
Cellular Senescence in Hepatocellular Carcinoma: Immune Microenvironment Insights via Machine Learning and In Vitro Experiments




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
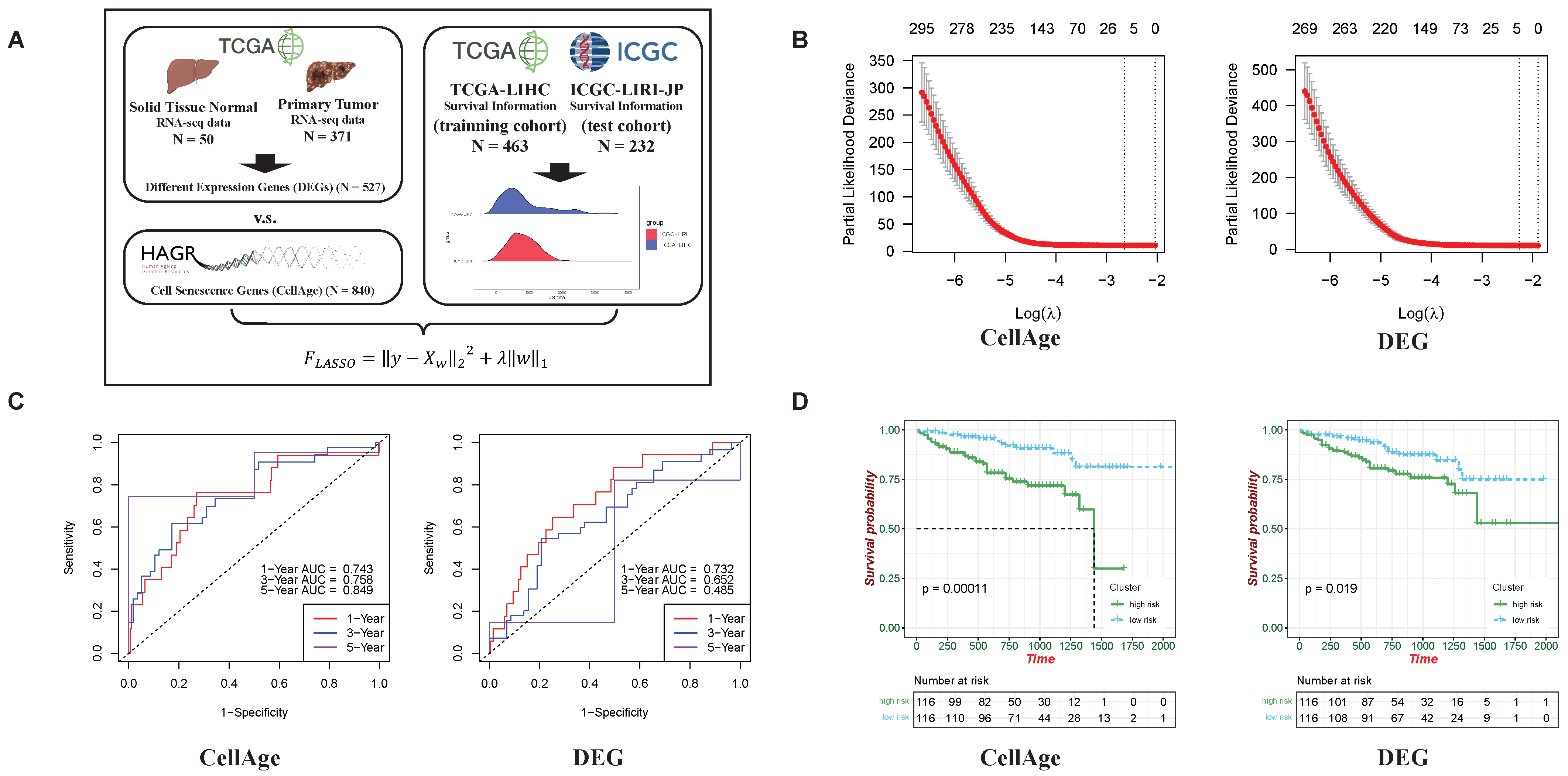
2.1. Stronger Predictive Abilities of Cellular Senescence-Related Genes on HCC Development than Differential Expression Genes (DEGs)
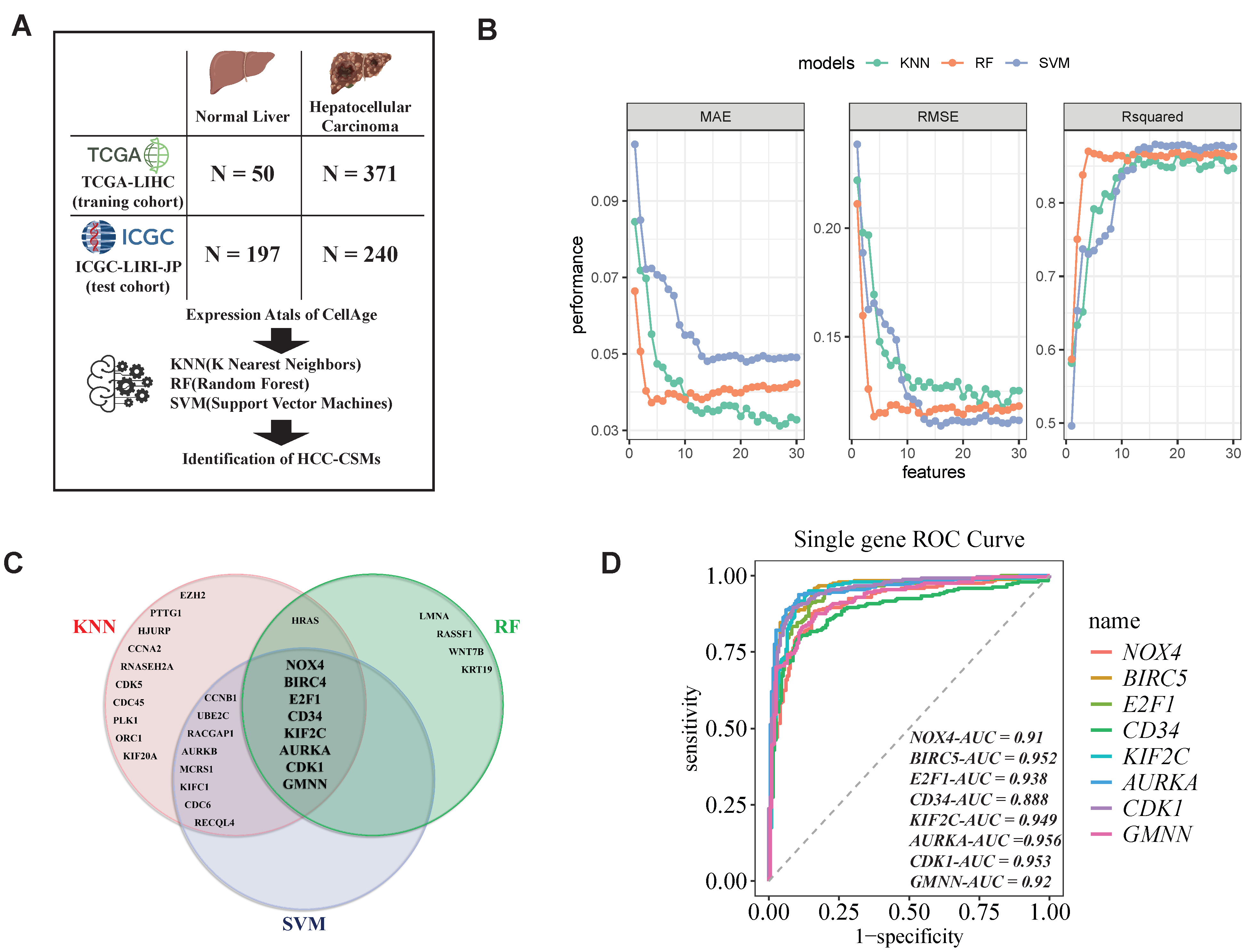
2.2. Identification of HCC-CSMs Using Multiple Machine Learning Algorithms
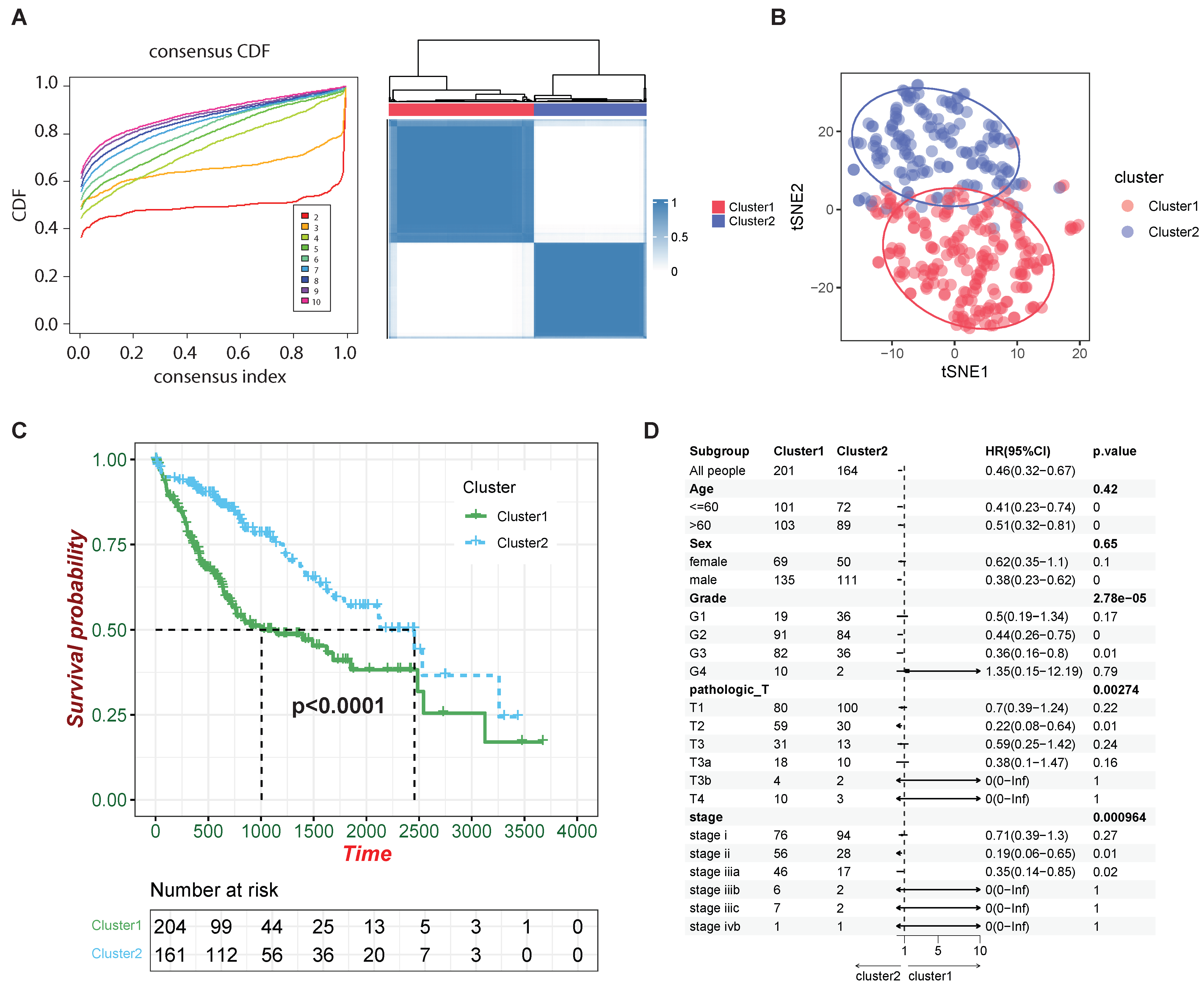
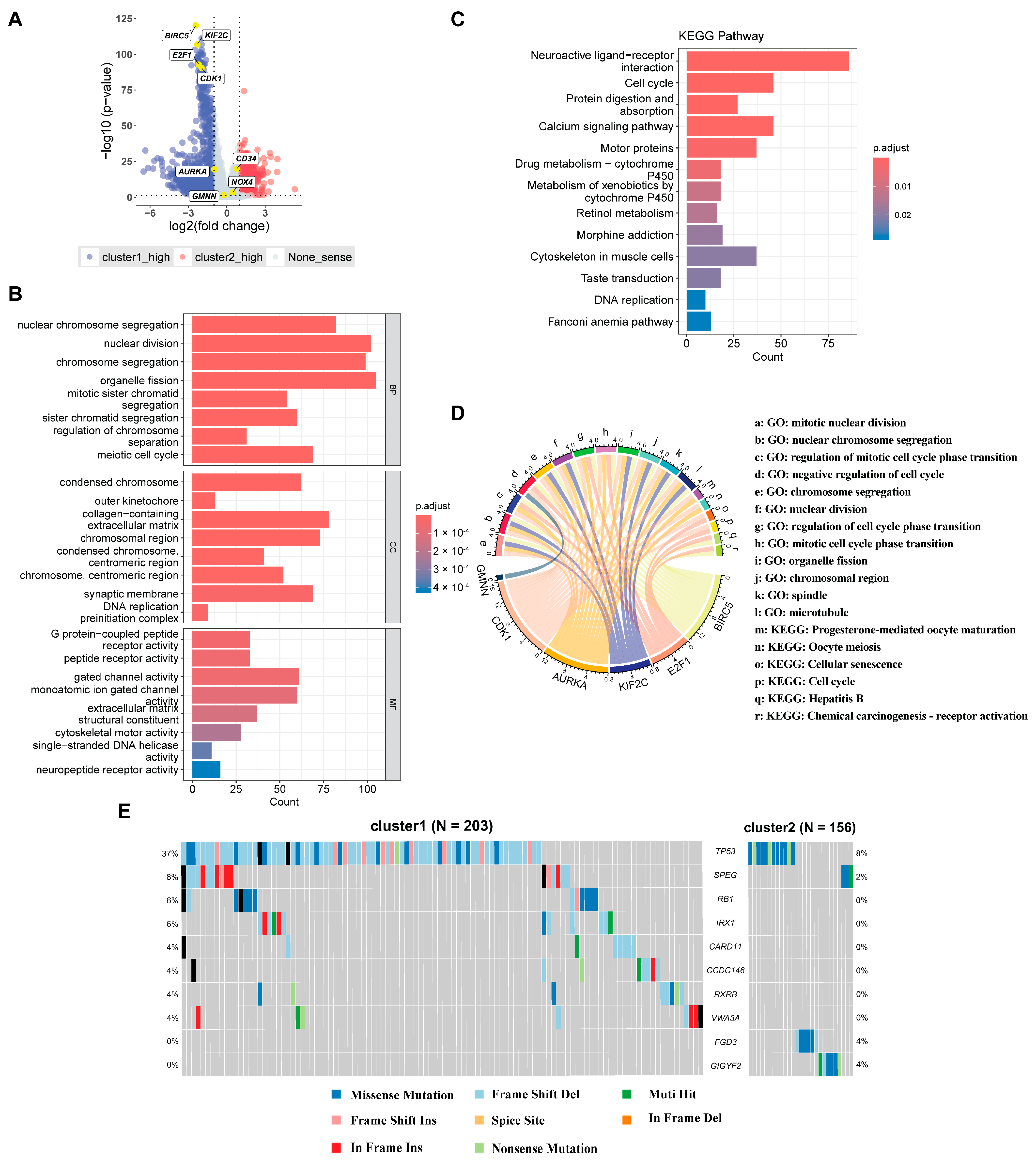
2.3. Examining the Clinical and Molecular Signatures of HCC Clusters Identified by HCC-CSMs
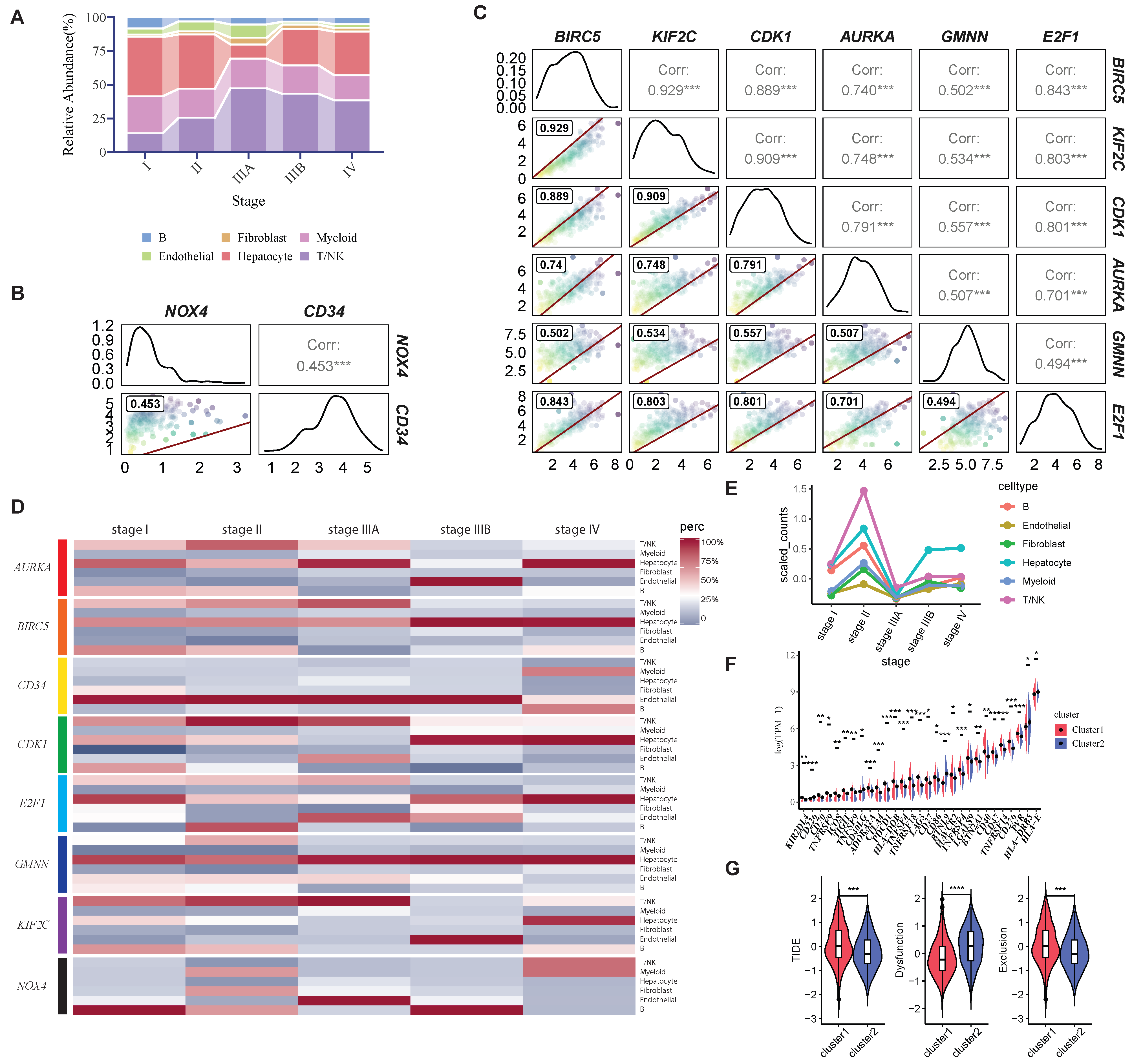
2.4. Exploration of Immune Microenvironment by HCC-CSMs
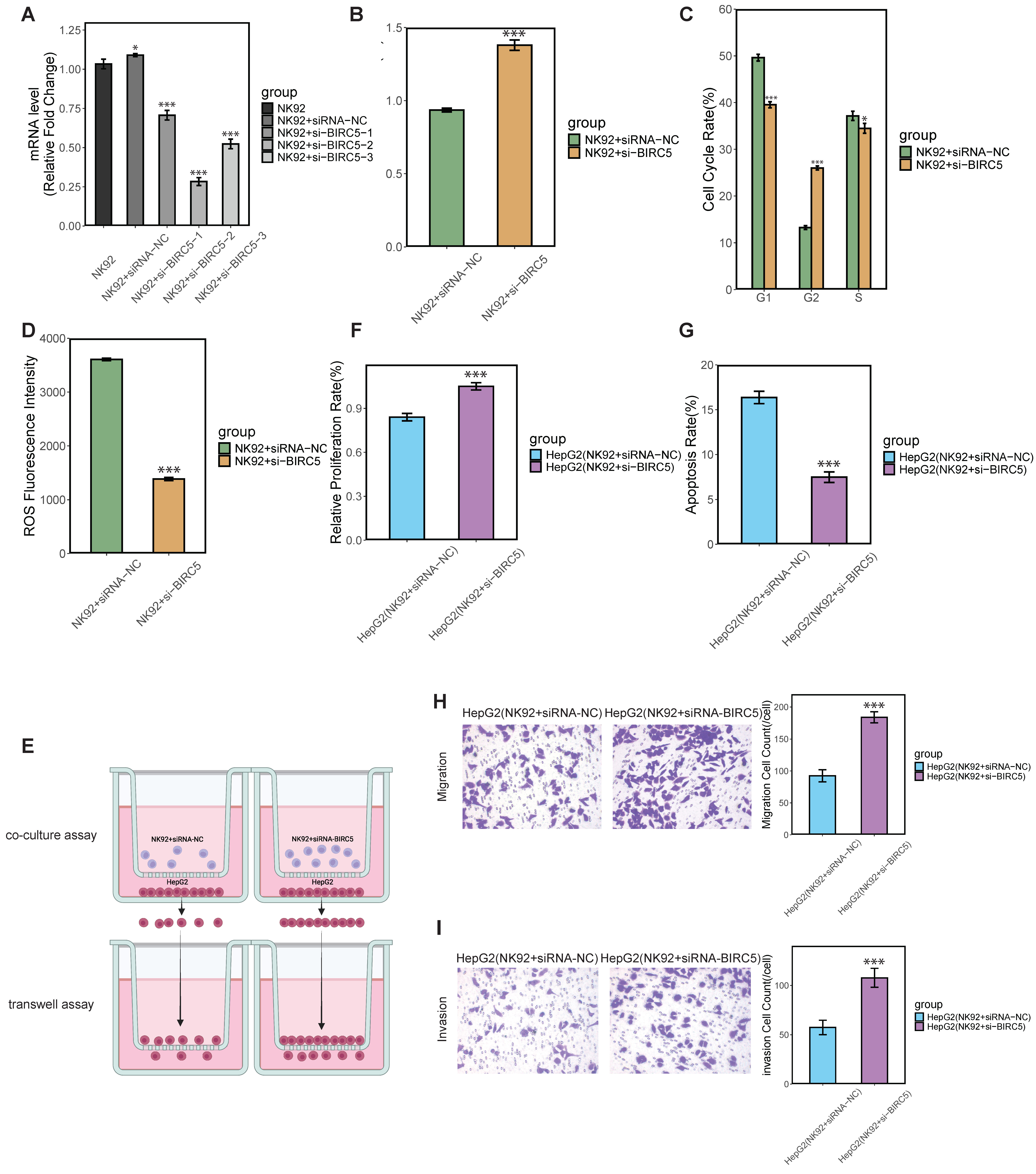
2.5. In Vitro Validation of BIRC5’s Role in Modulating NK Cell Proliferation and Cytotoxicity
3. Discussion
3.1. Cell Senescence Markers in HCC
3.2. Immunological Microenvironment and Therapeutic Strategies
4. Materials and Methods
4.1. Data Collection and Preprocessing
4.2. Feature Selection Method and Validation Based on LASSO Regression
4.3. Establishment and Validation of an HCC Diagnostic Model Based on Machine Learning Algorithms
4.4. Consensus Clustering for Subtyping and Prognostic Correlation
4.5. Differential Expression Gene Analysis and Gene Enrichment Analysis
4.6. Somatic Cell Mutation Analysis
4.7. Immune Infiltration Analysis
4.8. Single-Cell RNA-Seq Data Processing and Analysis
4.9. Cell Culture
