
Resilience of Spontaneously Hypertensive Rats to Secondary Insults After Traumatic Brain Injury: Immediate Seizures, Survival, and Stress Response

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
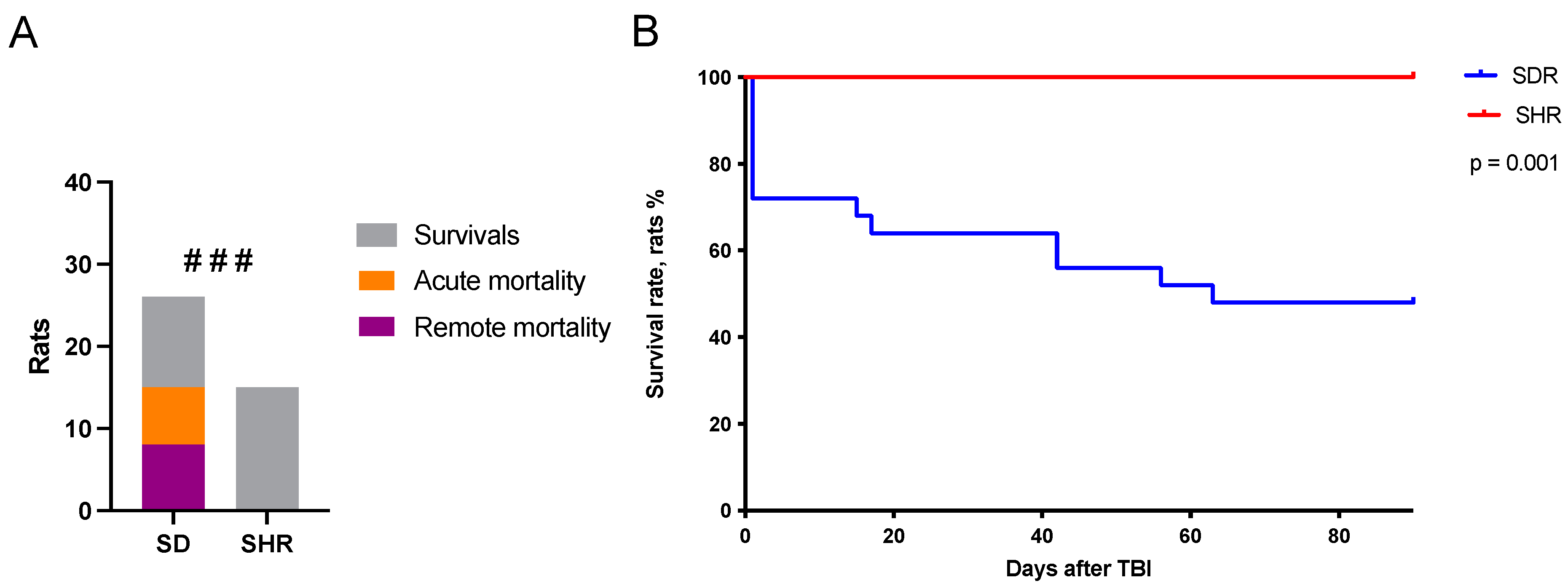
2.1. Survival Rates in SHRs and SDRs
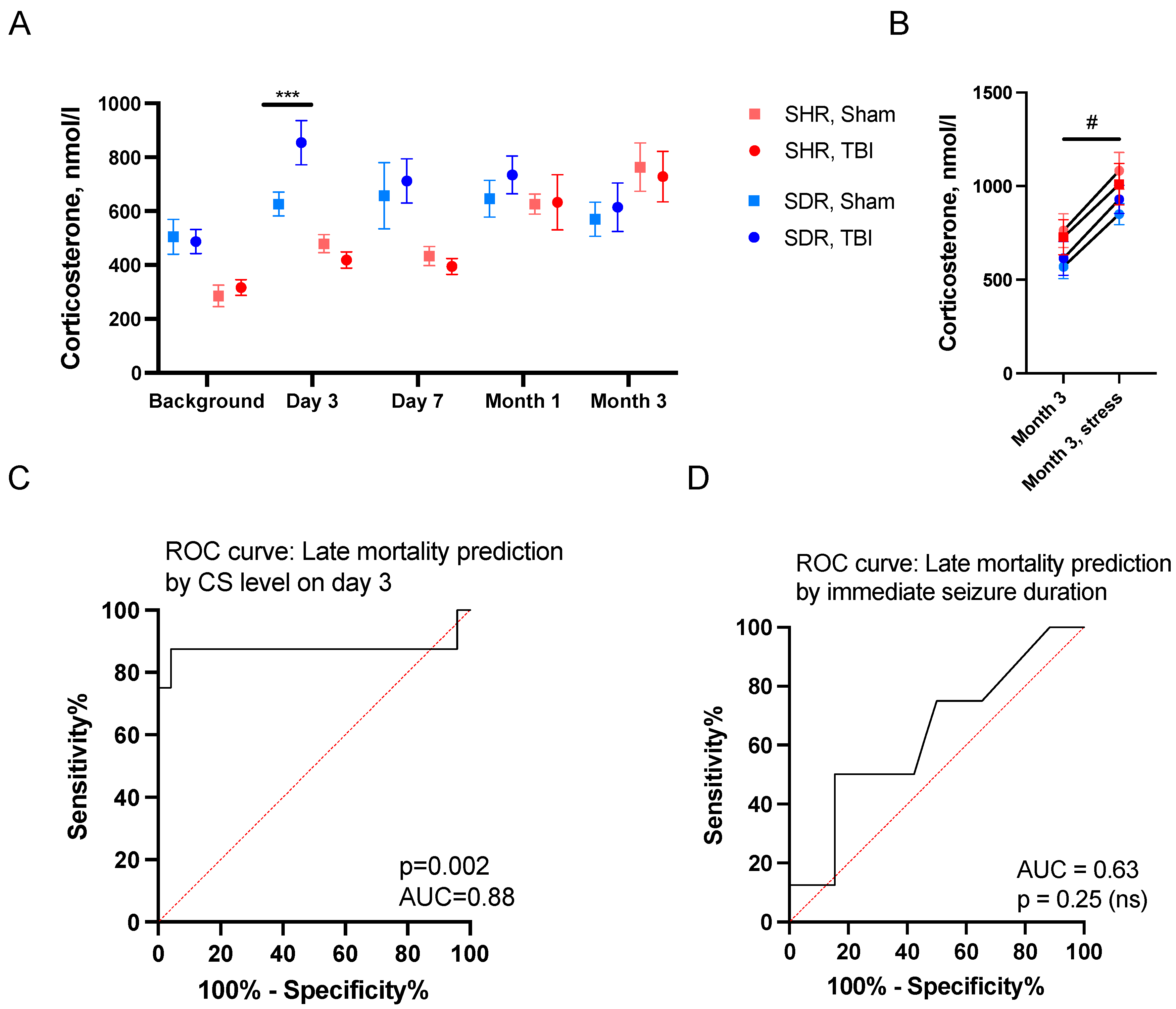
2.2. Predictors of Acute Mortality
2.3. Predictors of Remote Mortality
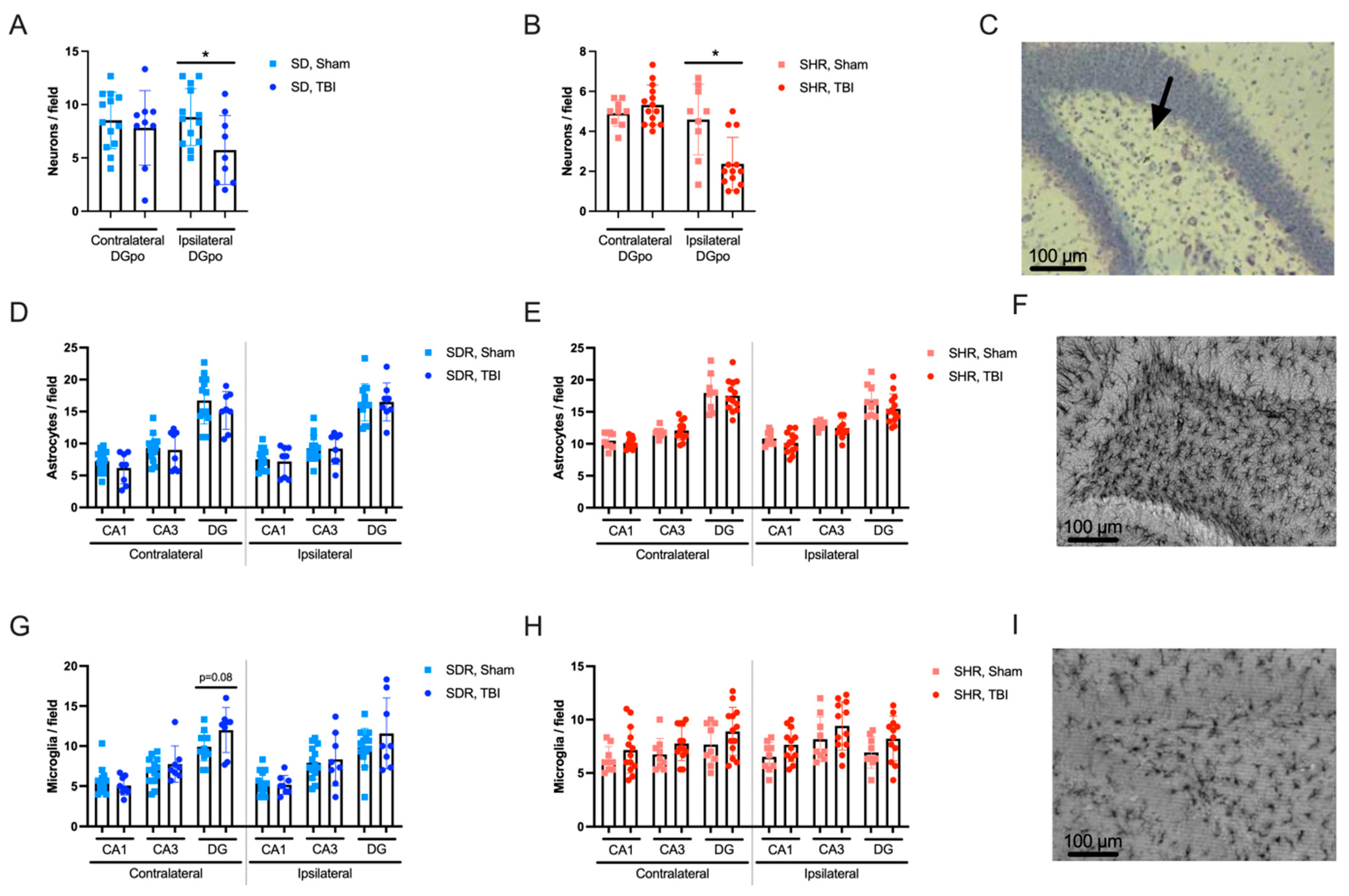
2.4. Effects of TBI on Hippocampal Morphology
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Experimental Design
4.3. Lateral Fluid-Percussion Injury
4.4. Immediate Seizure Visualization
4.5. Acute Stress Challenge
4.6. Histological Analyses
4.7. Measurement of CS in Blood Plasma
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AUC | area under the receiver operating characteristic (ROC) curve |
| BBB | blood–brain barrier |
| CS | corticosterone |
| DG | dentate gyrus |
| ELISA | enzyme-linked immunosorbent assay |
| GCs | glucocorticoids |
| GFAP | glial fibrillary acidic protein |
| Iba1 | ionized calcium-binding adaptor molecule 1 |
| IgG | immunoglobulin G |
| LFPI | lateral fluid-percussion injury |
| PBS | phosphate-buffered saline |
| PBST | PBS Triton X-100 solution |
| ROC | receiver operating characteristic |
| SHRs | spontaneously hypertensive rats |
| SDRs | Sprague Dawley rats |
| TBI | traumatic brain injury |
| WKY | Wistar Kyoto |
References
- Fordington, S.; Manford, M. A Review of Seizures and Epilepsy Following Traumatic Brain Injury. J. Neurol. 2020, 267, 3105–3111. [Google Scholar] [CrossRef] [PubMed]
- Alluri, H.; Wiggins-Dohlvik, K.; Davis, M.L.; Huang, J.H.; Tharakan, B. Blood–Brain Barrier Dysfunction Following Traumatic Brain Injury. Metab. Brain Dis. 2015, 30, 1093–1104. [Google Scholar] [CrossRef]
- Tomkins, O.; Shelef, I.; Kaizerman, I.; Eliushin, A.; Afawi, Z.; Misk, A.; Gidon, M.; Cohen, A.; Zumsteg, D.; Friedman, A. Blood-Brain Barrier Disruption in Post-Traumatic Epilepsy. J. Neurol. Neurosurg. Psychiatry 2008, 79, 774–777. [Google Scholar] [CrossRef] [PubMed]
- Shutter, L.A.; Narayan, R.K. Blood Pressure Management in Traumatic Brain Injury. Ann. Emerg. Med. 2008, 51, S37–S38. [Google Scholar] [CrossRef]
