Abstract
In modern pharmaceutical research and development (R&D), drug discovery remains a challenging process. Artificial intelligence (AI) has been extensively incorporated into various phases of drug discovery and development. AI enable effectively extract molecular structural features, perform in-depth analysis of drug–target interactions, and systematically model the relationships among drugs, targets, and diseases. These approaches improve prediction accuracy, accelerate discovery timelines, reduce costs from trial and error methods, and enhance success probabilities. This review summarizes recent advances in AI applications for drug design, including target identification, synthetic accessibility prediction, lead optimization, and ADMET property evaluation. Furthermore, it introduces various deep learning tools to guide researchers in selecting and implementing the most appropriate AI-driven strategies throughout the drug discovery process. We hope it can establish a conceptual framework intended to advance AI-driven methodologies in pharmaceutical research by comprehensively organizing novel perspectives and critical insights.
1. Introduction
The traditional drug discovery paradigm faces formidable challenges characterized by lengthy development cycles, prohibitive costs, and high preclinical trial failure rate [1]. The process from lead compound identification to regulatory approval typically spans over 12 years with cumulative expenditures exceeding $2.5 billion [2]. Clinical trial success probabilities decline precipitously from Phase I (52%) to Phase II (28.9%), culminating in an overall success rate of merely 8.1% [3,4,5,6,7]. Global efforts are intensifying to address the persistent inefficiency challenges in drug development. The strategy involves diversifying therapeutic targets to overcome the limitations of traditional approaches. This aims to reduce the preclinical attrition rate of candidate drugs, improve R&D efficiency, and optimize the cost-effectiveness ratio to alleviate the burden of high investment.
Computational molecular modeling has catalyzed a paradigm shift in pharmaceutical research. It enables the precise simulation of receptor–ligand interactions and the optimization of lead compounds [8,9]. For example, Talukder et al. integrated molecular docking, QSAR, and molecular dynamics to identify phytochemical inhibitors targeting EGFR in non-small cell lung cancer [10]. Similarly, Kaur et al. designed blood–brain barrier (BBB) permeable β-secretase enzyme (BACE-1) inhibitors for Alzheimer’s disease using 2D-QSAR and ADMET profiling. The novel molecules exhibited good potency, BBB permeability, excellent binding affinity, and stable conformations with BACE-1 [11]. Souza et al. investigated the ability of Artificial Intelligence (AI) to balance binding affinity with drug-likeness in the design of SARS-CoV-2 protease inhibitors using machine learning (ML) [12]. Moreover, Maliyakkal et al. employed QSAR-driven virtual screening to identify Trypanosoma cruzi inhibitors with high predicted efficacy [13]. This paradigm shift has catalyzed the rise of AI-driven drug discovery (AIDD). ML integrates multiple omics data and structural biology insights to provide information for experimental design. Modern drug development workflows increasingly rely on these predictive systems for critical tasks including target prioritization [14,15,16], high-throughput compound screening [17,18,19,20], synthetic route planning [21,22,23], and polymorph screening [24,25,26] (Figure 1). A representative research achievement from Insilico Medicine is rentosertib, an AI-discovered drug that has completed Phase II trials for pulmonary fibrosis, showcasing the power of its AI platform [27]. Parallel advancements in antimicrobial discovery reveal that computational drug design platforms can systematically decode structural determinants of antibiotic efficacy, particularly against drug resistant pathogens [28]. The field’s transformative potential was further validated by the 2024 Nobel Prize in Chemistry, awarded for AI-powered innovations in protein engineering [29].
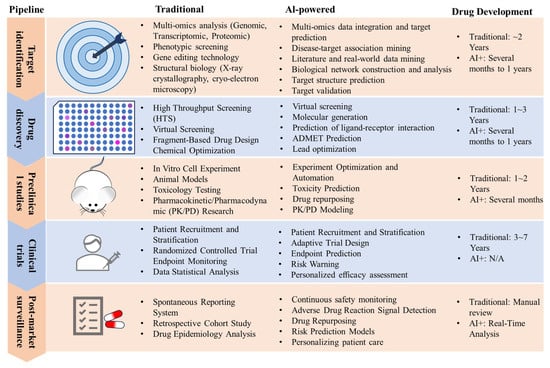
Figure 1.
Comparative analysis of AI applications across multistage drug development pipelines.
AIDD has experienced rapid advances, and Table 1 presents representative AI-designed small molecules currently progressing through clinical trials. A key technological strength lies in the capacity to decode intricate structure-activity relationships, facilitating de novo generation of bioactive compounds with optimized pharmacokinetic properties. The efficacy of these algorithms is intrinsically linked to the quality and volume of training data, particularly in deciphering latent patterns within complex biological datasets [30,31,32,33]. As deep learning architectures continue to evolve, the integration of AIDD into pharmaceutical pipelines is poised to significantly enhance development efficiency and substantially reduce costs.

Table 1.
Selected examples of AI-discovered/designed small molecules in clinical stages.
Table 1.
Selected examples of AI-discovered/designed small molecules in clinical stages.
| Small Molecule | Company | Target | Stage | Indication |
|---|---|---|---|---|
| REC-1245 [34] | Recursion | RBM39 | Phase 1 | Biomarker-enriched solid Tumors and lymphoma |
| REC-3565 [34] | Recursion | MALT1 | Phase 1 | B-Cell Malignancies |
| REC-4539 [34] | Recursion | LSD1 | Phase 1/2 | Small-Cell Lung Cancer |
| REC-4881 [34] | Recursion | MEK Inhibitor | Phase 2 | Familial adenomatous polyposis |
| REC-3964 [34] | Recursion | Selective C. diff Toxin Inhibitor | Phase 2 | Clostridioides difficile Infection |
| REC-7735 [34] | Recursion | PI3Kα H1047R | Preclinical | HER2−HR+ Breast cancer |
| REV102 [34] | Recursion | ENPP1 | Candidate profiling | Hypophosphatasia |
| ISM-6631 [35] | Insilico Medicine | Pan-TEAD | Phase 1 | Mesothelioma, and Solid Tumors |
| ISM-3412 [35] | Insilico Medicine | MAT2A | Phase 1 | MTAP−/− Cancers |
| INS018-055 [35] | Insilico Medicine | TNIK | Phase 2a | IPF |
| ISM-3091 [35] | Insilico Medicine | USP1 | Phase 1 | BRCA mutant cancer |
| ISM-8207 [35] | Insilico Medicine | QPCTL | Phase 1 | Solid Tumors |
| ISM-5043 [35] | Insilico Medicine | KAT6 | Phase 1 | Breast cancer |
| ISM-5939 [35] | Insilico Medicine | ENPP1 | IND Clearance | Solid Tumors |
| ISM-5411 [35] | Insilico Medicine | PHD | Phase 1 | IBD/Anemia of CKD |
| ISM-3312 [35] | Insilico Medicine | 3CLpro | Phase 1 | COVID-19 |
| RLY-4008 [36] | Relay Therapeutics | FGFR2 | Phase 1/2 | FGFR2-altered cholangiocarcinoma |
| RLY-8161 [36] | Relay Therapeutics | NRAS | Preclinical | Solid Tumors |
| RLY-2608 [36] | Relay Therapeutics | PI3Kα | Phase 1/2 | Advanced Breast Cancer |
| EXS4318 [37] | Exscientia | PKC-theta | Phase 1 | Inflammatory and immunologic diseases |
| GTAEXS617 [37] | Exscientia | CDK7 | Phase 1/2 | Solid Tumors |
| SIGX-1094 [38] | Signet Therapeutics | FAK | Phase 1/2 | Solid Tumors |
| BG-89894 [39] | BeiGene | Mat2A | Phase 1 | Advanced Solid Tumors |
| H002 [40] | RedCloud Bio | EGFR | Phase 1 | Non-Small Cell Lung Cancer |
| AC0682 [41] | Accutar Biotech | ER Degrader | Phase 1 | Breast Cancer |
| AC0682 [41] | Accutar Biotech | ER Degrader | Phase 1 | Breast Cancer |
| AC0176 [41] | Accutar Biotech | AR Degrader | Phase 1 | Prostate Cancer |
| AC0676 [41] | Accutar Biotech | BTK Degrader | Phase 1 | Hematology Oncology Indications |
| MDR-001 [42] | MindRank | GLP-1 | Phase 1/2 | Obesity/Type 2 Diabetes Mellitus |
| DF-006 [43] | Drug Farm | ALPK1 | Phase 1 | Hepatitis B/Hepatocellular cancer |
| LAM-001 [44] | OrphAI Therapeutics | mTOR | Phase 2 | PH/BOS |
| HLX-1502 [45] | Healx | N/A b | Phase 2 | Neurofibromatosis Type 1 |
| BGE-105 [46] | BioAge | APJ agonist | Phase 2 | Obesity/Type 2 diabetes |
| BXCL501 [47] | BioXcel Therapeutics | alpha-2 adrenergic | Phase 2/3 | Neurological Disorders |
| EVX-01 [48] | Evaxion Biotech | N/A b | Phase 2 | Metastatic melanoma |
| EVX-02 [48] | Evaxion Biotech | N/A b | Phase 1 | Adjuvant melanoma |
b, no marked information.
This review provides a systematic overview of recent advances in AI for medicinal chemistry design. While previous surveys have broadly outlined AI applications across the entire drug discovery pipeline, this work focuses specifically on two pivotal stages: drug–target interaction (DTI) prediction and lead optimization. We summarize a range of deep learning (DL) tools to assist researchers in selecting and implementing suitable AI-driven strategies within the drug discovery process. In addition to the core focus on DTI and lead optimization, discussions were included on target identification, synthetic feasibility prediction, virtual screening, and ADMET evaluation. It demonstrates that integrating these phases with AI reduces false positives, improves compound prioritization, and accelerates therapeutic design. The discussion also highlights the translational impact of current methods and identifies promising directions for future development.
2. AI Technology
AI develops systems capable of human-like reasoning and decision-making. Contemporary AI systems integrate ML and DL to address pharmaceutical challenges ranging from target validation to formulation optimization.
ML employs algorithmic frameworks to analyze high-dimensional datasets, identify latent patterns, and construct predictive models through iterative optimization processes [49]. ML has evolved into four principal paradigms: supervised learning, unsupervised learning, semi-supervised learning, and reinforcement learning [50]. Supervised learning employs labeled datasets for classification via algorithms like support vector machines (SVMs) and for regression via algorithms like support vector regression (SVR) and random forests (RFs) [51,52]. Unsupervised learning identifies latent data structures through clustering and dimensionality reduction techniques (such as principal component analysis and K-means clustering) to reveal underlying pharmacological patterns and streamline chemical descriptor analysis [53,54]. In contrast, t-distributed stochastic neighbor embedding (t-SNE) primarily serves as a nonlinear visualization tool, effectively mapping high-dimensional molecular features into low-dimensional spaces to facilitate the interpretation of chemical similarity and class separation [55]. Semi-supervised learning boosts drug–target interaction prediction by leveraging a small set of labeled data alongside a large pool of unlabeled data. This is achieved through model collaboration and by generating simulated data, which enhances prediction reliability [56,57]. Reinforcement learning optimizes molecular design via Markov decision processes, where agents iteratively refine policies to generate inhibitors and balance pharmacokinetic properties through reward-driven strategies [58,59]. A comparison of ML models are shown in Table 2.
Table 2.
Comparison of machine learning models.
Building upon these models, a broad spectrum of shallow learning models (such as SVM, decision Trees, RF, and artificial neural networks) has historically served as the computational backbone of data-driven drug discovery. These models provide strong interpretability, efficient training on moderate-sized datasets, and robust performance for QSAR modeling, activity prediction, and ADMET estimation. Their comparative strengths and limitations are summarized in Table 3, which outlines their respective mechanisms, advantages, and representative applications across medicinal chemistry tasks.
Table 3.
Comparative overview of commonly used shallow machine learning algorithms.
Despite the success of shallow learning models, the representational capacity of shallow models is limited when dealing with complex, nonlinear biological data or multimodal molecular information. This limitation has driven the evolution toward DL architectures. DL, as a subset of ML, processes information through multilayer neural networks. These networks automatically learn a hierarchy of features directly from raw data via a sequence of functional layers (e.g., convolutional layers, self-attention layers). The process begins with initial layers capturing low-level, local patterns (such as atomic properties in a molecule or edges in an image). These elementary features are then progressively combined and transformed through subsequent layers to form higher-level, more abstract representations. This multi-stage, hierarchical processing is powered by nonlinear transformations introduced by activation functions at each step, enabling DL models to approximate highly complex, nonlinear relationships that are inherent in multidimensional biological and chemical data [62]. Different models have different core mechanisms. Convolutional neural networks (CNNs) are designed to analyze grid-like data, owing to their unique convolutional architecture. This capability makes them particularly valuable for biomedical image processing tasks, such as cell morphology analysis [63]. Recurrent neural networks (RNNs), with their internal recurrent connections, effectively model sequential data. For instance, they can process SMILES strings for automated molecular generation [64]. Although RNNs once represented the state-of-the-art in this domain, they have gradually been supplanted by Transformer-based architectures. Transformers excel because they more effectively capture long-range dependencies and contextual relationships in complex datasets. Nonetheless, RNNs still demonstrate advantages in modeling local sequential patterns [65]. Graph neural networks (GNNs) analyze graph structures by propagating neighborhood information, excelling at molecular property prediction (Figure 2A) [66,67]. In contrast, Transformer models use self-attention to capture long-range dependencies in sequences, showing great promise for innovative tasks like designing antiviral analogs (Figure 2B) [66,67,68,69,70].In terms of new molecule generation, Generative adversarial networks (GANs) synthesize structurally diverse candidate molecules through an adversarial training framework involving a generator and a discriminator [71,72,73,74]. Variational autoencoders (VAEs) explore molecular structures within a latent space using an encoder–decoder architecture (Figure 2C) [75,76,77,78,79]. Large language models (LLMs) have shown remarkable capabilities in handling massive multimodal data, and their potential in generative drug design is being gradually uncovered. These models are expected to assist drug discovery by integrating structured knowledge extracted from scientific literature (Figure 2D) [80,81,82,83]. It should be noted that their direct application to generative drug design remains nascent. In the short term, their more established potential lies in supporting rational drug discovery workflows through literature mining, hypothesis generation, and knowledge integration [84]. These diverse DL architectures collectively aim to address critical bottlenecks in drug research, powerfully supporting core tasks such as toxicity prediction, molecular property optimization, and de novo drug design (for a detailed comparative summary of various models, see Table 4).
