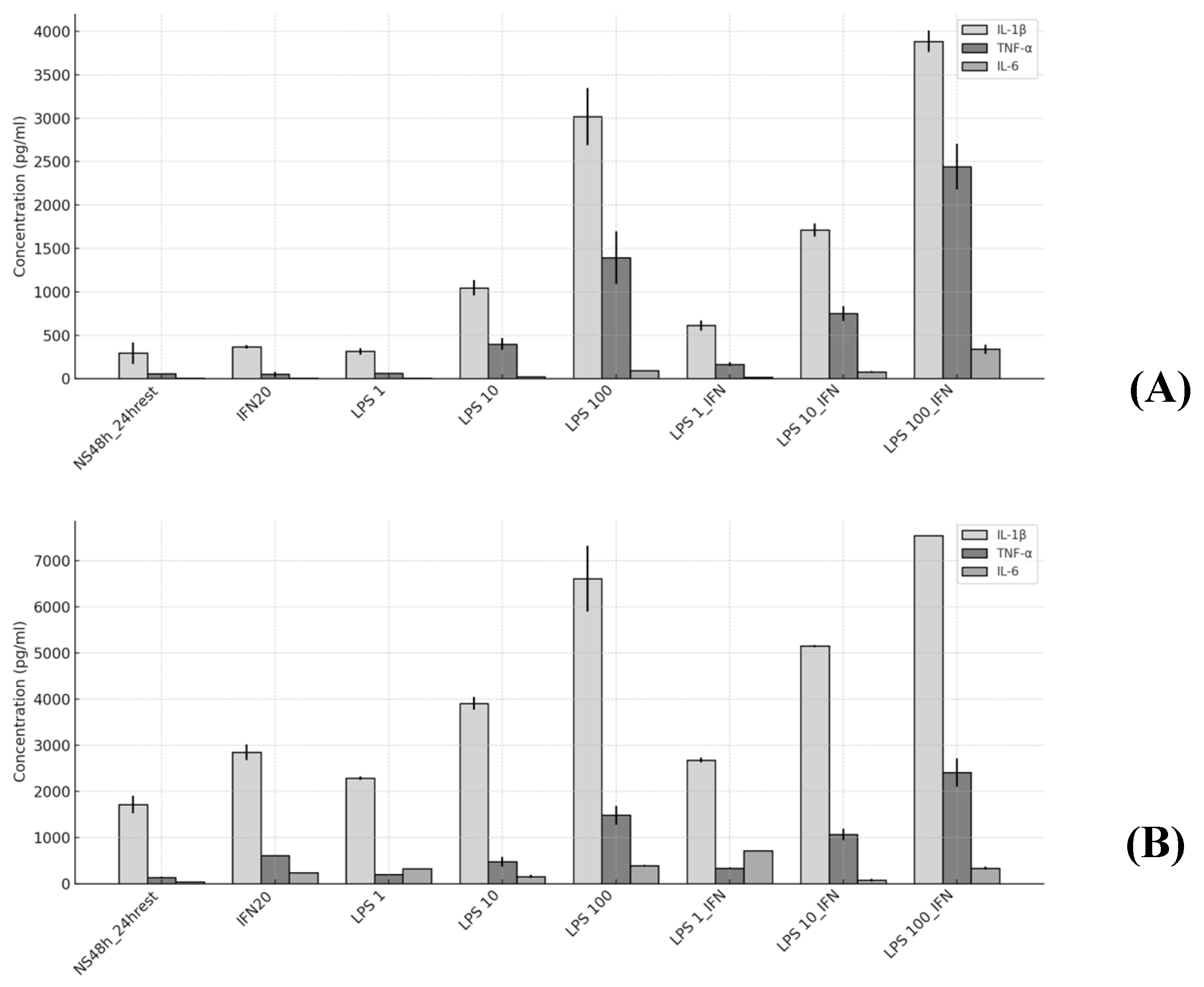
2.4. Infection Assay
3. Discussion
4. Materials and Methods
4.1. Microbial Growth Curve Optimization
4.2. Macrophage Cell Culture
Measurement of Cytokines
4.3. Infection Protocol of Activated Macrophages with P. gingivalis W83
4.4. Statistycal Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Papapanou, P.N.; Sanz, M.; Buduneli, N.; Dietrich, T.; Feres, M.; Fine, D.H.; Flemmig, T.F.; Garcia, R.; Giannobile, W.V.; Graziani, F.; et al. Periodontitis: Consensus report of workgroup 2 of the 2017 World Workshop on the Classification of Periodontal and Peri-Implant Diseases and Conditions. J. Periodontol. 2018, 89, S173–S182. [Google Scholar] [CrossRef]
- Sanz, M.; Beighton, D.; Curtis, M.A.; Cury, J.A.; Dige, I.; Dommisch, H.; Ellwood, R.; Giacaman, R.A.; Herrera, D.; Herzberg, M.C.; et al. Role of microbial biofilms in the maintenance of oral health and in the development of dental caries and periodontal diseases. Consensus report of group 1 of the Joint EFP/ORCA workshop on the boundaries between caries and periodontal disease. J. Clin. Periodontol. 2017, 44 (Suppl. 18), S5–S11. [Google Scholar] [CrossRef] [PubMed]
- Bostanci, N.; Belibasakis, G.N. Porphyromonas gingivalis: An invasive and evasive opportunistic oral pathogen. FEMS Microbiol. Lett. 2012, 333, 1–9. [Google Scholar] [CrossRef]
- Hajishengallis, G.; Darveau, R.P.; Curtis, M.A. The keystone-pathogen hypothesis. Nat. Rev. Microbiol. 2012, 10, 717–725. [Google Scholar] [CrossRef]
- Hajishengallis, G.; Lamont, R.J. Breaking bad: Manipulation of the host response by Porphyromonas gingivalis. Eur. J. Immunol. 2013, 44, 328–338. [Google Scholar] [CrossRef]
- Chopra, A.; Bhat, S.G.; Sivaraman, K. Porphyromonas gingivalis adopts intricate and unique molecular mechanisms to survive and persist within the host: A critical update. J. Oral Microbiol. 2020, 12, 1801090. [Google Scholar] [CrossRef]
- Hajishengallis, G. Periodontitis: From microbial immune subversion to systemic inflammation. Nat. Rev. Immunol. 2015, 15, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Mei, F.; Xie, M.; Huang, X.; Long, Y.; Lu, X.; Wang, X.; Chen, L. Porphyromonas gingivalis and Its Systemic Impact: Current Status. Pathogens 2020, 9, 944. [Google Scholar] [CrossRef]
- Curtis, M.; Aduse-Opoku, J.; Rangarajan, M. Cysteine Proteases of Porphyromonas Gingivalis. Crit. Rev. Oral Biol. Med. 2001, 12, 192–216. [Google Scholar] [CrossRef]
- Curtis, M.A.; Hanley, S.A.; Aduse-Opoku, J. The rag locus of Porphyromonas gingivalis: A novel pathogenicity island. J. Periodontal Res. 1999, 34, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Curtis, M.A.; Kuramitsu, H.K.; Lantz, M.; Macrina, F.L.; Nakayama, K.; Potempa, J.; Reynolds, E.C.; Aduse-Opoku, J. Molecular genetics and nomenclature of proteases of Porphyromonas gingivalis. J. Periodontal Res. 1999, 34, 464–472. [Google Scholar] [CrossRef]