- Safar, M.; Chamiot-Clerc, P.; Dagher, G.; Renaud, J.F. Pulse Pressure, Endothelium Function, and Arterial Stiffness in Spontaneously Hypertensive Rats. Hypertension 2001, 38, 1416–1421. [Google Scholar] [CrossRef] [PubMed]
- Rybnikova, E.A.; Vetrovoi, O.V.; Zenko, M.Y. Comparative Characterization of Rat Strains (Wistar, Wistar–Kyoto, Sprague Dawley, Long Evans, LT, SHR, BD-IX) by Their Behavior, Hormonal Level and Antioxidant Status. J. Evol. Biochem. Physiol. 2018, 54, 374–382. [Google Scholar] [CrossRef]
- Johnson, A.C.; Miller, J.E.; Cipolla, M.J. Memory Impairment in Spontaneously Hypertensive Rats Is Associated with Hippocampal Hypoperfusion and Hippocampal Vascular Dysfunction. J. Cereb. Blood Flow Metab. 2020, 40, 845–859. [Google Scholar] [CrossRef] [PubMed]
- Szarka, N.; Toth, L.; Czigler, A.; Kellermayer, Z.; Ungvari, Z.; Amrein, K.; Czeiter, E.; Bali, Z.K.; Tadepalli, S.A.; Wahr, M.; et al. Single Mild Traumatic Brain Injury Induces Persistent Disruption of the Blood-Brain Barrier, Neuroinflammation and Cognitive Decline in Hypertensive Rats. Int. J. Mol. Sci. 2019, 20, 3223. [Google Scholar] [CrossRef]
- Starzec, J.J.; Berger, D.F. Stress- and Age-Related Serum Glucose Changes in Spontaneously Hypertensive and Sprague-Dawley Rats. Bull. Psychon. Soc. 1986, 24, 222–224. [Google Scholar] [CrossRef]
- Pitkänen, A.; Kyyriäinen, J.; Andrade, P.; Pasanen, L.; Ndode-Ekane, X.E. Epilepsy After Traumatic Brain Injury. In Models of Seizures and Epilepsy; Elsevier: San Diego, CA, USA, 2017; pp. 661–681. ISBN 9780128040669. [Google Scholar]
- Komoltsev, I.G.; Gulyaeva, N.V. Brain Trauma, Glucocorticoids and Neuroinflammation: Dangerous Liaisons for the Hippocampus. Biomedicines 2022, 10, 1139. [Google Scholar] [CrossRef] [PubMed]
- Huusko, N.; Römer, C.; Ndode-Ekane, X.E.; Lukasiuk, K.; Pitkänen, A. Loss of Hippocampal Interneurons and Epileptogenesis: A Comparison of Two Animal Models of Acquired Epilepsy. Brain Struct. Funct. 2015, 220, 153–191. [Google Scholar] [CrossRef]
- Dubreuil, C.I.; Marklund, N.; Deschamps, K.; McIntosh, T.K.; McKerracher, L. Activation of Rho after Traumatic Brain Injury and Seizure in Rats. Exp. Neurol. 2006, 198, 361–369. [Google Scholar] [CrossRef]