4.10. SiRNA Sequence and Transfection Experimental Methods
4.11. RNA Isolation and qPCR
4.12. Cell Proliferation Assay
4.13. Cell Cycle Assay
4.14. Apoptotic Rate Assay
4.15. Detection of ROS
4.16. Transwell Assay
4.17. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Torimura, T.; Iwamoto, H. Treatment and the Prognosis of Hepatocellular Carcinoma in Asia. Liver Int. 2022, 42, 2042–2054. [Google Scholar] [CrossRef]
- El–Serag, H.B. Epidemiology of Hepatocellular Carcinoma. Clin. Liver Dis. 2001, 5, 87–107. [Google Scholar] [CrossRef]
- Fujiwara, N.; Friedman, S.L.; Goossens, N.; Hoshida, Y. Risk Factors and Prevention of Hepatocellular Carcinoma in the Era of Precision Medicine. J. Hepatol. 2018, 68, 526–549. [Google Scholar] [CrossRef] [PubMed]
- Masroor, M.S.; Parween, S.; Salim, M.; Prajapati, I.P. A Note on Hepatitis Viruses Causing Cancer in Human. Int. J. Biol. Innov. 2020, 2, 126–128. [Google Scholar] [CrossRef]
- Forner, A.; Reig, M.; Lope, C.R.d.; Bruix, J. Current Strategy for Staging and Treatment: The BCLC Update and Future Prospects. Semin. Liver Dis. 2010, 30, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.X.; Duda, D.G.; Sahani, D.V.; Jain, R.K. HCC and Angiogenesis: Possible Targets and Future Directions. Nat. Rev. Clin. Oncol. 2011, 8, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.M.; Ng, I.O.L. Molecular Pathogenesis of Hepatocellular Carcinoma. Liver Int. 2007, 28, 160–174. [Google Scholar] [CrossRef]
- Schmitt, C.A.; Wang, B.; Demaria, M. Senescence and Cancer—Role and Therapeutic Opportunities. Nat. Rev. Clin. Oncol. 2022, 19, 619–636. [Google Scholar] [CrossRef]