- Curtis, M.A.; Percival, R.S.; Devine, D.; Darveau, R.P.; Coats, S.R.; Rangarajan, M.; Tarelli, E.; Marsh, P.D. Temperature-Dependent Modulation of Porphyromonas gingivalis Lipid A Structure and Interaction with the Innate Host Defenses. Infect. Immun. 2011, 79, 1187–1193. [Google Scholar] [CrossRef]
- Curtis, M.A.; Thickett, A.; Slaney, J.M.; Rangarajan, M.; Aduse-Opoku, J.; Shepherd, P.; Paramonov, N.; Hounsell, E.F. Variable Carbohydrate Modifications to the Catalytic Chains of the RgpA and RgpB Proteases of Porphyromonas gingivalis W50. Infect. Immun. 1999, 67, 3816–3823. [Google Scholar] [CrossRef]
- Reyes, L.; Eiler-McManis, E.; Rodrigues, P.H.; Chadda, A.S.; Wallet, S.M.; Bélanger, M.; Barrett, A.G.; Alvarez, S.; Akin, D.; Dunn, W.A.; et al. Deletion of Lipoprotein PG0717 in Porphyromonas gingivalis W83 Reduces Gingipain Activity and Alters Trafficking in and Response by Host Cells. PLoS ONE 2013, 8, e74230. [Google Scholar] [CrossRef] [PubMed]
- Werheim, E.R.; Senior, K.G.; Shaffer, C.A.; Cuadra, G.A. Oral Pathogen Porphyromonas gingivalis Can Escape Phagocytosis of Mammalian Macrophages. Microorganisms 2020, 8, 1432. [Google Scholar] [CrossRef]
- Laine, M.L.; van Winkelhoff, A.J. Virulence of six capsular serotypes of Porphyromonas gingivalis in a mouse model. Oral Microbiol. Immunol. 1998, 13, 322–325. [Google Scholar] [CrossRef]
- Vernal, R.; Díaz-Zúñiga, J.; Melgar-Rodríguez, S.; Pujol, M.; Diaz-Guerra, E.; Silva, A.; Sanz, M.; Garcia-Sanz, J.A. Activation of RANKL-induced osteoclasts and memory T lymphocytes by Porphyromonas gingivalis is serotype dependant. J. Clin. Periodontol. 2014, 41, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Vernal, R.; León, R.; Silva, A.; Van Winkelhoff, A.J.; Garcia-Sanz, J.A.; Sanz, M. Differential cytokine expression by human dendritic cells in response to different Porphyromonas gingivalis capsular serotypes. J. Clin. Periodontol. 2009, 36, 823–829. [Google Scholar] [CrossRef]
- Nelson, K.E.; Fleischmann, R.D.; DeBoy, R.T.; Paulsen, I.T.; Fouts, D.E.; Eisen, J.A.; Daugherty, S.C.; Dodson, R.J.; Durkin, A.S.; Gwinn, M.; et al. Complete Genome Sequence of the Oral Pathogenic Bacterium Porphyromonas gingivalis Strain W83. J. Bacteriol. 2003, 185, 5591–5601. [Google Scholar] [CrossRef]
- Potempa, J.; Sroka, A.; Imamura, T.; Travis, J. Gingipains, the Major Cysteine Proteinases and Virulence Factors of Porphyromonas gingivalis: Structure, Function and Assembly of Multidomain Protein Complexes. Curr. Protein Pept. Sci. 2003, 4, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Glowczyk, I.; Wong, A.; Potempa, B.; Babyak, O.; Lech, M.; Lamont, R.J.; Potempa, J.; Koziel, J. Inactive Gingipains from P. gingivalis Selectively Skews T Cells toward a Th17 Phenotype in an IL-6 Dependent Manner. Front. Cell. Infect. Microbiol. 2017, 7, 140. [Google Scholar] [CrossRef]