- Rink, A.; Fung, K.; Trojanowski, J.Q.; Lee, V.M.; Neugebauer, E.; Mcintosh, T.K. Evidence of Apoptotic Cell Death after Experimental Traumatic Brain Injury in the Rat. Am. J. Pathol. 1995, 147, 1575–1583. [Google Scholar] [CrossRef] [PubMed]
- Raghupathi, R. Cell Death Mechanisms Following Traumatic Brain Injury. Brain Pathol. 2004, 14, 215–222. [Google Scholar] [CrossRef]
- Komoltsev, I.G.; Frankevich, S.O.; Shirobokova, N.I.; Volkova, A.A.; Onufriev, M.V.; Moiseeva, J.V.; Novikova, M.R.; Gulyaeva, N.V. Neuroinflammation and Neuronal Loss in the Hippocampus Are Associated with Immediate Posttraumatic Seizures and Corticosterone Elevation in Rats. Int. J. Mol. Sci. 2021, 22, 5883. [Google Scholar] [CrossRef] [PubMed]
- Komoltsev, I.G.; Sinkin, M.V.; Volkova, A.A.; Smirnova, E.A.; Novikova, M.R.; Kordonskaya, O.O.; Talypov, A.E.; Guekht, A.B.; Krylov, V.V.; Gulyaeva, N.V. A Translational Study on Acute Traumatic Brain Injury: High Incidence of Epileptiform Activity on Human and Rat Electrocorticograms and Histological Correlates in Rats. Brain Sci. 2020, 10, 570. [Google Scholar] [CrossRef] [PubMed]
- Gulyaeva, N.V. Biochemical Mechanisms and Translational Relevance of Hippocampal Vulnerability to Distant Focal Brain Injury: The Price of Stress Response. Biochemistry 2019, 84, 1306–1328. [Google Scholar] [CrossRef] [PubMed]
- Bolshakov, A.P.; Tret’yakova, L.V.; Kvichansky, A.A.; Gulyaeva, N.V. Glucocorticoids: Dr. Jekyll and Mr. Hyde of Hippocampal Neuroinflammation. Biochemistry 2021, 86, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Saichan, X.; Wei, C.; Qinglong, F.; Jun, W.; Lei, X. Plasma Cortisol as a Noninvasive Biomarker to Assess Severity and Prognosis of Patients with Craniocerebral Injury. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 3835–3838. [Google Scholar] [PubMed]
- Tanriverdi, F.; Schneider, H.J.; Aimaretti, G.; Masel, B.E.; Casanueva, F.F.; Kelestimur, F. Pituitary Dysfunction After Traumatic Brain Injury: A Clinical and Pathophysiological Approach. Endocr. Rev. 2015, 36, 305–342. [Google Scholar] [CrossRef] [PubMed]