- Wakita, M.; Takahashi, A.; Sano, O.; Loo, T.M.; Imai, Y.; Narukawa, M.; Iwata, H.; Matsudaira, T.; Kawamoto, S.; Ohtani, N.; et al. A BET Family Protein Degrader Provokes Senolysis by Targeting NHEJ and Autophagy in Senescent Cells. Nat. Commun. 2020, 11, 1935. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Tang, H.; Tu, B.; Zhu, W.G. SIRT7: A Sentinel of Genome Stability. Open Biol. 2021, 11, 210047. [Google Scholar] [CrossRef] [PubMed]
- Zane, L.; Sharma, V.; Misteli, T. Common Features of Chromatin in Aging and Cancer: Cause or Coincidence? Trends Cell Biol. 2014, 24, 686–694. [Google Scholar] [CrossRef] [PubMed]
- Fane, M.; Weeraratna, A.T. How the Ageing Microenvironment Influences Tumour Progression. Nat. Rev. Cancer 2019, 20, 89–106. [Google Scholar] [CrossRef]
- Partridge, L.; Deelen, J.; Slagboom, P.E. Facing Up to the Global Challenges of Ageing. Nature 2018, 561, 45–56. [Google Scholar] [CrossRef] [PubMed]
- D’Ambrosio, M.; Gil, J. Reshaping of the tumor microenvironment by cellular senescence: An opportunity for senotherapies. Dev. Cell 2023, 58, 1007–1021. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, L.E.; Maallin, S.; Haston, S.; Martinez-Barbera, J.P.; Hodivala-Dilke, K.M.; Pedrosa, A.R. Effects of senescence on the tumour microenvironment and response to therapy. FEBS J. 2024, 291, 2306–2319. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Yu, J.; Fu, Y.; He, X.; Ge, S.; Jia, R.; Zhuang, A.; Yang, Z.; Fan, X. The dual role of cellular senescence in human tumor progression and therapy. MedComm 2024, 5, e695. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Tang, Q.; Chen, M.; Chen, W.; Lu, Y.; Liu, Z.; He, Z. Hepatocellular Senescence: Immunosurveillance and Future Senescence-Induced Therapy in Hepatocellular Carcinoma. Front. Oncol. 2020, 10, 589908. [Google Scholar] [CrossRef]
- Lu, C.; Rong, D.; Zhang, B.; Zheng, W.; Wang, X.; Chen, Z.; Tang, W. Current perspectives on the immunosuppressive tumor microenvironment in hepatocellular carcinoma: Challenges and opportunities. Mol. Cancer 2019, 18, 130. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Brown, Z.J.; Huang, H.; Tsung, A. Metabolic reprogramming of immune cells: Shaping the tumor microenvironment in hepatocellular carcinoma. Cancer Med. 2021, 10, 6374–6383. [Google Scholar] [CrossRef]
- Cai, X.; Guillot, A.; Liu, H. Cellular Senescence in Hepatocellular Carcinoma: The Passenger or the Driver? Cells 2022, 12, 132. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Wang, Y.; Lu, S. Cellular Senescence Affects Energy Metabolism, Immune Infiltration and Immunotherapeutic Response in Hepatocellular Carcinoma. Sci. Rep. 2023, 13, 1137. [Google Scholar] [CrossRef] [PubMed]
- Eggert, T.; Wolter, K.; Ji, J.; Ma, C.; Yevsa, T.; Klotz, S.; Medina-Echeverz, J.; Longerich, T.; Forgues, M.; Reisinger, F.; et al. Distinct Functions of Senescence-Associated Immune Responses in Liver Tumor Surveillance and Tumor Progression. Cancer Cell 2016, 30, 533–547. [Google Scholar] [CrossRef] [PubMed]