In practice, many studies employ the same public datasets (such as MoleculeNet, TDC) to ensure fair model assessment and to compare results with prior work [85,86]. However, differences in research objectives, model architectures, and evaluation strategies often lead to diverse reporting practices. Increasingly, hybrid or ensemble models combine complementary strengths. For instance, GNN to capture 2D/3D molecular structures, recurrent architectures to handle sequential representations such as SMILES, and transformer-based models for long-range contextual dependencies [87]. These integrative approaches underscore the value of multi-perspective feature extraction over relying on a single model. Consequently, future benchmarking should evaluate not only raw performance but also the complementary strengths of different models.
Table 4.
Comparison of deep learning models.
Table 4.
Comparison of deep learning models.
| Model | Core Mechanism | Advantages | Disadvantages | Drug Discovery Applications |
|---|---|---|---|---|
| CNNs | Convolution-pooling stack [88] | Convolutional layers extract local features; pooling layers reduce dimensionality [89] | Relies on local receptive fields; struggles with global dependencies | Cellular morphology analysis, drug–drug interaction prediction [90] |
| RNNs | Recurrent connections | Process sequential data via cyclic connections [73,91,92,93] | Gradient vanishing/explosion; weak long-range dependency handling | SMILES-based molecular generation [94] |
| GNNs | Graph convolution | Aggregate neighborhood information through graph message-passing [95,96,97,98,99,100,101] | High computational complexity; hyperparameter sensitivity | Solubility prediction, binding affinity calculation [51,102,103] |
| Transformers | Self-attention mechanism [104] | Capture global dependencies via self-attention mechanisms [105,106] | High memory consumption; prone to overfitting on small datasets | Derivative design, multimodal data integration [69,107] |
| GANs | Generator-discriminator adversarial | Adversarial training between generator and discriminator | Training instability, mode collapse risk | Novel chemical entity synthesis [71] |
| VAEs | Encoder–decoder latent space | Compress molecular features via encoder–decoder architecture | Blurry generated samples, limited diversity | Targeted drug design [78] |
| LLMs | Multi-layer Transformer [108,109] | Multimodal Transformer-based joint reasoning | High training cost, requires domain-specific fine-tuning | Literature knowledge-driven molecular optimization [110] |
Convolutional neural networks, CNNs. Recurrent neural networks, RNNs. Graph neural networks, GNNs. Generative adversarial networks, GANs. Variational autoencoders, VAEs. Large language models, LLMs.

Figure 2.
Deep learning model architectures. (A), Directed message-passing neural network architecture [102]. (B), Transformer model architecture. Key hyperparameters include: N denotes the number of encoder/decoder layers, Muti-head attention represents the attention heads, and the dimension of input embeddings represents the length of the continuous, dense vector to which each discrete input token [104]. (C), VAE model architecture. (D), LLM model architecture. LLM processes diverse inputs through modality-specific encoders that convert heterogeneous data into latent representations. Cross-modal alignment is achieved via a query-based transformer (Q-Former) employing multi-head attention (MH-Attn) mechanisms. The central LLM performs joint reasoning enhanced by MLP-based feature fusion, and multimodal generators synthesize coordinated outputs such as text or images, completing the comprehension–generation cycle.
3. Applications in New Drug Development
The rapid expansion of bioactive compounds and biomedical multi-omics data, accelerated by technologies such as high-throughput virtual screening, has necessitated advanced computational approaches in drug discovery. AI plays a key role in multiple drug development stages, including molecular characterization, structural analysis of target proteins, and virtual screening of lead compounds. ML algorithms, with their predictive modeling capabilities, continuously optimize the entire drug design process. The following sections will highlight innovative applications of AI in these areas, illustrating how technological breakthroughs are driving further advances in new drug development.
3.1. Applications in Tertiary Structure of Target Proteins
The tertiary structure of a protein is formed by the folding of its amino acid sequence through complex atomic interactions, directly determining its biological function [111]. Since protein dysfunction is a key pathological mechanism in many diseases, accurately determining its three-dimensional structure is critical for structure-based drug discovery [112,113]. However, the immense computational cost of modeling complex molecular folding and dynamics renders traditional simulation-based methods a bottleneck in early drug development.
AlphaFold has revolutionized the field of structural biology by enabling data-driven prediction of protein conformations directly from sequence information [114,115]. AlphaFold2 achieves remarkable accuracy in predicting monomeric protein folds but is inherently limited to static conformations. It does not capture dynamic processes, such as allosteric transitions, conformational flexibility, or ligand-induced rearrangements. Consequently, its application in protein–ligand and protein–protein interaction studies remain constrained. On the other hand, more recent frameworks like AlphaFold-Multimer and AlphaFold3 can predict complexes of proteins, nucleic acids, and small molecules. However, they still cannot fully model dynamic interactions, which remain dependent on molecular dynamics simulations [116,117]. Despite these limitations, such models have shown practical value in structure-based drug discovery. For instance, through computational prioritization and preclinical validation, researchers successfully identified a potent lead compound that acts as a trace amine-associated receptor 1 (TAAR1) agonist [118]. Moreover, integration of structure prediction with generative AI systems further empowers de novo drug design. For example, the PCMol model enables simultaneous multi-target candidate molecule optimization while maintaining synthetic feasibility [119]. In summary, the synergy between structure prediction and AI-driven chemistry platforms can be directly applied to hit identification in drug discovery, offering viable development pathways for understudied targets [120].
Beyond AlphaFold, several other DL frameworks contribute to tertiary structure prediction and protein design. The RoseTTAFold model can design entirely novel proteins not observed in nature, expanding the druggable structural space [121,122]. Similarly, the trRosetta (transform-restrained Rosetta) server is a deep learning-driven platform for rapid and accurate de novo protein structure prediction. Unlike traditional homology modeling approaches, trRosetta excels in de novo prediction when no experimentally resolved templates are available [123]. Meanwhile, ESMFold, a Transformer-based protein language model, achieves rapid sequence to structure prediction through training at various scales. It is up to ten times faster than traditional methods and maintains accuracy even for understudied proteins with limited structural data [113]. Although its precision is slightly below mainstream benchmarks, this speed advantage makes such models critical for streamlining early-stage drug design. Overall, these technological advancements are accelerating structure-guided therapeutic development by enhancing computational efficiency and enabling structural innovation.
3.2. Applications in Target Identification
In precision medicine, the growing complexity of multifactorial diseases has created an urgent need for new molecular targets. However, the number of clinically validated candidate drug targets is still limited, patient applicability is narrow, and clinical-trial failure rates remain high [124]. Until 2022, fewer than 500 drug targets had been successfully established worldwide, a figure that falls far short of the needs for complex disease treatment [125,126]. Consequently, discovering and evaluating new therapeutic targets has become a central focus of drug development. AI has emerged as a key enabler in this area by systematically integrating multi-scale biomedical data to perform probabilistic target identification with enhanced mechanistic interpretability [14].
Modern target discovery relies on three core strategies: experimental validation, multi-omics integration, and computational modeling. Experimental approaches use functional genomics tools like CRISPR-based screens to directly validate connections between targets and diseases [127,128,129,130,131]. Multi-omics platforms integrate genomic, proteomic, and metabolomic data to systematically identify how genes influence disease characteristics [132,133,134,135,136,137]. Computational methods use ML and structural biology to prioritize drug targets. Techniques like inverse docking (which screens a single ligand against multiple protein targets) and pharmacophore analysis help identify promising candidates, reducing the need for experimental resources [138,139,140].
As biomedical datasets have grown, AI techniques leveraging large scale data analysis, pattern recognition and network construction have found broad application in target discovery. For example, AI analysis of transcriptome profiles from young and aged skeletal muscle identified key drivers of muscle aging [141]. At the same time, Transformer-based architectures have been particularly impactful for integrating multi-omics datasets. In single-cell transcriptomics, pre-trained transformer models learn gene relationships through attention mechanisms to capture gene–gene interactions. This enables researchers to accelerate the discovery of key network regulators and candidate therapeutic targets (Figure 3A) [142]. In parallel, building on pretrained protein language models have transformed target prioritization by integrating data across human, murine, and cellular contexts. Frameworks such as protein essentiality predictors use a multi-module structure that encodes protein sequences, weights biological context information, and classifies essentiality probabilities. These models generate protein essentiality scores that assist in identifying key therapeutic targets, cancer prognostic markers, and microprotein functions (Figure 3B) [143].
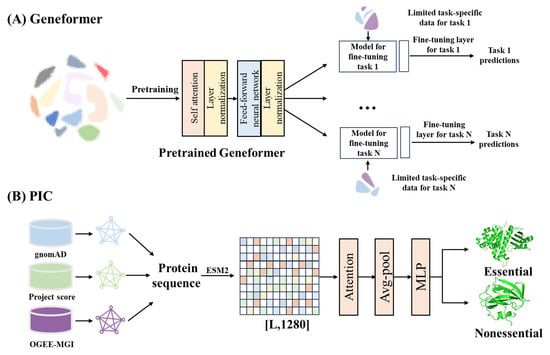
Figure 3.
Applications of AI in target discovery. (A), Transformer-based gene network model for single-cell transcriptomics [142]. (B), Protein essentiality prediction framework integrating multi-source biological data [143]. ESM2, Evolutionary scale modeling 2. MLP, Multilayer perceptron. Avg-pool, Average pooling. Attention, Attention mechanism.
Alongside these strategies, by applying RefMap to human induced pluripotent stem cells (iPSC) derived motor neurons, the researchers mapped noncoding regulatory regions to target genes, identifying 690 ALS-associated candidates and achieving a fivefold increase in recovered heritability [144]. Finally, AI-powered drug-repurposing platforms (such as deepDTnet, which embeds chemical, genomic, phenotypic, and cellular network data) have successfully predicted topotecan as a related orphan receptor-gamma t inhibitor and demonstrated its efficacy in a mouse model of multiple sclerosis [145].
As algorithmic sophistication converges with expanding biomedical datasets, AI-driven target discovery is transitioning from exploratory research to clinical implementation. Through their bridging of mechanistic insights and therapeutic design, these technologies promise to streamline drug development cycles and advance precision medicine.
3.3. Applications in Drug–Target Interaction Prediction
Accurate prediction of drug–target interactions is a cornerstone of modern drug discovery, providing key insights to understand binding mechanisms and directly guiding rational therapeutic design. Molecular docking is a core computational approach in this field, aiming to predict the preferred orientation and conformation of small-molecule ligands when bound to protein targets, while estimating their binding affinity through scoring functions. By combining search algorithms and energy-based scoring, docking enables virtual screening of large chemical libraries to identify potential binders efficiently [146]. Thanks to the close integration of ML and structural biology, this field has made significant progress in recent years. One of the prominent directions is the innovation of virtual screening methods. For example, molecular docking approaches optimized through active learning employ intelligent sampling strategies to effectively balance chemical space exploration with the identification of potentially highly active molecules. Such methods have demonstrated the ability to recover over 80% of experimentally confirmed active compounds while reducing computational costs by a factor of 14. Moreover, over 90% of active scaffolds were retained within the top 5% of model-predicted compounds, preserving the diversity of confirmed actives [147].
Innovative techniques for drug–target interaction (DTI) prediction are overcoming traditional limitations. To address the challenge of limited training data and poor generalization, ML based on pre-trained protein language models have significantly enhanced extrapolation capabilities by analyzing the structural determinants of molecular recognition. These systems have been shown to accurately distinguish true binders from decoy compounds. Furthermore, they offer interpretable visualizations of interactions, enabling the use of embeddings to represent the functional characteristics of human cell surface proteins [148].
The deep integration of DL and molecular docking has catalyzed a transformation in virtual screening platforms. These platforms have been extended to multi-target pharmacological studies and to the creation of customized, synergistic kinase inhibition regimens for aggressive cancers through transcriptome-driven models (Figure 4A) [149]. Traditional docking involves generating candidate poses and scoring them based on binding energy. DL now learns from docking outcomes to accelerate screening across vast chemical libraries. For instance, Deep Docking utilizes QSAR deep models to estimate the docking outcome for yet unprocessed entries. In this approach, a small subset of compounds is first docked. A deep neural network is then trained on this subset to predict the docking scores for the remaining library, which dramatically reduces computation time while maintaining high accuracy (Figure 4B) [150,151]. Similarly, receptor activation pattern analysis could guide the development of dual-target agonists with enhanced pharmacological properties [152].
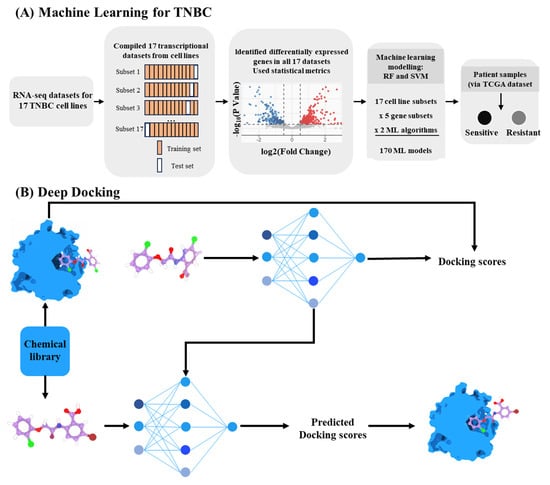
Figure 4.
AI applications in drug–target interaction prediction. (A), Transcriptome-guided framework for target prioritization [149]. (B), Deep Docking approach combining molecular docking and neural prediction [150].
However, systemic challenges still need to be overcome. Rare diseases and emerging pathogens often lack sufficient labeled data, limiting model generalizability. Current predictors often suffer from reduced reliability due to dataset bias. To mitigate these issues, transfer learning and zero-shot learning strategies have been increasingly adopted. For example, TxGNN, a graph neural network model developed for zero-shot drug repurposing, leverages large-scale medical knowledge graphs to predict indications and contraindications even for diseases without existing therapies [95]. Future advances will likely require systematic curation of balanced interaction datasets combined with bias-resistant and transfer-learning-enabled modeling architectures to improve predictive reliability. Recent DL models in the target prediction tasks were collected in Table 5.
Table 5.
DL Models for target prediction frameworks.
3.4. Applications in Virtual Screening of Lead Compounds
Virtual screening has emerged as a transformative approach in lead compound identification, overcoming the cost and scalability limitations of traditional high-throughput screening (HTS). This capability is achieved through the AI-driven prioritization of bioactive molecules from extensive chemical libraries [161]. The integration of structural informatics with DL has proven particularly impactful in antimicrobial discovery. For instance, directed message-passing neural network (D-MPNN) captures complex atomic relationships by transmitting messages along chemical bonds, generating molecular embeddings that encode both local and global structural information. This approach achieves high predictive accuracy for antimicrobial and anticancer lead discovery (Figure 5A) [51]. This computational framework has been further extended to develop narrow-spectrum therapeutics selectively targeting high-risk pathogens like Acinetobacter baumannii, demonstrating precision in addressing antimicrobial resistance [162]. Subsequent innovations combining structural feature analysis with bioactivity prediction enabled the discovery of Helicobacter pylori growth inhibitors [163]. Despite these advances, the limited interpretability of DL remains a critical challenge in virtual screening. Recent explainable AI frameworks address this gap by incorporating chemical substructure recognition and structural analysis into graph-based neural networks, thereby maintaining interpretable design principles while exploring novel molecular scaffolds [28].