- Martinez, C.d.J.H.; Glessner, J.; Finoti, L.S.; Silva, P.F.; Messora, M.; Della Coletta, R.; Hakonarson, H.; Palioto, D.B. Methylome-wide analysis in systemic microbial-induced experimental periodontal disease in mice with different susceptibility. Front. Cell. Infect. Microbiol. 2024, 14, 1369226. [Google Scholar] [CrossRef]
- Groeger, S.; Herrmann, J.M.; Chakraborty, T.; Domann, E.; Ruf, S.; Meyle, J. Porphyromonas gingivalis W83 Membrane Components Induce Distinct Profiles of Metabolic Genes in Oral Squamous Carcinoma Cells. Int. J. Mol. Sci. 2022, 23, 3442. [Google Scholar] [CrossRef] [PubMed]
- Almubarak, A.; Tanagala, K.K.K.; Papapanou, P.N.; Lalla, E.; Momen-Heravi, F. Disruption of Monocyte and Macrophage Homeostasis in Periodontitis. Front. Immunol. 2020, 11, 330. [Google Scholar] [CrossRef] [PubMed]
- Chapple, C.C.; Srivastava, M.; Hunter, N. Failure of macrophage activation in destructive periodontal disease. J. Pathol. 1998, 186, 281–286. [Google Scholar] [CrossRef]
- Hasturk, H.; Kantarci, A.; Van Dyke, T.E. Oral Inflammatory Diseases and Systemic Inflammation: Role of the Macrophage. Front. Immunol. 2012, 3, 118. [Google Scholar] [CrossRef]
- Veloso, P.; Fernández, A.; Astorga, J.; González-Quintanilla, D.; Castro, A.; Escobar, A.; Hoare, A.; Hernández, M. Lipopolysaccharide from Porphyromonas gingivalis, but Not from Porphyromonas endodontalis, Induces Macrophage M1 Profile. Int. J. Mol. Sci. 2022, 23, 10011. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Ding, L.; Liang, D.; Luo, L. Porphyromonas gingivalis inhibits M2 activation of macrophages by suppressing -ketoglutarate production in mice. Mol. Oral Microbiol. 2018, 33, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 Macrophages and the Th1/Th2 Paradigm. J. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef]
- Yasin, Z.N.M.; Idrus, F.N.M.; Hoe, C.H.; Yvonne-Tee, G.B. Macrophage polarization in THP-1 cell line and primary monocytes: A systematic review. Differentiation 2022, 128, 67–82. [Google Scholar] [CrossRef]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef]
- Curtis, M.A.; Zenobia, C.; Darveau, R.P. The Relationship of the Oral Microbiotia to Periodontal Health and Disease. Cell Host Microbe 2011, 10, 302–306. [Google Scholar] [CrossRef] [PubMed]