- Stepanichev, M.Y.; Mamedova, D.I.; Gulyaeva, N.V. Hippocampus under Pressure: Molecular Mechanisms of Development of Cognitive Impairments in SHR Rats. Biochemistry 2024, 89, 711–725. [Google Scholar] [CrossRef] [PubMed]
- Komoltsev, I.; Shalneva, D.; Kostyunina, O.; Volkova, A.; Frankevich, S.; Shirobokova, N.; Belikova, A.; Balan, S.; Chizhova, O.; Salyp, O.; et al. Delayed TBI-Induced Neuronal Death in the Ipsilateral Hippocampus and Behavioral Deficits in Rats: Influence of Corticosterone-Dependent Survivorship Bias? Int. J. Mol. Sci. 2023, 24, 4542. [Google Scholar] [CrossRef] [PubMed]
- Kolasa, M.; Faron-Górecka, A. Preclinical Models of Treatment-Resistant Depression: Challenges and Perspectives. Pharmacol. Rep. 2023, 75, 1326–1340. [Google Scholar] [CrossRef]
- Tchekalarova, J.; Krushovlieva, D.; Ivanova, P.; Kortenska, L. Spontaneously Hypertensive Rats vs. Wistar Kyoto and Wistar Rats: An Assessment of Anxiety, Motor Activity, Memory Performance, and Seizure Susceptibility. Physiol. Behav. 2023, 269, 114268. [Google Scholar] [CrossRef] [PubMed]
- Barone, F.C.; Price, W.J.; White, R.F.; Willette, R.N.; Feuerstein, G.Z. Genetic Hypertension and Increased Susceptibility to Cerebral Ischemia. Neurosci. Biobehav. Rev. 1992, 16, 219–233. [Google Scholar] [CrossRef]
- Schleiffer, R.; Gairard, A. Blood Pressure Effects of Calcium Intake in Experimental Models of Hypertension. Semin. Nephrol. 1995, 15, 526–535. [Google Scholar]
- Wookey, P.J.; Cao, Z.; Cooper, M.E. Interaction of the Renal Amylin and Renin-Angiotensin Systems in Animal Models of Diabetes and Hypertension. Miner. Electrolyte Metab. 1998, 24, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.J. Risk Factors, Endothelial Cell Turnover and Lipid Transport in Atherogenesis. Zhonghua Yi Xue Za Zhi 1996, 58, 309–316. [Google Scholar]
- Liu, K.L. Regulation of renal medullary circulation by the renin–angiotensin system in genetically hypertensive rats. Clin. Exp. Pharmacol. Physiol. 2009, 36, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Sagvolden, T.; Johansen, E.B.; Wøien, G.; Walaas, S.I.; Storm-Mathisen, J.; Bergersen, L.H.; Hvalby, Ø.; Jensen, V.; Aase, H.; Russell, V.A.; et al. The Spontaneously Hypertensive Rat Model of ADHD—The Importance of Selecting the Appropriate Reference Strain. Neuropharmacology 2009, 57, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Westfall, T.C.; Macarthur, H.; Byku, M.; Yang, C.