The therapeutic applications of such models span multiple disease domains, with notable progress in anticancer and metabolic drug development. ML approaches combining molecular feature encoding with dimensionality reduction techniques have identified validated antidiabetic natural [164]. In oncology, proteomics-driven prediction frameworks that integrate protein interaction networks demonstrate robust performance through noise-resistant algorithms. However, while these approaches show promise, their broader adoption requires addressing dataset limitations to mitigate overfitting risks inherent to proteomic-based deep learning models [165].
Finally, bioactivity prediction has been revolutionized through meta-learning architectures that mitigate data scarcity and experimental variability. A representative framework trained on cross-platform bioactivity data achieves robust generalization capabilities, enabling reliable predictions for novel drug development even with limited training samples (Figure 5B) [166]. Recent DL models in the virtual screening and bioactivity prediction tasks were collected in Table 6.
Table 6.
DL models for virtual screening and bioactivity prediction.
Table 6.
DL models for virtual screening and bioactivity prediction.
| No. | Model Name | Framework Description | Web |
|---|---|---|---|
| 1 | Chemprop | A deep learning package implementing Directed Message Passing Neural Networks (D-MPNNs) for molecular property prediction. It efficiently predicts physicochemical properties (such as logP, reaction barriers) and bioactivity, enabling rapid ADME/efficacy assessment in lead-compound screening to guide candidate optimization and reduce experimental cost [102]. | https://github.com/chemprop/chemprop (accessed on 20 August 2025) |
| 2 | iPADD | Screening molecular fingerprint features through feature selection strategies to predict the activity of anti-diabetic compounds using an XGBoost model [164]. | https://github.com/llllxw/iPADD/blob/main/README.md (accessed on 20 August 2025) |
| 3 | DRUMLR | An ensemble machine learning framework leverages proteomic and phosphoproteomic data to rank over 400 anticancer drugs by efficacy, enabling rapid prioritization of high-potential leads [165]. | https://github.com/CutillasLab/DRUMLR (accessed on 20 August 2025) |
| 4 | ActFound | A meta-learning and pairwise-learning bioactivity model that rapidly adapts to small assay datasets to predict relative activity differences with high precision, requiring minimal fine-tuning [166]. | https://github.com/BFeng14/ActFound (accessed on 20 August 2025) |
| 5 | TOML-BERT | This dual-level pretrained model combines self-supervised learning on molecular structures with domain-knowledge transfer using pseudo-labels. It integrates atomic and molecular-level tasks to achieve state-of-the-art ADMET prediction accuracy across ten drug datasets, especially when labeled data is scarc [167]. | https://github.com/yanjing-duan/TOML-BERT (accessed on 20 August 2025) |
| 6 | FP-GNN | A hybrid GNN model that fuses molecular-graph structural information with fingerprint-based substructure features, markedly improving prediction accuracy for molecular properties [168]. | https://github.com/idrugLab/FP-GNN (accessed on 20 August 2025) |
| 7 | ChemBERTa2 | A model pretrained on chemical molecular structures (SMILES) that efficiently predicts a wide range of physicochemical and bioactivity properties [169]. | https://github.com/miservilla/ChemBERTa (accessed on 20 August 2025) |
| 8 | Uni-Mol | This 3D molecular deep-learning framework is pretrained on extensive datasets of small molecules and protein binding pockets. It can directly predict molecular physicochemical properties, generate accurate 3D conformations, and simulate drug-target binding modes [170]. | https://github.com/deepmodeling/Uni-Mol/tree/main/unimol (accessed on 20 August 2025) |
| 9 | SPMM | A multimodal molecular model that jointly learns from both structural representations and associated properties, enabling bidirectional prediction and generation [171]. | https://github.com/jinhojsk515/SPMM/ (accessed on 20 August 2025) |
| 10 | MoLFormer | An efficient Transformer model pretrained on large-scale chemical SMILES datasets, capable of accurately predicting molecular properties to aid both drug discovery and materials design [172]. | https://github.com/IBM/molformer (accessed on 20 August 2025) |
TOML-BERT, Task-oriented multilevel learning based on BERT. FP-GNN, fingerprints and graph neural network. SPMM, Structure–Property Multi-Modal foundation model.

Figure 5.
AI applications in lead compound virtual screening. (A), Graph-based neural network architecture for antimicrobial discovery [51]. (B), Meta-learning framework addressing data scarcity across assays [166].
3.5. Application in the Generation of Lead Compounds
The identification and generation of lead compounds remain pivotal challenges in early drug discovery. Current estimates suggest the chemical space contains 1023–1060 potential drug-like molecules, yet only ~108 have been synthesized to date [173,174]. Lead compound generation and optimization constitute one of the most critical bottlenecks in early-stage drug discovery, since even subtle structural modifications can profoundly influence potency, selectivity, pharmacokinetics, and safety profiles [175]. Traditional approaches such as molecular docking, and free-energy perturbation (FEP) have provided valuable guidance but often rely on predefined chemical rules or approximated scoring functions, limiting their capacity to explore the vast chemical space and to capture complex structure–activity relationships [176].
Deep generative models trained on structural patterns and bioactivity relationships enable de novo design of novel molecular entities with optimized drug-like properties, effectively expanding the scope of virtual screening libraries. Equivariant diffusion models enable the generation of novel ligands conditioned on protein pockets [177]. Meanwhile, toxicity-controlled ligand generation frameworks integrate safety considerations directly into the design process [178]. Additionally, DiffGui facilitates the simultaneous generation of atoms and bonds, producing molecules that exhibit high structural rationality and desirable molecular properties [179]. Compared with classical QSAR or docking-based strategies, these models not only enhance hit rates and novelty but also improve synthetic feasibility and safety awareness, thereby redefining the state of the art in lead optimization.
VAEs encode molecules into a latent space and reconstruct new structures while optimizing predicted bioactivity. This approach has successfully developed novel compounds with high potency against parasitic enzymes, exhibiting IC50 values of 0.023 μM and 0.025 μM against P. falciparum 3D7 (Figure 6A) [180]. The SyntheMol platform, through combining a validated library of chemical transformations with Monte Carlo tree search, generated structurally novel antibiotics with strong activity against multidrug-resistant Acinetobacter baumannii [181].For tuberculosis treatment, a target-aware generative system successfully identified protease inhibitors with measurable efficacy against Mycobacterium tuberculosis, discovering 14 compounds with significant inhibitory activity against the ClpP protease of Mycobacterium tuberculosis. The most potent compound had an IC50 of 1.9 μM [182].
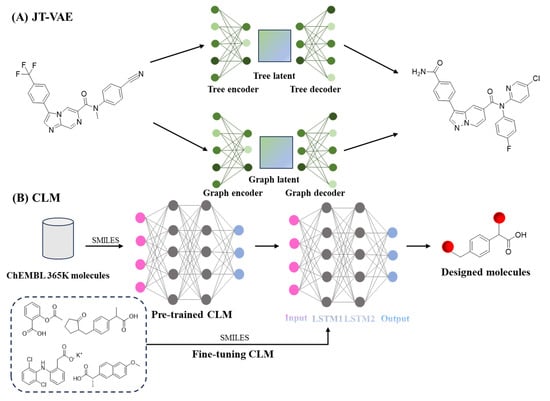
Figure 6.
Application of AI in lead compound generation. (A), Variational autoencoder-based generative framework [180]. (B), Chemical language model (CLM) for SMILES-based molecular design [94]. LSTM, long short-term memory.
Recent advances in chemical language modeling have further enhanced generative capabilities [183,184,185]. Chemical language models (CLMs), inspired by natural language processing, treat SMILES strings as sequences of tokens. Pretrained on large chemical datasets, these models learn the “grammar” of chemistry and can generate valid, synthetically accessible molecules. When fine-tuned on small datasets, CLMs can design dual-target ligands, such as multitarget compounds for metabolic disorders (Figure 6B) [94]. In a complementary approach, transcriptomic signature-driven generative models are designed to reconstruct drug-induced biological profiles. For example, they have been applied to repurpose therapies for pancreatic cancer by efficiently screening clinical compound libraries [186]. These synergistic innovations establish generative AI as a versatile platform for chemical space exploration. Their integration into high-throughput experimental workflows is accelerating therapeutic development across infectious diseases, metabolic disorders, and oncology. A summary of recent deep learning models for lead compound generation is provided in Table 7.
Table 7.
DL models for the generation of lead compounds.
3.6. Applications in Synthesis Prediction in Drug Discovery
The integration of computational synthesis prediction into drug discovery pipelines has emerged as a transformative strategy for addressing the inherent complexity and cost challenges in therapeutic development [201]. Researchers embed AI within the design-make-test-analyze (DMTA) cycle to accelerate synthetic route planning, while also ensuring practical feasibility through reaction condition optimization and outcome validation [23]. For instance, Thakkar et al. employ a Monte Carlo tree search guided by learned policies to break down target molecules into purchasable precursors, typically completing full routes in under one minute. Building on that approach, a lightweight classifier predicts retrosynthetic accessibility thousands of times faster than a full search by estimating whether a viable route exists [202].
Beyond route finding, predicting synthesis feasibility via learned metrics has matured significantly. DeepSA, trained on 3.6 million SMILES from public and proprietary sources, achieves an AUROC of 0.896 for synthesis accessibility. This model enables chemists to prioritize cost-effective and readily synthesizable molecules, reducing experimental attrition and accelerating lead optimization [22].AI-driven advances in reaction prediction prioritize spatial molecular interactions. In late-stage functionalization, where selective modification of complex molecular scaffolds is required, geometric deep learning plays a crucial role. With researchers integrating 3D atomic coordinates into GNNs, these models quantify both steric and electronic interactions to predict regioselectivity and reaction outcomes. This enables chemists to design site-selective transformations with improved efficiency and precision (Figure 7A) [203].
Complementary innovations address forward reaction prediction. Transformer-based models (e.g., T5Chem) treat reactions as text-to-text problems, achieving state-of-the-art accuracy in classification, retrosynthesis, and yield estimation on USPTO_500_MT data (SHAP analysis providing functional group-level interpretability) [204]. Graph-based reaction prediction systems, such as GraphRXN, represent another major innovation. Instead of relying on molecular descriptors, they directly encode reactant, reagent, and product graphs, node and edge features to learn reaction patterns. This end-to-end learning enables real-time product yield and regioselectivity prediction and allows seamless integration with robotic synthesis platforms, supporting fully automated chemical workflows (Figure 7B) [205].
With the maturation of multimodal data integration, AI-powered synthesis prediction stands poised to redefine pharmaceutical chemistry, transforming it into a fully data-driven discipline capable of intelligent molecular design and automated synthetic planning. Recent DL models in synthesis prediction tasks are collected in Table 8.
Table 8.
DL Models for the synthesis prediction tasks.
Table 8.
DL Models for the synthesis prediction tasks.
| No. | Model Name | Framework Description | Web |
|---|---|---|---|
| 1 | GSETransformer | An integrated model that combines graph neural networks and sequence processing to predict biosynthetic pathways of natural products. It accelerates route design in drug development and provides a visual interface that supports research workflow efficiency [206]. | https://github.com/momozhangcn/GSETRetro (accessed on 20 August 2025) |
| 2 | Molecular Transformer | An attention-based model that unifies reaction prediction and retrosynthetic analysis for drug molecules. It maintains strong accuracy on novel compounds and serves as an effective tool for planning pharmaceutical syntheses [207]. | https://github.com/pschwllr/MolecularTransformer (accessed on 20 August 2025) |
| 3 | DeepSA | A deep-learning chemical language model that predicts synthetic accessibility from molecular structure. It prioritizes easily synthesizable compounds and reduces both development time and cost [22]. | https://github.com/Shihang-Wang-58/DeepSA (accessed on 20 August 2025) |
| 4 | Retro-MTGR | A multitask learning framework that uses molecular structure features to predict key bond disconnections and leaving groups in single-step retrosynthesis. It provides an efficient tool for planning synthetic routes [208]. | https://github.com/zpczaizheli/Retro-MTGR (accessed on 20 August 2025) |
| 5 | LocalRetro | A retrosynthesis prediction model that integrates local molecular structure analysis with global attention mechanisms. It accurately designs synthetic routes for a broad range of drug-like molecules [209]. | https://github.com/kaist-amsg/LocalRetro (accessed on 20 August 2025) |
| 6 | RetroTRAE | An atom-environment-aware model that predicts reactants directly for single-step retrosynthesis. It learns chemical fragment patterns and achieves 61.6% accuracy on benchmark datasets, surpassing SMILES-based methods [210]. | https://github.com/knu-lcbc/RetroTRAE (accessed on 20 August 2025) |
| 7 | ReroSub | An end-to-end retrosynthesis model that automatically identifies conserved molecular substructures to simplify reaction prediction. It improves accuracy by over 5% compared with template-based approaches and removes the need for predefined reaction templates [211]. | https://github.com/fangleigit/RetroSub (accessed on 20 August 2025) |
| 8 | G2GT | A hybrid framework combining graph networks and Transformer architectures in an encoder–decoder design. It uses self-training to predict required reactants for a target molecule and guides retrosynthetic route construction with high precision [212]. | https://github.com/ZaiyunLin/G2GT_2 (accessed on 20 August 2025) |
| 9 | MolecularGET | A fusion model that combines graph neural networks with Transformer encoders to integrate structural and sequential chemical information. It enhances retrosynthesis prediction accuracy and accelerates route design [213]. | https://github.com/papercodekl/MolecularGET (accessed on 20 August 2025) |
| 10 | Graph2SMILES | A template-free neural model that predicts reactants or products directly from molecular graphs. It improves the accuracy of both retrosynthesis and forward reaction prediction without complex preprocessing [214]. | https://github.com/coleygroup/Graph2SMILES (accessed on 20 August 2025) |
| 11 | Graph2Edits | An edit-based architecture that applies stepwise graph modifications to infer feasible reactants from products. It achieves 55.1% accuracy in single-step prediction and performs well in complex multi-center transformations [208]. | https://github.com/Jamson-Zhong/Graph2Edits (accessed on 20 August 2025) |
| 12 | RetroPrime | A two-stage Transformer model that first decomposes a target molecule and then generates plausible reactant sets. It improves retrosynthesis accuracy while increasing diversity and chemical feasibility of predicted routes [215]. | https://github.com/wangxr0526/RetroPrime (accessed on 20 August 2025) |
| 13 | RAscore | A machine learning classifier that estimates synthetic accessibility scores for molecules. It enables large-scale prescreening to enrich chemical libraries with easily synthesizable compounds [202]. | https://github.com/reymond-group/Rascore (accessed on 20 August 2025) |
| 14 | T5Chem | A sequence-to-sequence model based on SMILES representation that unifies multiple chemistry tasks, including reaction prediction, retrosynthesis, classification, and yield estimation. It also supports model interpretability studies [204]. | https://github.com/HelloJocelynLu/t5chem (accessed on 20 August 2025) |
| 15 | GraphRXN | A graph neural network framework that analyzes 2D molecular structures of reactants and products to predict reaction outcomes. It demonstrates strong performance when trained with high-throughput experimental data [205]. | https://github.com/jidushanbojue/GraphRXN (accessed on 20 August 2025) |
| 16 | Geometric deep learning | A 3D-geometry-based framework that integrates structural information with high-throughput experimentation to predict late-stage functionalization outcomes such as yield and regioselectivity. It accelerates physicochemical property optimization for complex drug candidates [203]. | https://github.com/ETHmodlab/lsfml (accessed on 20 August 2025) |
GSETransformer, graph-sequence enhanced transformer. Retro-MTGR, a Multi-Task Graph Representation learning framework for Retrosynthesis prediction. LocalRetro, a local retrosynthesis framework. G2T2, a graph-to-graph transformation model. Molecular GET, molecular graph enhanced transformer. RAscore, retrosynthetic accessibility score. T5Chem, “Text-to-Text Transfer Transformer” (T5) framework.