- Huda, N.; Liu, G.; Hong, H.; Yan, S.; Khambu, B.; Yin, X.M. Hepatic Senescence, the Good and the Bad. World J. Gastroenterol. 2019, 25, 5069–5081. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Du, R.; Gui, H.; Zhou, M.; Zhong, W.Z.; Mao, C.; Ma, J. Identification of Potential Hub Genes Related to the Progression and Prognosis of Hepatocellular Carcinoma Through Integrated Bioinformatics Analysis. Oncol. Rep. 2019, 43, 133–146. [Google Scholar] [CrossRef]
- Chen, X.; Xia, Z.; Wan, Y.; Huang, P. Identification of Hub Genes and Candidate Drugs in Hepatocellular Carcinoma by Integrated Bioinformatics Analysis. Medicine 2021, 100, e27117. [Google Scholar] [CrossRef]
- Xue, S.; Ge, W.; Wang, K.; Mao, T.; Zhang, X.; Xu, H.; Wang, Y.; Yao, J.; Li, S.; Ming, Y.; et al. Association of Aging-Related Genes With Prognosis and Immune Infiltration in Pancreatic Adenocarcinoma. Front. Cell Dev. Biol. 2022, 10, 942225. [Google Scholar] [CrossRef]
- Greener, J.G.; Kandathil, S.M.; Moffat, L.; Jones, D.T. A guide to machine learning for biologists. Nat. Rev. Mol. Cell Biol. 2022, 23, 40–55. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, K.; Poirion, O.B.; Lu, L.; Garmire, L.X. Deep Learning-Based Multi-Omics Integration Robustly Predicts Survival in Liver Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 1248–1259. [Google Scholar] [CrossRef]