- Zenobia, C.; Hajishengallis, G. Porphyromonas gingivalis virulence factors involved in subversion of leukocytes and microbial dysbiosis. Virulence 2015, 6, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Bereta, G.; Goulas, T.; Madej, M.; Bielecka, E.; Solà, M.; Potempa, J.; Gomis-Rüth, F.X. Structure, function, and inhibition of a genomic/clinical variant of Porphyromonas gingivalis peptidylarginine deiminase. Protein Sci. 2019, 28, 478–486. [Google Scholar] [CrossRef]
- A Klein, B.; Tenorio, E.L.; Lazinski, D.W.; Camilli, A.; Duncan, M.J.; Hu, L.T. Identification of essential genes of the periodontal pathogen Porphyromonas gingivalis. BMC Genom. 2012, 13, 578. [Google Scholar] [CrossRef] [PubMed]
- Sisk-Hackworth, L.; Ortiz-Velez, A.; Reed, M.B.; Kelley, S.T. Compositional Data Analysis of Periodontal Disease Microbial Communities. Front. Microbiol. 2021, 12, 617949. [Google Scholar] [CrossRef]
- Xu, X.; Tong, T.; Yang, X.; Pan, Y.; Lin, L.; Li, C. Differences in survival, virulence and biofilm formation between sialidase-deficient and W83 wild-type Porphyromonas gingivalis strains under stressful environmental conditions. BMC Microbiol. 2017, 17, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Pollack-Milgate, S.; Saitia, S.; Tang, J.X. Rapid growth rate of Enterobacter sp. SM3 determined using several methods. BMC Microbiol. 2024, 24, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Tedesco, S.; De Majo, F.; Kim, J.; Trenti, A.; Trevisi, L.; Fadini, G.P.; Bolego, C.; Zandstra, P.W.; Cignarella, A.; Vitiello, L. Convenience versus Biological Significance: Are PMA-Differentiated THP-1 Cells a Reliable Substitute for Blood-Derived Macrophages When Studying in Vitro Polarization? Front. Pharmacol. 2018, 9, 71. [Google Scholar] [CrossRef]
- Huang, Y.; Tian, C.; Li, Q.; Xu, Q. TET1 Knockdown Inhibits Porphyromonas gingivalis LPS/IFN-γ-Induced M1 Macrophage Polarization through the NF-κB Pathway in THP-1 Cells. Int. J. Mol. Sci. 2019, 20, 2023. [Google Scholar] [CrossRef]
- Vallés, G.; Bensiamar, F.; Maestro-Paramio, L.; García-Rey, E.; Vilaboa, N.; Saldaña, L. Influence of inflammatory conditions provided by macrophages on osteogenic ability of mesenchymal stem cells. Stem Cell Res. Ther. 2020, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Guo, H.; Song, A.; Huang, J.; Zhang, Y.; Jin, S.; Li, S.; Zhang, L.; Yang, C.; Yang, P. Progranulin inhibits LPS-induced macrophage M1 polarization via NF-кB and MAPK pathways. BMC Immunol. 2020, 21, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G. Local destruction from distant action. J. Leukoc. Biol. 2020, 108, 1033–1035. [Google Scholar] [CrossRef]
- Hajishengallis, G.; Chavakis, T.; Lambris, J.D. Current understanding of periodontal disease pathogenesis and targets for host-modulation therapy. Periodontology 2000 2020, 84, 14–34. [Google Scholar] [CrossRef] [PubMed]