-L.; Murray, J. Interactions of Neuropeptide Y, Catecholamines, and Angiotensin at the Vascular Neuroeffector Junction. In Advances in Pharmacology; Academic Press: New York, NY, USA, 2013; Volume 68, pp. 115–139. [Google Scholar]
- Prasad, K. Importance of Flaxseed and Its Components in the Management of Hypertension. Int. J. Angiol. 2019, 28, 153–160. [Google Scholar] [CrossRef]
- Komoltsev, I.G.; Tret’yakova, L.V.; Frankevich, S.O.; Shirobokova, N.I.; Volkova, A.A.; Butuzov, A.V.; Novikova, M.R.; Kvichansky, A.A.; Moiseeva, Y.V.; Onufriev, M.V.; et al. Neuroinflammatory Cytokine Response, Neuronal Death, and Microglial Proliferation in the Hippocampus of Rats During the Early Period After Lateral Fluid Percussion-Induced Traumatic Injury of the Neocortex. Mol. Neurobiol. 2021, 59, 1151–1167. [Google Scholar] [CrossRef] [PubMed]
- Félétou, M.; Verbeuren, T.J.; Vanhoutte, P.M. Endothelium-dependent Contractions in SHR: A Tale of Prostanoid TP and IP Receptors. Br. J. Pharmacol. 2009, 156, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Gluais, P.; Lonchampt, M.; Morrow, J.D.; Vanhoutte, P.M.; Feletou, M. Acetylcholine-Induced Endothelium-Dependent Contractions in the SHR Aorta: The Janus Face of Prostacyclin. Br. J. Pharmacol. 2005, 146, 834–845. [Google Scholar] [CrossRef] [PubMed]
- Lendvai-Emmert, D.; Magyar-Sumegi, Z.D.; Hegedus, E.; Szarka, N.; Fazekas, B.; Amrein, K.; Czeiter, E.; Buki, A.; Ungvari, Z.; Toth, P. Mild Traumatic Brain Injury-Induced Persistent Blood–Brain Barrier Disruption Is Prevented by Cyclosporine A Treatment in Hypertension. Front. Neurol. 2023, 14, 1252796. [Google Scholar] [CrossRef]
- Johnson, A.C. Hippocampal Vascular Supply and Its Role in Vascular Cognitive Impairment. Stroke 2023, 54, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Simon, R.P. Epileptic Sudden Death: Animal Models. Epilepsia 1997, 38, S35–S37. [Google Scholar] [CrossRef]
- Gulyaeva, N.V. Functional Neurochemistry of the Ventral and Dorsal Hippocampus: Stress, Depression, Dementia and Remote Hippocampal Damage. Neurochem. Res. 2019, 44, 1306–1322. [Google Scholar] [CrossRef] [PubMed]