- Shen, J.; Hu, J.; Wu, J.; Luo, X.; Li, Y.; Li, J. Molecular characterization of long-term survivors of hepatocellular carcinoma. Aging 2021, 13, 7517–7537. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Huang, T.; Li, Q.; Han, J.; Luo, X. Preliminary study on establishing the discriminant equation in pathological grading of hepatocellular carcinoma using immunohistochemical marker parameters. J. Cancer Res. Ther. 2022, 18, 1320–1327. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Shi, Y.; Li, Y.; Wu, S.; Yang, R.; Liu, Q.; Sun, L. DNA damage response-related signatures characterize the immune landscape and predict the prognosis of HCC via integrating single-cell and bulk RNA-sequencing. Int. Immunopharmacol. 2024, 137, 112475. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xu, W.; Kang, W.; Wong, S.H.; Wang, M.; Zhou, Y.; Fang, X.; Zhang, X.; Yang, H.; Wong, C.H.; et al. Genomic analysis of liver cancer unveils novel driver genes and distinct prognostic features. Theranostics 2018, 8, 1740–1751. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Zhang, M.; Lu, H.; Chen, Y.; Sun, W.; Zhu, J.; Chen, Z. Identification and evaluation of plasma exosome RNA biomarkers for non-invasive diagnosis of hepatocellular carcinoma using RNA-seq. BMC Cancer 2024, 24, 1552. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, H.; Xu, Y.; Liu, Y.Z.; Al-Adra, D.P.; Yeh, M.M.; Zhang, Z. Five Critical Gene-Based Biomarkers With Optimal Performance for Hepatocellular Carcinoma. Cancer Inform. 2023, 22, 11769351231190477. [Google Scholar] [CrossRef]