Figure 7.
Applications of AI in chemical synthesis. (A), Geometric deep learning for regioselectivity prediction and late-stage functionalization [203]. (B), Graph-based and transformer models for reaction and yield prediction [205]. MLP, Multilayer perceptron.
3.7. Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) Prediction in Drug Discovery
The accurate prediction of ADMET properties has become indispensable in modern drug discovery. AI has transformed this domain by enabling systematic analysis of molecular structure-pharmacokinetic relationships, overcoming the cost limitation of traditional experimental approaches. For instance, Bayesian pharmacokinetic (bPK) frameworks combine physicochemical and biological descriptors into composite metrics that quantitatively estimate molecular developability. These models outperform single-endpoint predictors by enabling multi-parameter optimization of absorption, bioavailability, and clearance profiles, effectively prioritizing compounds likely to succeed in downstream stages (Figure 8A) [216]. To address the interpretability–accuracy trade-off, fragment-based explainable AI (FBXAI) models have emerged. These methods decompose molecules into functional fragments and apply attention-based deep networks to learn how each substructure contributes to ADMET scores. Using domain-aware fragmentation and visualization tools such as Grad-CAM, the framework finds key functional domains responsible for properties like toxicity or metabolic stability (Figure 8B) [217].
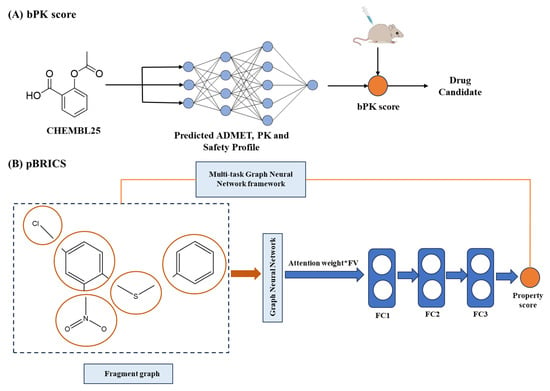
Figure 8.
Applications of AI in ADMET. (A), Bayesian pharmacokinetic (bPK) framework integrating multi-descriptor data for compound prioritization [216]. (B), Fragment-based explainable model linking molecular substructures to pharmacokinetic properties [217]. FV, feature vector. FC, fully connected layers.
Recent advances in toxicity prediction emphasize the integration of mechanistic interpretability with computational precision. Unified modeling approaches achieve superior discriminative power across diverse toxicity scores through DL-enhanced molecular descriptor analysis [218,219]. For example, specialized cardiac safety frameworks rigorously benchmark structural feature representations to elucidate drivers of multi-ion channel cardiotoxicity, significantly enhancing prediction reliability through structurally dissimilar validation sets [52]. Complementary platforms employ interpretable pattern recognition to decode structural determinants across diverse ADMET endpoints [220]. Furthermore, the field continues to evolve through large-scale open-source data initiatives that systematically address historical limitations in dataset diversity and balance, establishing comprehensive benchmarks for next-generation ADMET prediction [221]. Recent DL models in ADMET prediction tasks are collected in Table 9.
Table 9.
DL Models for the ADMET prediction tasks.
4. Future Challenges
Over the past decade, AI has driven transformative advances in drug discovery, particularly in compound property prediction and virtual screening, demonstrating its potential to shorten drug discovery cycles and reduce costs. However, AI is not a panacea for pharmaceutical R&D, as its integration faces multifaceted technical limitations, safety concerns, and ethical dilemmas that demand urgent resolution.
AI model performance is highly dependent on high-quality and sufficient datasets. Despite the vast scale of existing chemical libraries, rigorously curated datasets with robust biological, pharmacological, and clinical annotations remain scarce. This challenge is particularly acute in drug discovery, where the procurement of high-quality training data is hindered by extreme costs, stringent privacy regulations, and restrictive data-sharing agreements, especially for rare diseases and novel target studies [183]. Furthermore, biological variability poses a major obstacle to reproducibility and generalization. This variability, stemming from differences in experimental conditions, patient demographics, and genetic backgrounds, introduces significant noise and can lead to substantial fluctuations in molecular response data. Without careful normalization and multi-omics integration, AI models may learn these variations instead of the underlying biological signals, resulting in predictions that are biased, non-reproducible, and poorly generalizable to new populations or experimental settings.
Another critical issue is the lack of negative and failed experimental data in public databases. Because most published datasets overrepresent successful experiments, AI models are often unable to learn from failure modes, which are crucial for predicting adverse effects, toxicity, and off-target interactions [228]. Therefore, future efforts should emphasize the systematic curation of both positive and negative results, along with the establishment of shared, standardized data infrastructures to enhance cross-study comparability and robustness [229].
The black-box nature of DL models represents another major barrier to their adoption in regulated biomedical environments [230]. While DL architectures such as convolutional and Transformer-based networks have significantly advanced molecular representation learning, their lack of interpretability remains a fundamental limitation [228]. In particular, large models are prone to high-risk modeling illusions due to the inherent bias of the attention mechanism that leads to the generation process. In addition, the black-box nature of large models makes it difficult to meet the interpretable requirements of vertical applications. Developing inherently explainable AI frameworks capable of elucidating structure-activity relationships and toxicity pathways is essential to bridge this gap. Recent advances in attention mechanisms and causal inference models offer promising pathways toward interpretable predictions that align with domain expertise [28].
Computational efficiency represents another major obstacle. Although modern graphics processing units (GPUs) have improved training performance, state-of-the-art deep learning architectures, particularly large Transformer models, demand massive computational and energy resources, creating both environmental and practical challenges [231]. To mitigate these costs, future research should prioritize green computing strategies. This includes developing lightweight yet high-performing architectures to reduce carbon footprints without sacrificing predictive power. Furthermore, decentralized frameworks like federated learning can enhance sustainability by minimizing redundant data transfers and enabling secure, collaborative model development across institutions without centralizing sensitive data.
In summary, while AI-driven drug discovery has achieved significant progress, its continued success depends on addressing data scarcity, biological variability, model interpretability, and sustainability challenges. With the ongoing evolution of algorithmic efficiency, interdisciplinary data integration, and open-source model development, AI holds great promise to fundamentally reshape drug discovery—making it faster, greener, and more reliable for global health advancement.
Author Contributions
Q.W. and B.S.: Data curation, Writing—original draft; Y.Y., T.V. and J.S.: Writing—review & editing; C.D. and H.J.: Conceptualization, Funding acquisition, Writing—review & editing, Supervision. All authors have read and agreed to the published version of the manuscript.
Funding
This study was funded supported by the National Key Research and Development Program of China (Grant No. 2023YFD1802300) and the National High-Level Talents Special Support Program.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable. No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Jung, Y.L.; Yoo, H.S.; Hwang, J. Artificial intelligence-based decision support model for new drug development planning. Expert Syst. Appl. 2022, 198, 116825. [Google Scholar] [CrossRef] [PubMed]
- Wouters, O.J.; McKee, M.; Luyten, J. Estimated Research and Development Investment Needed to Bring a New Medicine to Market, 2009–2018. JAMA 2020, 323, 844–853. [Google Scholar] [CrossRef] [PubMed]
- Harrison, R.K. Phase II and phase III failures: 2013–2015. Nat. Rev. Drug Discov. 2016, 15, 817–818. [Google Scholar] [CrossRef]
- Dowden, H.; Munro, J. Trends in clinical success rates and therapeutic focus. Nat. Rev. Drug Discov. 2019, 18, 495–496. [Google Scholar] [CrossRef]
- Smietana, K.; Siatkowski, M.; Møller, M. Trends in clinical success rates. Nat. Rev. Drug Discov. 2016, 15, 379–380. [Google Scholar] [CrossRef]
- Sun, D.; Gao, W.; Hu, H.; Zhou, S. Why 90% of clinical drug development fails and how to improve it? Acta Pharm. Sin. B 2022, 12, 3049–3062. [Google Scholar] [CrossRef] [PubMed]
- Biotechnology Innovation Organization; Informa Pharma Intelligence; Quantitative Life Sciences Advisors. Clinical Development Success Rates and Contributing Factors 2011–2020. 2021. Available online: https://www.bio.org/clinical-development-success-rates-and-contributing-factors-2011-2020 (accessed on 20 February 2025).
- Peng, C.; Zhao, S.; Tang, L.; Wang, K.; Wang, Y.; Ding, L. A simplified and reliable LC-tandem mass spectrometry method for determination of ulipristal acetate in human plasma and its application to a pharmacokinetic study in healthy Chinese volunteers. Biomed. Chromatogr. 2020, 34, e4908. [Google Scholar] [CrossRef]
- Lyu, J.; Wang, S.; Balius, T.E.; Singh, I.; Levit, A.; Moroz, Y.S.; O’Meara, M.J.; Che, T.; Algaa, E.; Tolmachova, K.; et al. Ultra-large library docking for discovering new chemotypes. Nature 2019, 566, 224–229. [Google Scholar] [CrossRef]
- Talukder, M.E.K.; Atif, M.F.; Siddiquee, N.H.; Rahman, S.; Rafi, N.I.; Israt, S.; Shahir, N.F.; Islam, M.T.; Samad, A.; Wani, T.A.; et al. Molecular docking, QSAR, and simulation analyses of EGFR-targeting phytochemicals in non-small cell lung cancer. J. Mol. Struct. 2025, 1321, 139924. [Google Scholar] [CrossRef]
- Kaur, N.; Gupta, S.; Pal, J.; Bansal, Y.; Bansal, G. Design of BBB permeable BACE-1 inhibitor as potential drug candidate for Alzheimer disease: 2D-QSAR, molecular docking, ADMET, molecular dynamics, MMGBSA. Comput. Biol. Chem. 2025, 116, 108371. [Google Scholar] [CrossRef]
- Souza, A.S.; Amorim, V.M.F.; Soares, E.P.; de Souza, R.F.; Guzzo, C.R. Antagonistic Trends Between Binding Affinity and Drug-Likeness in SARS-CoV-2 Mpro Inhibitors Revealed by Machine Learning. Viruses 2025, 17, 935. [Google Scholar] [CrossRef]
- Maliyakkal, N.; Kumar, S.; Bhowmik, R.; Vishwakarma, H.C.; Yadav, P.; Mathew, B. Two-dimensional QSAR-driven virtual screening for potential therapeutics against Trypanosoma cruzi. Front. Chem. 2025, 13, 1600945. [Google Scholar] [CrossRef] [PubMed]
- Pun, F.W.; Ozerov, I.V.; Zhavoronkov, A. AI-powered therapeutic target discovery. Trends Pharmacol. Sci. 2023, 44, 561–572. [Google Scholar] [CrossRef]
- Cassan, O.; Lèbre, S.; Martin, A. Inferring and analyzing gene regulatory networks from multi-factorial expression data: A complete and interactive suite. BMC Genom. 2021, 22, 387. [Google Scholar] [CrossRef] [PubMed]
- Nogales, C.; Mamdouh, Z.M.; List, M.; Kiel, C.; Casas, A.I.; Schmidt, H.H.H.W. Network pharmacology: Curing causal mechanisms instead of treating symptoms. Trends Pharmacol. Sci. 2022, 43, 136–150. [Google Scholar] [CrossRef]
- Zhuo, C.; Gao, J.; Li, A.; Liu, X.; Zhao, Y. A Machine Learning Method for RNA–Small Molecule Binding Preference Prediction. J. Chem. Inf. Model. 2024, 64, 7386–7397. [Google Scholar] [CrossRef]
- Zhou, J.-B.; Tang, D.; He, L.; Lin, S.; Lei, J.H.; Sun, H.; Xu, X.; Deng, C.-X. Machine learning model for anti-cancer drug combinations: Analysis, prediction, and validation. Pharmacol. Res. 2023, 194, 106830. [Google Scholar] [CrossRef]
- He, D.; Liu, Q.; Mi, Y.; Meng, Q.; Xu, L.; Hou, C.; Wang, J.; Li, N.; Liu, Y.; Chai, H.; et al. De Novo Generation and Identification of Novel Compounds with Drug Efficacy Based on Machine Learning. Adv. Sci. 2024, 11, 2307245. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Xu, T.; Yu, Y.; Zhao, P.; Chen, X.; Han, J.; Xie, Z.; Li, H.; Zhong, W.; Wong, K.-C.; et al. A dual diffusion model enables 3D molecule generation and lead optimization based on target pockets. Nat. Commun. 2024, 15, 2657. [Google Scholar] [CrossRef]
- Zhou, G.; Rusnac, D.-V.; Park, H.; Canzani, D.; Nguyen, H.M.; Stewart, L.; Bush, M.F.; Nguyen, P.T.; Wulff, H.; Yarov-Yarovoy, V.; et al. An artificial intelligence accelerated virtual screening platform for drug discovery. Nat. Commun. 2024, 15, 7761. [Google Scholar] [CrossRef]
- Wang, S.; Wang, L.; Li, F.; Bai, F. DeepSA: A deep-learning driven predictor of compound synthesis accessibility. J. Cheminform. 2023, 15, 103. [Google Scholar] [CrossRef] [PubMed]
- Struble, T.J.; Alvarez, J.C.; Brown, S.P.; Chytil, M.; Cisar, J.; DesJarlais, R.L.; Engkvist, O.; Frank, S.A.; Greve, D.R.; Griffin, D.J.; et al. Current and Future Roles of Artificial Intelligence in Medicinal Chemistry Synthesis. J. Med. Chem. 2020, 63, 8667–8682. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Wang, N.; Zhou, J.; Ouyang, D. Organic crystal structure prediction via coupled generative adversarial networks and graph convolutional networks. Innovation 2024, 5, 100562. [Google Scholar] [CrossRef]
- Yang, Z.; Zhao, Y.-M.; Wang, X.; Liu, X.; Zhang, X.; Li, Y.; Lv, Q.; Chen, C.Y.-C.; Shen, L. Scalable crystal structure relaxation using an iteration-free deep generative model with uncertainty quantification. Nat. Commun. 2024, 15, 8148. [Google Scholar] [CrossRef]
- Ryan, K.; Lengyel, J.; Shatruk, M. Crystal Structure Prediction via Deep Learning. J. Am. Chem. Soc. 2018, 140, 10158–10168. [Google Scholar] [CrossRef] [PubMed]
- Ren, F.; Aliper, A.; Chen, J.; Zhao, H.; Rao, S.; Kuppe, C.; Ozerov, I.V.; Zhang, M.; Witte, K.; Kruse, C.; et al. A small-molecule TNIK inhibitor targets fibrosis in preclinical and clinical models. Nat. Biotechnol. 2025, 43, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Wong, F.; Zheng, E.J.; Valeri, J.A.; Donghia, N.M.; Anahtar, M.N.; Omori, S.; Li, A.; Cubillos-Ruiz, A.; Krishnan, A.; Jin, W.; et al. Discovery of a structural class of antibiotics with explainable deep learning. Nature 2024, 626, 177. [Google Scholar] [CrossRef]
- Nobel Prize in Chemistry 2024. Available online: https://www.nature.com/collections/edjcfdihdi (accessed on 16 December 2024).