- Agha, A.; Rogers, B.; Mylotte, D.; Taleb, F.; Tormey, W.; Phillips, J.; Thompson, C.J. Neuroendocrine Dysfunction in the Acute Phase of Traumatic Brain Injury. Clin. Endocrinol. 2004, 60, 584–591. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, W.T.; Smyth, A.; Gilchrist, M.D. Animal Models of Traumatic Brain Injury: A Critical Evaluation. Pharmacol. Ther. 2011, 130, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Jarrahi, A.; Braun, M.; Ahluwalia, M.; Gupta, R.V.; Wilson, M.; Munie, S.; Ahluwalia, P.; Vender, J.R.; Vale, F.L.; Dhandapani, K.M.; et al. Revisiting Traumatic Brain Injury: From Molecular Mechanisms to Therapeutic Interventions. Biomedicines 2020, 8, 389. [Google Scholar] [CrossRef] [PubMed]
- Webster, J.B.; Bell, K.R. Primary Adrenal Insufficiency Following Traumatic Brain Injury: A Case Report and Review of the Literature. Arch. Phys. Med. Rehabil. 1997, 78, 314–318. [Google Scholar] [CrossRef] [PubMed]
- Grady, M.S.; Charleston, J.S.; Maris, D.; Witgen, B.M.; Lifshitz, J. Neuronal and Glial Cell Number in the Hippocampus after Experimental Traumatic Brain Injury: Analysis by Stereological Estimation. J. Neurotrauma 2003, 20, 929–941. [Google Scholar] [CrossRef] [PubMed]
- Gulyaeva, N.V. Stress-Associated Molecular and Cellular Hippocampal Mechanisms Common for Epilepsy and Comorbid Depressive Disorders. Biochemistry 2021, 86, 641–656. [Google Scholar] [CrossRef] [PubMed]
- Aungst, S.L.; Kabadi, S.V.; Thompson, S.M.; Stoica, B.A.; Faden, A.I. Repeated Mild Traumatic Brain Injury Causes Chronic Neuroinflammation, Changes in Hippocampal Synaptic Plasticity, and Associated Cognitive Deficits. J. Cereb. Blood Flow Metab. 2014, 34, 1223–1232. [Google Scholar] [CrossRef]
- Aimaretti, G.; Ghigo, E. Traumatic Brain Injury and Hypopituitarism. Sci. World J. 2005, 5, 777–781. [Google Scholar] [CrossRef] [PubMed]
- Olivera, S.; Graham, D. Sex Differences in Preclinical Models of Hypertension. J. Hum. Hypertens. 2022, 37, 619–625. [Google Scholar] [CrossRef]