- Gwon, M.R.; Cho, J.H.; Kim, J.R. Mitotic centromere-associated kinase (MCAK/Kif2C) regulates cellular senescence in human primary cells through a p53-dependent pathway. FEBS Lett. 2012, 586, 4148–4156. [Google Scholar] [CrossRef]
- Unruhe, B.; Schröder, E.; Wünsch, D.; Knauer, S.K. An Old Flame Never Dies: Survivin in Cancer and Cellular Senescence. Gerontology 2016, 62, 173–181. [Google Scholar] [CrossRef]
- Goga, A.; Yang, D.; Tward, A.D.; Morgan, D.O.; Bishop, J.M. Inhibition of CDK1 as a potential therapy for tumors over-expressing MYC. Nat. Med. 2007, 13, 820–827. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.; Li, Z.; Feng, Z.; Yuan, Z.; Dai, Y.; Wu, X.; Zhang, F.; Wang, Y.; Zhou, Y.; Liu, L.; et al. Metformin Improves the Senescence of Renal Tubular Epithelial Cells in a High-Glucose State Through E2F1. Front. Pharmacol. 2022, 13, 926211. [Google Scholar] [CrossRef] [PubMed]
- Meng, P.; Ghosh, R. Transcription Addiction: Can We Garner the Yin and Yang Functions of E2F1 for Cancer Therapy? Cell Death Dis. 2014, 5, e1360. [Google Scholar] [CrossRef]
- Mao, R.; Liu, J.; Liu, G.; Jin, S.; Xue, Q.; Ma, L.; Fu, Y.; Zhao, N.; Xing, J.; Li, L.; et al. Whole genome sequencing of matched tumor, adjacent non-tumor tissues and corresponding normal blood samples of hepatocellular carcinoma patients revealed dynamic changes of the mutations profiles during hepatocarcinogenesis. Oncotarget 2017, 8, 26185–26199. [Google Scholar] [CrossRef]
- Guichard, C.; Amaddeo, G.; Imbeaud, S.; Ladeiro, Y.; Pelletier, L.; Maad, I.B.; Calderaro, J.; Bioulac-Sage, P.; Letexier, M.; Degos, F.; et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat. Genet. 2012, 44, 694–698. [Google Scholar] [CrossRef]