- Van de Sande, B.; Lee, J.S.; Mutasa-Gottgens, E.; Naughton, B.; Bacon, W.; Manning, J.; Wang, Y.; Pollard, J.; Mendez, M.; Hill, J.; et al. Applications of single-cell RNA sequencing in drug discovery and development. Nat. Rev. Drug Discov. 2023, 22, 496–520. [Google Scholar] [CrossRef]
- Yang, F.; Wang, W.; Wang, F.; Fang, Y.; Tang, D.; Huang, J.; Lu, H.; Yao, J. scBERT as a large-scale pretrained deep language model for cell type annotation of single-cell RNA-seq data. Nat. Mach. Intell. 2022, 4, 852–866. [Google Scholar] [CrossRef]
- Chen, J.; Wang, X.; Ma, A.; Wang, Q.-E.; Liu, B.; Li, L.; Xu, D.; Ma, Q. Deep transfer learning of cancer drug responses by integrating bulk and single-cell RNA-seq data. Nat. Commun. 2022, 13, 6494. [Google Scholar] [CrossRef]
- Hou, R.; Xie, C.; Gui, Y.; Li, G.; Li, X. Machine-Learning-Based Data Analysis Method for Cell-Based Selection of DNA-Encoded Libraries. ACS Omega 2023, 8, 19057–19071. [Google Scholar] [CrossRef] [PubMed]
- Recursion. Recursion’s Drug Pipeline. Available online: https://www.recursion.com/pipeline (accessed on 20 February 2025).
- Insilico Medicine. Insilico Medicine’s Drug Pipeline. Available online: https://insilico.com/ (accessed on 20 February 2025).
- Relay Therapeutics. Relay Therapeutics’s Drug Pipeline. Available online: https://relaytx.com/pipeline/ (accessed on 20 February 2025).
- Exscientia. Exscientia’s Drug Pipeline. Available online: https://www.exscientia.com/pipeline/ (accessed on 20 February 2025).
- Signet Therapeutics. Signet Therapeutics’ Drug Pipeline. Available online: https://www.signettx.com/about/ (accessed on 20 February 2025).
- BeiGene. BeiGene’s Drug Pipeline. Available online: https://www.beonemedicines.com.cn/science/pipeline/ (accessed on 20 February 2025).
- RedCloud Bio. RedCloud Bio’s Drug Pipeline. Available online: http://www.redcloudbio.com/en/h-col-105.html (accessed on 20 February 2025).
- Accutar Biotech. Accutar Biotech’s Drug Pipeline. Available online: https://www.accutarbio.com/workflow/ (accessed on 20 February 2025).
- MindRank. MindRank’s Drug Pipeline. Available online: https://www.mindrank.ai/zh-CN/pipeline (accessed on 20 February 2025).
- Drug Farm. Drug Farm’s Drug Pipeline. Available online: https://www.drug-farm.com/pipeline (accessed on 20 February 2025).
- OrphAI Therapeutics. OrphAI Therapeutics’s Drug Pipline. Available online: https://www.orphai-therapeutics.com/pipeline (accessed on 20 February 2025).
- Healx. Healx’s Drug Pipeline. Available online: https://healx.ai/pipeline/ (accessed on 20 February 2025).
- BioAge. BioAge’s Drug Pipeline. Available online: https://bioagelabs.com/apj (accessed on 20 February 2025).
- BioXcel Therapeutics. BioXcel Therapeutics’s Drug Pipeline. Available online: https://www.bioxceltherapeutics.com/our-pipeline/ (accessed on 20 February 2025).
- Evaxion Biotech. Evaxion Biotech’s Drug Pipeline. Available online: https://evaxion.ai/pipeline (accessed on 20 February 2025).
- Deo, R.C. Machine Learning in Medicine. Circulation 2015, 132, 1920–1930. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Gradus, J.L.; Rosellini, A.J. Supervised Machine Learning: A Brief Primer. Behav. Ther. 2020, 51, 675–687. [Google Scholar] [CrossRef]
- Stokes, J.M.; Yang, K.; Swanson, K.; Jin, W.; Cubillos-Ruiz, A.; Donghia, N.M.; MacNair, C.R.; French, S.; Carfrae, L.A.; Bloom-Ackerman, Z.; et al. A deep learning approach to antibiotic discovery. Cell 2020, 180, 688–702. [Google Scholar] [CrossRef]
- Arab, I.; Egghe, K.; Laukens, K.; Chen, K.; Barakat, K.; Bittremieux, W. Benchmarking of Small Molecule Feature Representations for hERG, Nav1.5, and Cav1.2 Cardiotoxicity Prediction. J. Chem. Inf. Model. 2024, 64, 2515–2527. [Google Scholar] [CrossRef]
- Singh, S.; Kaur, N.; Gehlot, A. Application of artificial intelligence in drug design: A review. Comput. Biol. Med. 2024, 179, 108810. [Google Scholar] [CrossRef]
- Glielmo, A.; Husic, B.E.; Rodriguez, A.; Clementi, C.; Noé, F.; Laio, A. Unsupervised Learning Methods for Molecular Simulation Data. Chem. Rev. 2021, 121, 9722–9758. [Google Scholar] [CrossRef]
- Cihan Sorkun, M.; Mullaj, D.; Koelman, J.M.V.A.; Er, S. ChemPlot, a Python Library for Chemical Space Visualization. Chem. –Methods 2022, 2, e202200005. [Google Scholar] [CrossRef]
- van Engelen, J.E.; Hoos, H.H. A survey on semi-supervised learning. Mach. Learn. 2020, 109, 373–440. [Google Scholar] [CrossRef]
- Niu, Q.; Li, H.; Tong, L.; Liu, S.; Zong, W.; Zhang, S.; Tian, S.; Wang, J.; Liu, J.; Li, B.; et al. TCMFP: A novel herbal formula prediction method based on network target’s score integrated with semi-supervised learning genetic algorithms. Brief. Bioinform. 2023, 24, bbad102. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Lu, L.; Li, J.; Jiang, J.; Zhang, J.; Zhou, S.; Wen, H.; Cai, H.; Luo, X.; Li, Z.; et al. Synthetically Feasible De Novo Molecular Design of Leads Based on a Reinforcement Learning Model: AI-Assisted Discovery of an Anti-IBD Lead Targeting CXCR4. J. Med. Chem. 2024, 67, 10057–10075. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hu, Z.; Chang, J.; Yu, B. Thinking on the Use of Artificial Intelligence in Drug Discovery. J. Med. Chem. 2025, 68, 4996–4999. [Google Scholar] [CrossRef] [PubMed]
- Botvinick, M.; Ritter, S.; Wang, J.X.; Kurth-Nelson, Z.; Blundell, C.; Hassabis, D. Reinforcement Learning, Fast and Slow. Trends Cogn. Sci. 2019, 23, 408–422. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Tu, S.; Xu, L. Multilevel Attention Network with Semi-supervised Domain Adaptation for Drug-Target Prediction. Proc. AAAI Conf. Artif. Intell. 2024, 38, 329–337. [Google Scholar] [CrossRef]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef]
- Kusumoto, D.; Seki, T.; Sawada, H.; Kunitomi, A.; Katsuki, T.; Kimura, M.; Ito, S.; Komuro, J.; Hashimoto, H.; Fukuda, K.; et al. Anti-senescent drug screening by deep learning-based morphology senescence scoring. Nat. Commun. 2021, 12, 257. [Google Scholar] [CrossRef]
- Grebner, C.; Matter, H.; Plowright, A.T.; Hessler, G. Automated De Novo Design in Medicinal Chemistry: Which Types of Chemistry Does a Generative Neural Network Learn? J. Med. Chem. 2020, 63, 8809–8823. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, Z.; Zeng, X.; Li, Y.; Li, P.; Ye, X.; Sakurai, T. Molecular language models: RNNs or transformer? Brief. Funct. Genom. 2023, 22, 392–400. [Google Scholar] [CrossRef]
- Shor, B.; Schneidman-Duhovny, D. DockFormer: Efficient Multi-Modal Receptor-Ligand Interaction Prediction using Pair Transformer. bioRxiv 2024. [Google Scholar] [CrossRef]
- Su, X.; Hu, P.; You, Z.-H.; Yu, P.S.; Hu, L. Dual-Channel Learning Framework for Drug-Drug Interaction Prediction via Relation-Aware Heterogeneous Graph Transformer. Proc. AAAI Conf. Artif. Intell. 2024, 38, 249–256. [Google Scholar] [CrossRef]
- Teng, S.; Yin, C.; Wang, Y.; Chen, X.; Yan, Z.; Cui, L.; Wei, L. MolFPG: Multi-level fingerprint-based Graph Transformer for accurate and robust drug toxicity prediction. Comput. Biol. Med. 2023, 164, 106904. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.-W.; Chen, D.; Liu, J. TopoFormer: Multiscale Topology-enabled Structure-to-Sequence Transformer for Protein-Ligand Interaction Predictions. Nat. Mach. Intell. 2024, 6, 799–810. [Google Scholar]
- Jiang, L.; Jiang, C.; Yu, X.; Fu, R.; Jin, S.; Liu, X. DeepTTA: A transformer-based model for predicting cancer drug response. Brief. Bioinform. 2022, 23, bbac100. [Google Scholar] [CrossRef]
- Zhang, O.; Lin, H.; Zhang, H.; Zhao, H.; Huang, Y.; Hsieh, C.-Y.; Pan, P.; Hou, T. Deep Lead Optimization: Leveraging Generative AI for Structural Modification. J. Am. Chem. Soc. 2024, 146, 31357–31370. [Google Scholar] [CrossRef]
- Wang, F.; Feng, X.; Kong, R.; Chang, S.; Wang, F.; Feng, X.; Kong, R.; Chang, S. Generating new protein sequences by using dense network and attention mechanism. Math. Biosci. Eng. 2023, 20, 4178–4197. [Google Scholar] [CrossRef]
- Zeng, X.; Wang, F.; Luo, Y.; Kang, S.-g.; Tang, J.; Lightstone, F.C.; Fang, E.F.; Cornell, W.; Nussinov, R.; Cheng, F. Deep generative molecular design reshapes drug discovery. Cell Rep. Med. 2022, 3, 100794. [Google Scholar] [CrossRef]
- Guimaraes, G.L.; Sanchez-Lengeling, B.; Outeiral, C.; Farias, P.L.C.; Aspuru-Guzik, A. Objective-Reinforced Generative Adversarial Networks (ORGAN) for Sequence Generation Models. arXiv 2017, arXiv:1705.10843. [Google Scholar]
- Hu, C.; Li, S.; Yang, C.; Chen, J.; Xiong, Y.; Fan, G.; Liu, H.; Hong, L. ScaffoldGVAE: Scaffold generation and hopping of drug molecules via a variational autoencoder based on multi-view graph neural networks. J. Cheminform. 2023, 15, 91. [Google Scholar] [CrossRef]
- Wang, M.; Hsieh, C.-Y.; Wang, J.; Wang, D.; Weng, G.; Shen, C.; Yao, X.; Bing, Z.; Li, H.; Cao, D.; et al. RELATION: A Deep Generative Model for Structure-Based De Novo Drug Design. J. Med. Chem. 2022, 65, 9478–9492. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.; Liu, X.; Tan, X.; Li, X.; Jiang, J.; Xiong, Z.; Xu, T.; Jiang, H.; Qiao, N.; Zheng, M. Generative Models for De Novo Drug Design. J. Med. Chem. 2021, 64, 14011–14027. [Google Scholar] [CrossRef] [PubMed]
- Ragoza, M.; Masuda, T.; Koes, D.R. Generating 3D molecules conditional on receptor binding sites with deep generative models. Chem. Sci. 2022, 13, 2701–2713. [Google Scholar] [CrossRef]
- Skalic, M.; Jiménez, J.; Sabbadin, D.; De Fabritiis, G. Shape-Based Generative Modeling for de Novo Drug Design. J. Chem. Inf. Model. 2019, 59, 1205–1214. [Google Scholar] [CrossRef] [PubMed]
- DeepSeek-AI; Guo, D.; Yang, D.; Zhang, H.; Song, J.-M.; Zhang, R.; Xu, R.; Zhu, Q.; Ma, S.; Wang, P.; et al. DeepSeek-R1: Incentivizing Reasoning Capability in LLMs via Reinforcement Learning. arXiv 2025, arXiv:abs/2501.12948. [Google Scholar]
- Anthropic, S. Model Card Addendum: Claude 3.5 Haiku and Upgraded Claude 3.5 Sonnet. 2024. Available online: https://assets.anthropic.com/m/1cd9d098ac3e6467/original/Claude-3-Model-Card-October-Addendum.pdf (accessed on 20 February 2025).