- Späni, C.B.; Braun, D.J.; Van Eldik, L.J. Sex-Related Responses after Traumatic Brain Injury: Considerations for Preclinical Modeling. Front. Neuroendocrinol. 2018, 50, 52–66. [Google Scholar] [CrossRef]
- Gupte, R.P.; Brooks, W.M.; Vukas, R.R.; Pierce, J.D.; Harris, J.L. Sex Differences in Traumatic Brain Injury: What We Know and What We Should Know. J. Neurotrauma 2019, 36, 3063–3091. [Google Scholar] [CrossRef] [PubMed]
- Yankelevitch-Yahav, R.; Franko, M.; Huly, A.; Doron, R. The Forced Swim Test as a Model of Depressive-like Behavior. J. Vis. Exp. 2015, 2015, 52587. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Komoltsev, I.; Kostyunina, O.; Kostrukov, P.; Bashkatova, D.; Shalneva, D.; Frankevich, S.; Salyp, O.; Shirobokova, N.; Volkova, A.; Soloveva, A.; et al. Resilience of Spontaneously Hypertensive Rats to Secondary Insults After Traumatic Brain Injury: Immediate Seizures, Survival, and Stress Response. Int. J. Mol. Sci. 2025, 26, 829. https://doi.org/10.3390/ijms26020829
Komoltsev I, Kostyunina O, Kostrukov P, Bashkatova D, Shalneva D, Frankevich S, Salyp O, Shirobokova N, Volkova A, Soloveva A, et al. Resilience of Spontaneously Hypertensive Rats to Secondary Insults After Traumatic Brain Injury: Immediate Seizures, Survival, and Stress Response. International Journal of Molecular Sciences. 2025; 26(2):829. https://doi.org/10.3390/ijms26020829
Chicago/Turabian StyleKomoltsev, Ilia, Olga Kostyunina, Pavel Kostrukov, Daria Bashkatova, Daria Shalneva, Stepan Frankevich, Olga Salyp, Natalia Shirobokova, Aleksandra Volkova, Aleksandra Soloveva, and et al. 2025. "Resilience of Spontaneously Hypertensive Rats to Secondary Insults After Traumatic Brain Injury: Immediate Seizures, Survival, and Stress Response" International Journal of Molecular Sciences 26, no. 2: 829. https://doi.org/10.3390/ijms26020829
APA StyleKomoltsev, I., Kostyunina, O., Kostrukov, P., Bashkatova, D., Shalneva, D., Frankevich, S., Salyp, O., Shirobokova, N., Volkova, A., Soloveva, A., Novikova, M., & Gulyaeva, N. (2025). Resilience of Spontaneously Hypertensive Rats to Secondary Insults After Traumatic Brain Injury: Immediate Seizures, Survival, and Stress Response. International Journal of Molecular Sciences, 26(2), 829. https://doi.org/10.3390/ijms26020829