- Vandendriessche, S.; Cambier, S.; Proost, P.; Marques, P.E. Complement Receptors and Their Role in Leukocyte Recruitment and Phagocytosis. Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Ebersole, J.L.; Dawson, D.; Emecen-Huja, P.; Nagarajan, R.; Howard, K.; Grady, M.E.; Thompson, K.; Peyyala, R.; Al-Attar, A.; Lethbridge, K.; et al. The periodontal war: Microbes and immunity. Periodontology 2000 2017, 75, 52–115. [Google Scholar] [CrossRef] [PubMed]
- Kinane, D.F.; Lappin, D.F. Immune Processes in Periodontal Disease: A Review. Ann. Periodontol. 2002, 7, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Levy, B.D. Resolvins in inflammation: Emergence of the pro-resolving superfamily of mediators. J. Clin. Investig. 2018, 128, 2657–2669. [Google Scholar] [CrossRef]
- Lam, R.S.; O’Brien-Simpson, N.M.; Holden, J.A.; Lenzo, J.C.; Fong, S.B.; Reynolds, E.C. Unprimed, M1 and M2 Macrophages Differentially Interact with Porphyromonas gingivalis. PLoS ONE 2016, 11, e0158629. [Google Scholar] [CrossRef]
- Maeß, M.B.; Wittig, B.; Cignarella, A.; Lorkowski, S. Reduced PMA enhances the responsiveness of transfected THP-1 macrophages to polarizing stimuli. J. Immunol. Methods 2014, 402, 76–81. [Google Scholar] [CrossRef]
- Shiratori, H.; Feinweber, C.; Luckhardt, S.; Linke, B.; Resch, E.; Geisslinger, G.; Weigert, A.; Parnham, M.J. THP-1 and human peripheral blood mononuclear cell-derived macrophages differ in their capacity to polarize in vitro. Mol. Immunol. 2017, 88, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Schmidt, S.V.; Sander, J.; Draffehn, A.; Krebs, W.; Quester, I.; De Nardo, D.; Gohel, T.D.; Emde, M.; Schmidleithner, L.; et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 2014, 40, 274–288. [Google Scholar] [CrossRef]
- Choudhuri, S.; Chowdhury, I.H.; Garg, N.J. Mitochondrial Regulation of Macrophage Response Against Pathogens. Front. Immunol. 2021, 11. [Google Scholar] [CrossRef]
- Boutaga, K.; Van Winkelhoff, A.J.; Vandenbroucke-Grauls, C.M.J.E.; Savelkoul, P.H.M. Comparison of real-time PCR and culture for detection of Porphyromonas gingivalis in subgingival plaque samples. J. Clin. Microbiol. 2003, 41, 4950–4954. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, M.; Llama-Palacios, A.; Fernández, E.; Figuero, E.; Marín, M.; León, R.; Blanc, V.; Herrera, D.; Sanz, M. An in vitro biofilm model associated to dental implants: Structural and quantitative analysis of in vitro biofilm formation on different dental implant surfaces. Dent. Mater. 2014, 30, 1161–1171. [Google Scholar] [CrossRef] [PubMed]
- Marin, M.J.; Ambrosio, N.; Herrera, D.; Sanz, M.; Figuero, E. Validation of a multiplex qPCR assay for the identification and quantification of Aggregatibacter actinomycetemcomitans and Porphyromonas gingivalis: In vitro and subgingival plaque samples. Arch. Oral Biol. 2018, 88, 47–53. [Google Scholar] [CrossRef]