- Dubey, A.; Jauhri, A.; Pandey, A.; Kadian, A.; Al-Dahle, A.; Letman, A.; Mathur, A.; Schelten, A.; Yang, A.; Fan, A.; et al. The Llama 3 Herd of Models. arXiv 2024, arXiv:2407.21783. [Google Scholar] [CrossRef]
- El-Kishky, A.; Wei, A.; Saraiva, A.; Minaev, B.; Selsam, D.; Dohan, D.; Song, F.; Lightman, H.; Clavera, I.; Pachocki, J.W.; et al. Competitive Programming with Large Reasoning Models. arXiv 2025, arXiv:2502.06807. [Google Scholar]
- Lin, A.; Ye, J.; Qi, C.; Zhu, L.; Mou, W.; Gan, W.; Zeng, D.; Tang, B.; Xiao, M.; Chu, G.; et al. Bridging artificial intelligence and biological sciences: A comprehensive review of large language models in bioinformatics. Brief. Bioinform. 2025, 26, bbaf357. [Google Scholar] [CrossRef]
- Wu, Z.; Ramsundar, B.; Feinberg, E.N.; Gomes, J.; Geniesse, C.; Pappu, A.S.; Leswing, K.; Pande, V. MoleculeNet: A benchmark for molecular machine learning. Chem. Sci. 2018, 9, 513–530. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Fu, T.; Gao, W.; Zhao, Y.; Roohani, Y.; Leskovec, J.; Coley, C.W.; Xiao, C.; Sun, J.; Zitnik, M. Artificial intelligence foundation for therapeutic science. Nat. Chem. Biol. 2022, 18, 1033–1036. [Google Scholar] [CrossRef]
- Xu, L.-C.; Tang, M.-J.; An, J.; Cao, F.; Qi, Y. A unified pre-trained deep learning framework for cross-task reaction performance prediction and synthesis planning. Nat. Mach. Intell. 2025, 7, 1561–1571. [Google Scholar] [CrossRef]
- He, K.; Zhang, X.; Ren, S.; Sun, J. Deep Residual Learning for Image Recognition. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition (CVPR), Las Vegas, NV, USA, 27–30 June 2016. [Google Scholar]
- Dou, B.; Zhu, Z.; Merkurjev, E.; Ke, L.; Chen, L.; Jiang, J.; Zhu, Y.; Liu, J.; Zhang, B.; Wei, G.-W. Machine Learning Methods for Small Data Challenges in Molecular Science. Chem. Rev. 2023, 123, 8736–8780. [Google Scholar] [CrossRef]
- Asfand-e-yar, M.; Hashir, Q.; Shah, A.A.; Malik, H.A.M.; Alourani, A.; Khalil, W. Multimodal CNN-DDI: Using multimodal CNN for drug to drug interaction associated events. Sci. Rep. 2024, 14, 4076. [Google Scholar] [CrossRef]
- Chen, S.; Li, T.; Yang, L.; Zhai, F.; Jiang, X.; Xiang, R.; Ling, G. Artificial intelligence-driven prediction of multiple drug interactions. Brief. Bioinform. 2022, 23, bbac427. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, M.; Carvalho, F.G.; Ribeiro, B.; Arrais, J.P. Predicting drug activity against cancer through genomic profiles and SMILES. Artif. Intell. Med. 2024, 150, 102820. [Google Scholar] [CrossRef]
- Hochreiter, S.; Schmidhuber, J. Long Short-Term Memory. Neural Comput. 1997, 9, 1735–1780. [Google Scholar] [CrossRef] [PubMed]
- Isigkeit, L.; Hörmann, T.; Schallmayer, E.; Scholz, K.; Lillich, F.F.; Ehrler, J.H.M.; Hufnagel, B.; Büchner, J.; Marschner, J.A.; Pabel, J.; et al. Automated design of multi-target ligands by generative deep learning. Nat. Commun. 2024, 15, 7946. [Google Scholar] [CrossRef]
- Huang, K.; Chandak, P.; Wang, Q.; Havaldar, S.; Vaid, A.; Leskovec, J.; Nadkarni, G.N.; Glicksberg, B.S.; Gehlenborg, N.; Zitnik, M. A foundation model for clinician-centered drug repurposing. Nat. Med. 2024, 30, 3601–3613. [Google Scholar] [CrossRef]
- Lim, J.; Ryu, S.; Park, K.; Choe, Y.J.; Ham, J.; Kim, W.Y. Predicting Drug–Target Interaction Using a Novel Graph Neural Network with 3D Structure-Embedded Graph Representation. J. Chem. Inf. Model. 2019, 59, 3981–3988. [Google Scholar] [CrossRef]
- Sun, Y.; Li, Y.Y.; Leung, C.K.; Hu, P. iNGNN-DTI: Prediction of drug–target interaction with interpretable nested graph neural network and pretrained molecule models. Bioinformatics 2024, 40, btae135. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Chen, L.; Zhong, F.; Wang, D.; Jiang, J.; Zhang, S.; Jiang, H.; Zheng, M.; Li, X. Graph neural network approaches for drug-target interactions. Curr. Opin. Struct. Biol. 2022, 73, 102327. [Google Scholar] [CrossRef]
- Ng, S.S.S.; Lu, Y. Evaluating the Use of Graph Neural Networks and Transfer Learning for Oral Bioavailability Prediction. J. Chem. Inf. Model. 2023, 63, 5035–5044. [Google Scholar] [CrossRef]
- Yang, Z.; Zhong, W.; Zhao, L.; Chen, C.Y.-C. MGraphDTA: Deep multiscale graph neural network for explainable drug–target binding affinity prediction. Chem. Sci. 2022, 13, 816–833. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Hua, L.; Ma, Z.; Hu, W.; Liu, Y.; Zhu, J. Conformalized Graph Learning for Molecular ADMET Property Prediction and Reliable Uncertainty Quantification. J. Chem. Inf. Model. 2024, 64, 8705–8717. [Google Scholar] [CrossRef]
- Heid, E.; Greenman, K.P.; Chung, Y.; Li, S.-C.; Graff, D.E.; Vermeire, F.H.; Wu, H.; Green, W.H.; McGill, C.J. Chemprop: A Machine Learning Package for Chemical Property Prediction. J. Chem. Inf. Model. 2024, 64, 9–17. [Google Scholar] [CrossRef]
- Bai, P.; Miljković, F.; John, B.; Lu, H. Interpretable bilinear attention network with domain adaptation improves drug–target prediction. Nat. Mach. Intell. 2023, 5, 126–136. [Google Scholar] [CrossRef]
- Vaswani, A. Attention is all you need. Adv. Neural Inf. Process. Syst. 2017, 30, 5998–6008. [Google Scholar]
- Radford, A.; Narasimhan, K. Improving Language Understanding by Generative Pre-Training. 2018. Available online: https://www.semanticscholar.org/paper/Improving-Language-Understanding-by-Generative-Radford-Narasimhan/cd18800a0fe0b668a1cc19f2ec95b5003d0a5035 (accessed on 15 February 2025).
- Devlin, J.; Chang, M.-W.; Lee, K.; Toutanova, K. BERT: Pre-training of Deep Bidirectional Transformers for Language Understanding. In Proceedings of the 2019 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies, Volume 1 (Long and Short Papers), Minneapolis, MN, USA, 2–7 June 2019; pp. 4171–4186. [Google Scholar]
- Mao, J.; Wang, J.; Zeb, A.; Cho, K.-H.; Jin, H.; Kim, J.; Lee, O.; Wang, Y.; No, K.T. Transformer-Based Molecular Generative Model for Antiviral Drug Design. J. Chem. Inf. Model. 2024, 64, 2733–2745. [Google Scholar] [CrossRef] [PubMed]
- OpenAi; Achiam, J.; Adler, S.; Agarwal, S.; Ahmad, L.; Akkaya, I.; Aleman, F.L.; Almeida, D.; Altenschmidt, J.; Altman, S.; et al. GPT-4 Technical Report. arXiv 2024, arXiv:2303.08774. [Google Scholar]
- Brown, T.B.; Mann, B.; Ryder, N.; Subbiah, M.; Kaplan, J.; Dhariwal, P.; Neelakantan, A.; Shyam, P.; Sastry, G.; Askell, A.; et al. Language models are few-shot learners. In Proceedings of the 34th International Conference on Neural Information Processing Systems, Vancouver, BC, Canada, 6–12 December 2020; p. 159. [Google Scholar]
- Liu, S.; Wang, J.; Yang, Y.; Wang, C.; Liu, L.; Guo, H.; Xiao, C. Chatgpt-powered conversational drug editing using retrieval and domain feedback. arXiv 2023, arXiv:2305.18090. [Google Scholar]
- Greenblatt, J.F.; Alberts, B.M.; Krogan, N.J. Discovery and significance of protein-protein interactions in health and disease. Cell 2024, 187, 6501–6517. [Google Scholar] [CrossRef]
- Lim, Y.; Tamayo-Orrego, L.; Schmid, E.; Tarnauskaite, Z.; Kochenova, O.V.; Gruar, R.; Muramatsu, S.; Lynch, L.; Schlie, A.V.; Carroll, P.L.; et al. In silico protein interaction screening uncovers DONSON’s role in replication initiation. Science 2023, 381, eadi3448. [Google Scholar] [CrossRef]
- Lin, Z.; Akin, H.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; Smetanin, N.; Verkuil, R.; Kabeli, O.; Shmueli, Y.; et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 2023, 379, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Žídek, A.; Nelson, A.W.R.; Bridgland, A.; et al. Improved protein structure prediction using potentials from deep learning. Nature 2020, 577, 706–710. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Evans, R.; O’Neill, M.; Pritzel, A.; Antropova, N.; Senior, A.; Green, T.; Žídek, A.; Bates, R.; Blackwell, S.; Yim, J.; et al. Protein complex prediction with AlphaFold-Multimer. bioRxiv 2021. [Google Scholar] [CrossRef]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef]
- Díaz-Holguín, A.; Saarinen, M.; Vo, D.D.; Sturchio, A.; Branzell, N.; Cabeza de Vaca, I.; Hu, H.; Mitjavila-Domènech, N.; Lindqvist, A.; Baranczewski, P.; et al. AlphaFold accelerated discovery of psychotropic agonists targeting the trace amine–associated receptor 1. Sci. Adv. 2024, 10, eadn1524. [Google Scholar] [CrossRef]
- Bernatavicius, A.; Šícho, M.; Janssen, A.P.A.; Hassen, A.K.; Preuss, M.; van Westen, G.J.P. AlphaFold Meets De Novo Drug Design: Leveraging Structural Protein Information in Multitarget Molecular Generative Models. J. Chem. Inf. Model. 2024, 64, 8113–8122. [Google Scholar] [CrossRef]
- Ren, F.; Ding, X.; Zheng, M.; Korzinkin, M.; Cai, X.; Zhu, W.; Mantsyzov, A.; Aliper, A.; Aladinskiy, V.; Cao, Z.; et al. AlphaFold accelerates artificial intelligence powered drug discovery: Efficient discovery of a novel CDK20 small molecule inhibitor. Chem. Sci. 2023, 14, 1443–1452. [Google Scholar] [CrossRef]
- Michaud, J.M.; Madani, A.; Fraser, J.S. A language model beats alphafold2 on orphans. Nat. Biotechnol. 2022, 40, 1576–1577. [Google Scholar] [CrossRef]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Su, H.; Wang, W.; Ye, L.; Wei, H.; Peng, Z.; Anishchenko, I.; Baker, D.; Yang, J. The trRosetta server for fast and accurate protein structure prediction. Nat. Protoc. 2021, 16, 5634–5651. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhang, Y.; Lian, X.; Li, F.; Wang, C.; Zhu, F.; Qiu, Y.; Chen, Y. Therapeutic target database update 2022: Facilitating drug discovery with enriched comparative data of targeted agents. Nucleic Acids Res. 2022, 50, D1398–D1407. [Google Scholar] [CrossRef]
- Finan, C.; Gaulton, A.; Kruger, F.A.; Lumbers, R.T.; Shah, T.; Engmann, J.; Galver, L.; Kelley, R.; Karlsson, A.; Santos, R.; et al. The druggable genome and support for target identification and validation in drug development. Sci. Transl. Med. 2017, 9, eaag1166. [Google Scholar] [CrossRef] [PubMed]
- Kana, O.; Brylinski, M. Elucidating the druggability of the human proteome with eFindSite. J. Comput. Aided Mol. Des. 2019, 33, 509–519. [Google Scholar] [CrossRef]
- Li, X.; Wang, S.; Xie, Y.; Jiang, H.; Guo, J.; Wang, Y.; Peng, Z.; Hu, M.; Wang, M.; Wang, J.; et al. Deacetylation induced nuclear condensation of HP1gamma promotes multiple myeloma drug resistance. Nat. Commun. 2023, 14, 1290. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, S.; Chen, L.; Liu, S.; Ma, J.; Cao, Z.; Li, Q. DUT enhances drug resistance to proteasome inhibitors via promoting mitochondrial function in multiple myeloma. Carcinogenesis 2022, 43, 1030–1038. [Google Scholar] [CrossRef]
- Qi, T.F.; Tang, F.; Yin, J.; Miao, W.; Wang, Y. Parallel-reaction monitoring revealed altered expression of a number of epitranscriptomic reader, writer, and eraser proteins accompanied with colorectal cancer metastasis. Proteomics 2023, 23, e2200059. [Google Scholar] [CrossRef] [PubMed]
- Nidhi, S.; Anand, U.; Oleksak, P.; Tripathi, P.; Lal, J.A.; Thomas, G.; Kuca, K.; Tripathi, V. Novel CRISPR-Cas Systems: An Updated Review of the Current Achievements, Applications, and Future Research Perspectives. Int. J. Mol. Sci. 2021, 22, 3327. [Google Scholar] [CrossRef]
- Ramkumar, P.; Abarientos, A.B.; Tian, R.; Seyler, M.; Leong, J.T.; Chen, M.; Choudhry, P.; Hechler, T.; Shah, N.; Wong, S.W.; et al. CRISPR-based screens uncover determinants of immunotherapy response in multiple myeloma. Blood Adv. 2020, 4, 2899–2911. [Google Scholar] [CrossRef]
- Raivola, J.; Dini, A.; Karvonen, H.; Piki, E.; Salokas, K.; Niininen, W.; Kaleva, L.; Zhang, K.; Arjama, M.; Gudoityte, G.; et al. Multiomics characterization implicates PTK7 in ovarian cancer EMT and cell plasticity and offers strategies for therapeutic intervention. Cell Death Dis. 2022, 13, 714. [Google Scholar] [CrossRef]
- Gulfidan, G.; Soylu, M.; Demirel, D.; Erdonmez, H.B.C.; Beklen, H.; Ozbek Sarica, P.; Arga, K.Y.; Turanli, B. Systems biomarkers for papillary thyroid cancer prognosis and treatment through multi-omics networks. Arch Biochem. Biophys. 2022, 715, 109085. [Google Scholar] [CrossRef] [PubMed]
- Assum, I.; Krause, J.; Scheinhardt, M.O.; Muller, C.; Hammer, E.; Borschel, C.S.; Volker, U.; Conradi, L.; Geelhoed, B.; Zeller, T.; et al. Tissue-specific multi-omics analysis of atrial fibrillation. Nat. Commun. 2022, 13, 441. [Google Scholar] [CrossRef]
- Abell, N.S.; DeGorter, M.K.; Gloudemans, M.J.; Greenwald, E.; Smith, K.S.; He, Z.; Montgomery, S.B. Multiple causal variants underlie genetic associations in humans. Science 2022, 375, 1247–1254. [Google Scholar] [CrossRef]
- Namba, S.; Konuma, T.; Wu, K.H.; Zhou, W.; Global Biobank Meta-analysis, I.; Okada, Y. A practical guideline of genomics-driven drug discovery in the era of global biobank meta-analysis. Cell Genom. 2022, 2, 100190. [Google Scholar] [CrossRef]
- Deelen, J.; Evans, D.S.; Arking, D.E.; Tesi, N.; Nygaard, M.; Liu, X.; Wojczynski, M.K.; Biggs, M.L.; van der Spek, A.; Atzmon, G.; et al. Publisher Correction: A meta-analysis of genome-wide association studies identifies multiple longevity genes. Nat. Commun. 2021, 12, 2463. [Google Scholar] [CrossRef]
- Vamathevan, J.; Clark, D.; Czodrowski, P.; Dunham, I.; Ferran, E.; Lee, G.; Li, B.; Madabhushi, A.; Shah, P.; Spitzer, M.; et al. Applications of machine learning in drug discovery and development. Nat. Rev. Drug. Discov. 2019, 18, 463–477. [Google Scholar] [CrossRef]
- Lo, Y.C.; Senese, S.; Damoiseaux, R.; Torres, J.Z. 3D Chemical Similarity Networks for Structure-Based Target Prediction and Scaffold Hopping. ACS Chem. Biol. 2016, 11, 2244–2253. [Google Scholar] [CrossRef]
- Wolber, G.; Seidel, T.; Bendix, F.; Langer, T. Molecule-pharmacophore superpositioning and pattern matching in computational drug design. Drug Discov. Today 2008, 13, 23–29. [Google Scholar] [CrossRef]
- Mamoshina, P.; Volosnikova, M.; Ozerov, I.V.; Putin, E.; Skibina, E.; Cortese, F.; Zhavoronkov, A. Machine Learning on Human Muscle Transcriptomic Data for Biomarker Discovery and Tissue-Specific Drug Target Identification. Front. Genet. 2018, 9, 242. [Google Scholar] [CrossRef] [PubMed]
- Theodoris, C.V.; Xiao, L.; Chopra, A.; Chaffin, M.D.; Al Sayed, Z.R.; Hill, M.C.; Mantineo, H.; Brydon, E.M.; Zeng, Z.; Liu, X.S.; et al. Transfer learning enables predictions in network biology. Nature 2023, 618, 616–624. [Google Scholar] [CrossRef] [PubMed]
- Kang, B.; Fan, R.; Cui, C.; Cui, Q. Comprehensive prediction and analysis of human protein essentiality based on a pretrained large language model. Nat. Comput. Sci. 2025, 5, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Cooper-Knock, J.; Weimer, A.K.; Shi, M.; Moll, T.; Marshall, J.N.G.; Harvey, C.; Nezhad, H.G.; Franklin, J.; Souza, C.D.S.; et al. Genome-wide identification of the genetic basis of amyotrophic lateral sclerosis. Neuron 2022, 110, 992–1008.e11. [Google Scholar] [CrossRef]
- Zeng, X.; Zhu, S.; Lu, W.; Liu, Z.; Huang, J.; Zhou, Y.; Fang, J.; Huang, Y.; Guo, H.; Li, L.; et al. Target identification among known drugs by deep learning from heterogeneous networks. Chem. Sci. 2020, 11, 1775–1797. [Google Scholar] [CrossRef]
- Zhou, L.; Lian, G.; Zhou, T.; Cai, Z.; Yang, S.; Li, W.; Cheng, L.; Ye, Y.; He, M.; Lu, J.; et al. Palmitoylation of GPX4 via the targetable ZDHHC8 determines ferroptosis sensitivity and antitumor immunity. Nat. Cancer 2025, 6, 768–785. [Google Scholar] [CrossRef]
- Yang, Y.; Yao, K.; Repasky, M.P.; Leswing, K.; Abel, R.; Shoichet, B.K.; Jerome, S.V. Efficient Exploration of Chemical Space with Docking and Deep Learning. J. Chem. Theory Comput. 2021, 17, 7106–7119. [Google Scholar] [CrossRef]
- Singh, R.; Sledzieski, S.; Bryson, B.; Cowen, L.; Berger, B. Contrastive learning in protein language space predicts interactions between drugs and protein targets. Proc. Natl. Acad. Sci. USA 2023, 120, e2220778120. [Google Scholar] [CrossRef]
- Schade, A.E.; Perurena, N.; Yang, Y.; Rodriguez, C.L.; Krishnan, A.; Gardner, A.; Loi, P.; Xu, Y.; Nguyen, V.T.M.; Mastellone, G.M.; et al. AKT and EZH2 inhibitors kill TNBCs by hijacking mechanisms of involution. Nature 2024, 635, 755–763. [Google Scholar] [CrossRef]
- Gentile, F.; Yaacoub, J.C.; Gleave, J.; Fernandez, M.; Ton, A.T.; Ban, F.; Stern, A.; Cherkasov, A. Artificial intelligence-enabled virtual screening of ultra-large chemical libraries with deep docking. Nat. Protoc. 2022, 17, 672–697. [Google Scholar] [CrossRef] [PubMed]
- Acharya, A.; Agarwal, R.; Baker, M.B.; Baudry, J.; Bhowmik, D.; Boehm, S.; Byler, K.G.; Chen, S.Y.; Coates, L.; Cooper, C.J.; et al. Supercomputer-Based Ensemble Docking Drug Discovery Pipeline with Application to Covid-19. J. Chem. Inf. Model. 2020, 60, 5832–5852. [Google Scholar] [CrossRef] [PubMed]
- Puszkarska, A.M.; Taddese, B.; Revell, J.; Davies, G.; Field, J.; Hornigold, D.C.; Buchanan, A.; Vaughan, T.J.; Colwell, L.J. Machine learning designs new GCGR/GLP-1R dual agonists with enhanced biological potency. Nat. Chem. 2024, 16, 1436–1444. [Google Scholar] [CrossRef]
- Trinh, T.C.; Falson, P.; Tran-Nguyen, V.K.; Boumendjel, A. Ligand-Based Drug Discovery Leveraging State-of-the-Art Machine Learning Methodologies Exemplified by Cdr1 Inhibitor Prediction. J. Chem. Inf. Model. 2025, 65, 4027–4042. [Google Scholar] [CrossRef]
- Hansson, F.G.; Madsen, N.G.; Hansen, L.G.; Jakociunas, T.; Lengger, B.; Keasling, J.D.; Jensen, M.K.; Acevedo-Rocha, C.G.; Jensen, E.D. Labels as a feature: Network homophily for systematically annotating human GPCR drug-target interactions. Nat. Commun. 2025, 16, 4121. [Google Scholar] [CrossRef] [PubMed]
- Hadipour, H.; Li, Y.Y.; Sun, Y.; Deng, C.; Lac, L.; Davis, R.; Cardona, S.T.; Hu, P. GraphBAN: An inductive graph-based approach for enhanced prediction of compound-protein interactions. Nat. Commun. 2025, 16, 2541. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Song, G.; Zhu, H.; Lei, C.; Sun, X.; Wang, K.; Qin, L.; Chen, Y.; Tang, J.; Li, M. DTIAM: A unified framework for predicting drug-target interactions, binding affinities and drug mechanisms. Nat. Commun. 2025, 16, 2548. [Google Scholar] [CrossRef]
- Liu, X.; Li, Q.; Yan, X.; Wang, L.; Qiu, J.; Yao, X.; Liu, H. A Specialized and Enhanced Deep Generation Model for Active Molecular Design Targeting Kinases Guided by Affinity Prediction Models and Reinforcement Learning. J. Chem. Inf. Model. 2025, 65, 3294–3308. [Google Scholar] [CrossRef]
- Yang, H.; Chen, Y.; Zuo, Y.; Deng, Z.; Pan, X.; Shen, H.B.; Choi, K.S.; Yu, D.J. MINDG: A drug-target interaction prediction method based on an integrated learning algorithm. Bioinformatics 2024, 40, btae147. [Google Scholar] [CrossRef]
- Moon, S.; Hwang, S.-Y.; Lim, J.; Kim, W.Y. PIGNet2: A versatile deep learning-based protein–ligand interaction prediction model for binding affinity scoring and virtual screening. Digit. Discov. 2024, 3, 287–299. [Google Scholar] [CrossRef]
- Cai, H.; Shen, C.; Jian, T.; Zhang, X.; Chen, T.; Han, X.; Yang, Z.; Dang, W.; Hsieh, C.Y.; Kang, Y.; et al. CarsiDock: A deep learning paradigm for accurate protein-ligand docking and screening based on large-scale pre-training. Chem. Sci. 2024, 15, 1449–1471. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Stokes, J.M.; Eastman, R.T.; Itkin, Z.; Zakharov, A.V.; Collins, J.J.; Jaakkola, T.S.; Barzilay, R. Deep learning identifies synergistic drug combinations for treating COVID-19. Proc. Natl. Acad. Sci. USA 2021, 118, e2105070118. [Google Scholar] [CrossRef]
- Liu, G.; Catacutan, D.B.; Rathod, K.; Swanson, K.; Jin, W.; Mohammed, J.C.; Chiappino-Pepe, A.; Syed, S.A.; Fragis, M.; Rachwalski, K.; et al. Deep learning-guided discovery of an antibiotic targeting Acinetobacter baumannii. Nat. Chem. Biol. 2023, 19, 1342–1350. [Google Scholar] [CrossRef]
- Guo, X.; Zhao, X.; Lu, X.; Zhao, L.; Zeng, Q.; Chen, F.; Zhang, Z.; Xu, M.; Feng, S.; Fan, T.; et al. A deep learning-driven discovery of berberine derivatives as novel antibacterial against multidrug-resistant Helicobacter pylori. Signal Transduct. Target. Ther. 2024, 9, 183. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-W.; Shi, T.-Y.; Gao, D.; Ma, C.-Y.; Lin, H.; Yan, D.; Deng, K.-J. iPADD: A Computational Tool for Predicting Potential Antidiabetic Drugs Using Machine Learning Algorithms. J. Chem. Inf. Model. 2023, 63, 4960–4969. [Google Scholar] [CrossRef]
- Gerdes, H.; Casado, P.; Dokal, A.; Hijazi, M.; Akhtar, N.; Osuntola, R.; Rajeeve, V.; Fitzgibbon, J.; Travers, J.; Britton, D.; et al. Drug ranking using machine learning systematically predicts the efficacy of anti-cancer drugs. Nat. Commun. 2021, 12, 1850. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Liu, Z.; Huang, N.; Xiao, Z.; Zhang, H.; Mirzoyan, S.; Xu, H.; Hao, J.; Xu, Y.; Zhang, M.; et al. A bioactivity foundation model using pairwise meta-learning. Nat. Mach. Intell. 2024, 6, 962–974. [Google Scholar] [CrossRef]
- Duan, Y.; Yang, X.; Zeng, X.; Wang, W.; Deng, Y.; Cao, D. Enhancing Molecular Property Prediction through Task-Oriented Transfer Learning: Integrating Universal Structural Insights and Domain-Specific Knowledge. J. Med. Chem. 2024, 67, 9575–9586. [Google Scholar] [CrossRef]
- Cai, H.; Zhang, H.; Zhao, D.; Wu, J.; Wang, L. FP-GNN: A versatile deep learning architecture for enhanced molecular property prediction. Brief. Bioinform. 2022, 23, bbac408. [Google Scholar] [CrossRef]
- Ahmad, W.; Simon, E.; Chithrananda, S.; Grand, G.; Ramsundar, B. Chemberta-2: Towards chemical foundation models. arXiv 2022, arXiv:2209.01712. [Google Scholar] [CrossRef]
- Zhou, G.; Gao, Z.; Ding, Q.; Zheng, H.; Xu, H.; Wei, Z.; Zhang, L.; Ke, G. Uni-mol: A universal 3d molecular representation learning framework. ChemRxiv 2023. [Google Scholar] [CrossRef]
- Chang, J.; Ye, J.C. Bidirectional generation of structure and properties through a single molecular foundation model. Nat. Commun. 2024, 15, 2323. [Google Scholar] [CrossRef]
- Ross, J.; Belgodere, B.; Chenthamarakshan, V.; Padhi, I.; Mroueh, Y.; Das, P. Large-scale chemical language representations capture molecular structure and properties. Nat. Mach. Intell. 2022, 4, 1256–1264. [Google Scholar] [CrossRef]
- Virshup, A.M.; Contreras-García, J.; Wipf, P.; Yang, W.; Beratan, D.N. Stochastic Voyages into Uncharted Chemical Space Produce a Representative Library of All Possible Drug-Like Compounds. J. Am. Chem. Soc. 2013, 135, 7296–7303. [Google Scholar] [CrossRef]
- Polishchuk, P.G.; Madzhidov, T.I.; Varnek, A. Estimation of the size of drug-like chemical space based on GDB-17 data. J. Comput.-Aided Mol. Des. 2013, 27, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Tropsha, A.; Isayev, O.; Varnek, A.; Schneider, G.; Cherkasov, A. Integrating QSAR modelling and deep learning in drug discovery: The emergence of deep QSAR. Nat. Rev. Drug Discov. 2024, 23, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Carvalho Martins, L.; Cino, E.A.; Ferreira, R.S. PyAutoFEP: An Automated Free Energy Perturbation Workflow for GROMACS Integrating Enhanced Sampling Methods. J. Chem. Theory Comput. 2021, 17, 4262–4273. [Google Scholar] [CrossRef]
- Schneuing, A.; Harris, C.; Du, Y.; Didi, K.; Jamasb, A.; Igashov, I.; Du, W.; Gomes, C.; Blundell, T.L.; Lio, P.; et al. Structure-based drug design with equivariant diffusion models. Nat. Comput. Sci. 2024, 4, 899–909. [Google Scholar] [CrossRef]