- Marin, M.J.; Ambrosio, N.; O’Connor, A.; Herrera, D.; Sanz, M.; Figuero, E. Validation of a multiplex qPCR assay for detection and quantification of Aggregatibacter actinomycetemcomitans, Porphyromonas gingivalis and Tannerella forsythia in subgingival plaque samples. A comparison with anaerobic culture. Arch. Oral Biol. 2019, 102, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.D. Chapter 1: Continuous Population Models for Single Species. In Mathematical Biology: I. An Introduction, 3rd ed.; Springer: New York, NY, USA, 2002; pp. XXIII, 551. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| T (hours) | OD (550 nm) (SD) | CFU/mL (SD) |
|---|---|---|
| 0 (0 h) | 0.233 (0.038) | 3.70 × 108 (6.52 × 107) |
| 1 (2 h) | 0.248 (0.022) | 4.75 × 108 (1.43 × 108) |
| 2 (4 h) | 0.327 (0.017) | 6.43 × 108 (9.55 × 107) |
| 3 (6 h) | 0.487 (0.043) | 9.43 × 108 (3.47 × 108) |
| 4 (8 h) | 0.674 (0.035) | 9.80 × 108 (2.62 × 108) |
| 5 (10 h) | 0.870 (0.039) | 1.29 × 109 (4.93 × 108) |
| 6 (12 h) | 1.042 (0.043) | 1.21 × 109 (6.02 × 108) |
| IL-1 β (pg/mL) | TNF-α (pg/mL) | IL-6 (pg/mL) | |
|---|---|---|---|
| NS 48 h_24 h rest (Baseline) | 1.00 | 1.00 | 1.00 |
| IFN-γ | 1.25 | 0.86 | 1.06 |
| * | 1.66 | 4.44 | 5.79 |
| LPS 1 | 1.07 | 1.06 | 0.94 |
| * | 1.33 | 1.46 | 7.88 |
| LPS 10 | 3.57 | 6.83 | 3.29 |
| * | 2.27 | 3.47 | 3.90 |
| LPS 100 | 10.31 | 23.91 | 13.34 |
| * | 3.84 | 10.81 | 9.53 |
| LPS 1_IFN- γ | 2.09 | 2.81 | 2.79 |
| * | 1.56 | 2.43 | 17.54 |
| LPS 10_IFN- γ | 5.84 | 12.86 | 11.42 |
| * | 2.99 | 7.80 | 1.89 |
| LPS 100_IFN- γ | 13.26 | 41.83 | 49.93 |
| * | 4.38 | 17.55 | 8.26 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
La Rosa, M.; Spagnolo, A.; Gamonal, J.D.; Marín, M.J.; Figuero, E.; Sanz, M. In Vitro Infection of Human Macrophages with Porphyromonas gingivalis W83. Int. J. Mol. Sci. 2025, 26, 1054. https://doi.org/10.3390/ijms26031054
La Rosa M, Spagnolo A, Gamonal JD, Marín MJ, Figuero E, Sanz M. In Vitro Infection of Human Macrophages with Porphyromonas gingivalis W83. International Journal of Molecular Sciences. 2025; 26(3):1054. https://doi.org/10.3390/ijms26031054
Chicago/Turabian StyleLa Rosa, Martina, Alessandra Spagnolo, Juan Daniel Gamonal, Maria Jose Marín, Elena Figuero, and Mariano Sanz. 2025. "In Vitro Infection of Human Macrophages with Porphyromonas gingivalis W83" International Journal of Molecular Sciences 26, no. 3: 1054. https://doi.org/10.3390/ijms26031054
APA StyleLa Rosa, M., Spagnolo, A., Gamonal, J. D., Marín, M. J., Figuero, E., & Sanz, M. (2025). In Vitro Infection of Human Macrophages with Porphyromonas gingivalis W83. International Journal of Molecular Sciences, 26(3), 1054. https://doi.org/10.3390/ijms26031054