- Li, P.; Zhang, K.; Liu, T.; Lu, R.; Chen, Y.; Yao, X.; Gao, L.; Zeng, X. A deep learning approach for rational ligand generation with toxicity control via reactive building blocks. Nat. Comput. Sci. 2024, 4, 851–864. [Google Scholar] [CrossRef]
- Hu, Q.; Sun, C.; He, H.; Xu, J.; Liu, D.; Zhang, W.; Shi, S.; Zhang, K.; Li, H. Target-aware 3D molecular generation based on guided equivariant diffusion. Nat. Commun. 2025, 16, 7928. [Google Scholar] [CrossRef]
- Godinez, W.J.; Ma, E.J.; Chao, A.T.; Pei, L.; Skewes-Cox, P.; Canham, S.M.; Jenkins, J.L.; Young, J.M.; Martin, E.J.; Guiguemde, W.A. Design of potent antimalarials with generative chemistry. Nat. Mach. Intell. 2022, 4, 180–186. [Google Scholar] [CrossRef]
- Swanson, K.; Liu, G.; Catacutan, D.B.; Arnold, A.; Zou, J.; Stokes, J.M. Generative AI for designing and validating easily synthesizable and structurally novel antibiotics. Nat. Mach. Intell. 2024, 6, 338–353. [Google Scholar] [CrossRef]
- Wu, K.; Xia, Y.; Deng, P.; Liu, R.; Zhang, Y.; Guo, H.; Cui, Y.; Pei, Q.; Wu, L.; Xie, S.; et al. TamGen: Drug design with target-aware molecule generation through a chemical language model. Nat. Commun. 2024, 15, 9360. [Google Scholar] [CrossRef]
- Skinnider, M.A.; Stacey, R.G.; Wishart, D.S.; Foster, L.J. Chemical language models enable navigation in sparsely populated chemical space. Nat. Mach. Intell. 2021, 3, 759–770. [Google Scholar] [CrossRef]
- Flam-Shepherd, D.; Zhu, K.; Aspuru-Guzik, A. Language models can learn complex molecular distributions. Nat. Commun. 2022, 13, 3293. [Google Scholar] [CrossRef]
- Grisoni, F. Chemical language models for de novo drug design: Challenges and opportunities. Curr. Opin. Struct. Biol. 2023, 79, 102527. [Google Scholar] [CrossRef]
- Tong, X.; Qu, N.; Kong, X.; Ni, S.; Zhou, J.; Wang, K.; Zhang, L.; Wen, Y.; Shi, J.; Zhang, S.; et al. Deep representation learning of chemical-induced transcriptional profile for phenotype-based drug discovery. Nat. Commun. 2024, 15, 5378. [Google Scholar] [CrossRef]
- Tan, Y.; Dai, L.; Huang, W.; Guo, Y.; Zheng, S.; Lei, J.; Chen, H.; Yang, Y. DRlinker: Deep Reinforcement Learning for Optimization in Fragment Linking Design. J. Chem. Inf. Model. 2022, 62, 5907–5917. [Google Scholar] [CrossRef]
- Guo, J.; Knuth, F.; Margreitter, C.; Janet, J.P.; Papadopoulos, K.; Engkvist, O.; Patronov, A. Link-INVENT: Generative linker design with reinforcement learning. Digit. Discov. 2023, 2, 392–408. [Google Scholar] [CrossRef]
- Zheng, S.; Lei, Z.; Ai, H.; Chen, H.; Deng, D.; Yang, Y. Deep scaffold hopping with multimodal transformer neural networks. J. Cheminform. 2021, 13, 87. [Google Scholar] [CrossRef] [PubMed]
- Bagal, V.; Aggarwal, R.; Vinod, P.K.; Priyakumar, U.D. MolGPT: Molecular Generation Using a Transformer-Decoder Model. J. Chem. Inf. Model. 2022, 62, 2064–2076. [Google Scholar] [CrossRef] [PubMed]
- Arus-Pous, J.; Patronov, A.; Bjerrum, E.J.; Tyrchan, C.; Reymond, J.L.; Chen, H.; Engkvist, O. SMILES-based deep generative scaffold decorator for de-novo drug design. J. Cheminform. 2020, 12, 38. [Google Scholar] [CrossRef]
- Langevin, M.; Minoux, H.; Levesque, M.; Bianciotto, M. Scaffold-Constrained Molecular Generation. J. Chem. Inf. Model. 2020, 60, 5637–5646. [Google Scholar] [CrossRef]
- Fialkova, V.; Zhao, J.; Papadopoulos, K.; Engkvist, O.; Bjerrum, E.J.; Kogej, T.; Patronov, A. LibINVENT: Reaction-based Generative Scaffold Decoration for in Silico Library Design. J. Chem. Inf. Model. 2022, 62, 2046–2063. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Chen, S.; Lei, J.; Yang, Y. DiffDec: Structure-Aware Scaffold Decoration with an End-to-End Diffusion Model. J. Chem. Inf. Model. 2024, 64, 2554–2564. [Google Scholar] [CrossRef]
- Green, H.; Koes, D.R.; Durrant, J.D. DeepFrag: A deep convolutional neural network for fragment-based lead optimization. Chem. Sci. 2021, 12, 8036–8047. [Google Scholar] [CrossRef]
- Hadfield, T.E.; Imrie, F.; Merritt, A.; Birchall, K.; Deane, C.M. Incorporating Target-Specific Pharmacophoric Information into Deep Generative Models for Fragment Elaboration. J. Chem. Inf. Model. 2022, 62, 2280–2292. [Google Scholar] [CrossRef] [PubMed]
- Imrie, F.; Hadfield, T.E.; Bradley, A.R.; Deane, C.M. Deep generative design with 3D pharmacophoric constraints. Chem. Sci. 2021, 12, 14577–14589. [Google Scholar] [CrossRef]
- Liu, X.; Ye, K.; van Vlijmen, H.W.T.; AP, I.J.; van Westen, G.J.P. DrugEx v3: Scaffold-constrained drug design with graph transformer-based reinforcement learning. J. Cheminform. 2023, 15, 24. [Google Scholar] [CrossRef]
- Loeffler, H.H.; He, J.; Tibo, A.; Janet, J.P.; Voronov, A.; Mervin, L.H.; Engkvist, O. Reinvent 4: Modern AI-driven generative molecule design. J. Cheminform. 2024, 16, 20. [Google Scholar] [CrossRef]
- Wang, M.; Li, S.; Wang, J.; Zhang, O.; Du, H.; Jiang, D.; Wu, Z.; Deng, Y.; Kang, Y.; Pan, P.; et al. ClickGen: Directed exploration of synthesizable chemical space via modular reactions and reinforcement learning. Nat. Commun. 2024, 15, 10127. [Google Scholar] [CrossRef]
- Segler, M.H.S.; Preuss, M.; Waller, M.P. Planning chemical syntheses with deep neural networks and symbolic AI. Nature 2018, 555, 604–610. [Google Scholar] [CrossRef] [PubMed]
- Thakkar, A.; Chadimová, V.; Bjerrum, E.J.; Engkvist, O.; Reymond, J.-L. Retrosynthetic accessibility score (RAscore)—rapid machine learned synthesizability classification from AI driven retrosynthetic planning. Chem. Sci. 2021, 12, 3339–3349. [Google Scholar] [CrossRef]
- Nippa, D.F.; Atz, K.; Hohler, R.; Muller, A.T.; Marx, A.; Bartelmus, C.; Wuitschik, G.; Marzuoli, I.; Jost, V.; Wolfard, J.; et al. Enabling late-stage drug diversification by high-throughput experimentation with geometric deep learning. Nat. Chem. 2024, 16, 239–248. [Google Scholar] [CrossRef]
- Lu, J.; Zhang, Y. Unified Deep Learning Model for Multitask Reaction Predictions with Explanation. J. Chem. Inf. Model. 2022, 62, 1376–1387. [Google Scholar] [CrossRef]
- Li, B.; Su, S.; Zhu, C.; Lin, J.; Hu, X.; Su, L.; Yu, Z.; Liao, K.; Chen, H. A deep learning framework for accurate reaction prediction and its application on high-throughput experimentation data. J. Cheminform 2023, 15, 72. [Google Scholar] [CrossRef] [PubMed]
- Cong, S.; Zhang, M.; Song, Y.; Chang, S.; Tian, J.; Zeng, H.; Ji, H. Graph-sequence enhanced transformer for template-free prediction of natural product biosynthesis. Patterns 2025, 6, 101259. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.A.; Yang, Q.; Sresht, V.; Bolgar, P.; Hou, X.; Klug-McLeod, J.L.; Butler, C.R. Molecular Transformer unifies reaction prediction and retrosynthesis across pharma chemical space. Chem. Commun. 2019, 55, 12152–12155. [Google Scholar] [CrossRef]
- Zhao, P.C.; Wei, X.X.; Wang, Q.; Wang, Q.H.; Li, J.N.; Shang, J.; Lu, C.; Shi, J.Y. Single-step retrosynthesis prediction via multitask graph representation learning. Nat. Commun. 2025, 16, 814. [Google Scholar] [CrossRef]
- Chen, S.; Jung, Y. Deep Retrosynthetic Reaction Prediction using Local Reactivity and Global Attention. JACS Au 2021, 1, 1612–1620. [Google Scholar] [CrossRef]
- Ucak, U.V.; Ashyrmamatov, I.; Ko, J.; Lee, J. Retrosynthetic reaction pathway prediction through neural machine translation of atomic environments. Nat. Commun. 2022, 13, 1186. [Google Scholar] [CrossRef]
- Fang, L.; Li, J.; Zhao, M.; Tan, L.; Lou, J.G. Single-step retrosynthesis prediction by leveraging commonly preserved substructures. Nat. Commun. 2023, 14, 2446. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Yin, S.; Shi, L.; Zhou, W.; Zhang, Y.J. G2GT: Retrosynthesis Prediction with Graph-to-Graph Attention Neural Network and Self-Training. J. Chem. Inf. Model. 2023, 63, 1894–1905. [Google Scholar] [CrossRef]
- Mao, K.; Xiao, X.; Xu, T.; Rong, Y.; Huang, J.; Zhao, P. Molecular graph enhanced transformer for retrosynthesis prediction. Neurocomputing 2021, 457, 193–202. [Google Scholar] [CrossRef]
- Tu, Z.; Coley, C.W. Permutation Invariant Graph-to-Sequence Model for Template-Free Retrosynthesis and Reaction Prediction. J. Chem. Inf. Model. 2022, 62, 3503–3513. [Google Scholar] [CrossRef]
- Wang, X.; Li, Y.; Qiu, J.; Chen, G.; Liu, H.; Liao, B.; Hsieh, C.-Y.; Yao, X. Retroprime: A diverse, plausible and transformer-based method for single-step retrosynthesis predictions. Chem. Eng. J. 2021, 420, 129845. [Google Scholar] [CrossRef]
- Beckers, M.; Sturm, N.; Sirockin, F.; Fechner, N.; Stiefl, N. Prediction of Small-Molecule Developability Using Large-Scale In Silico ADMET Models. J. Med. Chem. 2023, 66, 14047–14060. [Google Scholar] [CrossRef]
- Vangala, S.R.; Krishnan, S.R.; Bung, N.; Srinivasan, R.; Roy, A. pBRICS: A Novel Fragmentation Method for Explainable Property Prediction of Drug-like Small Molecules. J. Chem. Inf. Model. 2023, 63, 5066–5076. [Google Scholar] [CrossRef]
- Jamrozik, E.; Śmieja, M.; Podlewska, S. ADMET-PrInt: Evaluation of ADMET Properties: Prediction and Interpretation. J. Chem. Inf. Model. 2024, 64, 1425–1432. [Google Scholar] [CrossRef] [PubMed]
- Mamada, H.; Takahashi, M.; Ogino, M.; Nomura, Y.; Uesawa, Y. Predictive Models Based on Molecular Images and Molecular Descriptors for Drug Screening. ACS Omega 2023, 8, 37186–37195. [Google Scholar] [CrossRef] [PubMed]
- Komissarov, L.; Manevski, N.; Groebke Zbinden, K.; Schindler, T.; Zitnik, M.; Sach-Peltason, L. Actionable Predictions of Human Pharmacokinetics at the Drug Design Stage. Mol. Pharm. 2024, 21, 4356–4371. [Google Scholar] [CrossRef]
- Niu, Z.; Xiao, X.; Wu, W.; Cai, Q.; Jiang, Y.; Jin, W.; Wang, M.; Yang, G.; Kong, L.; Jin, X.; et al. PharmaBench: Enhancing ADMET benchmarks with large language models. Sci. Data 2024, 11, 985. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.; Walther, P.; Leitz, J.; Mukherjee, S.; Wu, J.C.; Shivnaraine, R.V.; Zou, J. ADMET-AI: A machine learning ADMET platform for evaluation of large-scale chemical libraries. Bioinformatics 2024, 40, btae416. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Fu, L.; Shi, S.; Yi, J.; Wang, N.; He, Y.; Wu, Z.; Peng, J.; Deng, Y.; Wang, W.; Wu, C.; et al. ADMETlab 3.0: An updated comprehensive online ADMET prediction platform enhanced with broader coverage, improved performance, API functionality and decision support. Nucleic Acids Res. 2024, 52, W422–W431. [Google Scholar] [CrossRef]
- Pires, D.E.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Schyman, P.; Liu, R.; Desai, V.; Wallqvist, A. vNN Web Server for ADMET Predictions. Front. Pharmacol. 2017, 8, 889. [Google Scholar] [CrossRef]
- Yi, J.C.; Yang, Z.Y.; Zhao, W.T.; Yang, Z.J.; Zhang, X.C.; Wu, C.K.; Lu, A.P.; Cao, D.S. ChemMORT: An automatic ADMET optimization platform using deep learning and multi-objective particle swarm optimization. Brief. Bioinform. 2024, 25, bbae008. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Yang, X.; Wang, Y.; Yu, Y.; Huang, N.; Li, G.; Li, X.; Wu, J.C.; Yang, S. Artificial intelligence in drug development. Nat. Med. 2025, 31, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.; Lee, N.; Frieske, R.; Yu, T.; Su, D.; Xu, Y.; Ishii, E.; Bang, Y.J.; Madotto, A.; Fung, P. Survey of Hallucination in Natural Language Generation. ACM Comput. Surv. 2023, 55, 248. [Google Scholar] [CrossRef]
- Wellawatte, G.P.; Gandhi, H.A.; Seshadri, A.; White, A.D. A Perspective on Explanations of Molecular Prediction Models. J. Chem. Theory Comput. 2023, 19, 2149–2160. [Google Scholar] [CrossRef]
- Frye, L.; Bhat, S.; Akinsanya, K.; Abel, R. From computer-aided drug discovery to computer-driven drug discovery. Drug Discov. Today Technol. 2021, 39, 111–117. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).