- Nault, J.C.; Martin, Y.; Caruso, S.; Hirsch, T.Z.; Bayard, Q.; Calderaro, J.; Charpy, C.; Copie-Bergman, C.; Ziol, M.; Bioulac-Sage, P.; et al. Clinical Impact of Genomic Diversity From Early to Advanced Hepatocellular Carcinoma. Hepatology 2020, 71, 164–182. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental Regulation of Tumor Progression and Metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Pei, S.; Zhang, P.; Yang, L.; Kang, Y.; Chen, H.; Zhao, S.; Dai, Y.; Zheng, M.; Xia, Y.; Xie, H. Exploring the Role of Sphingolipid-Related Genes in Clinical Outcomes of Breast Cancer. Front. Immunol. 2023, 14, 1116839. [Google Scholar] [CrossRef]
- Lei, Y.; Tang, R.; Xu, J.; Wang, W.; Zhang, B.; Liu, J.; Yu, X.; Shi, S. Applications of Single-Cell Sequencing in Cancer Research: Progress and Perspectives. J. Hematol. Oncol. 2021, 14, 91. [Google Scholar] [CrossRef]
- Zona, S.; Bella, L.; Burton, M.; Moraes, G.N.d.; Lam, E.W.F. FOXM1: An Emerging Master Regulator of DNA Damage Response and Genotoxic Agent Resistance. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2014, 1839, 1316–1322. [Google Scholar] [CrossRef] [PubMed]
- Kreis, N.N.; Moon, H.H.; Wordeman, L.; Louwen, F.; Solbach, C.; Yuan, J.; Ritter, A. KIF2C/MCAK a prognostic biomarker and its oncogenic potential in malignant progression, and prognosis of cancer patients: A systematic review and meta-analysis as biomarker. Crit. Rev. Clin. Lab. Sci. 2024, 61, 404–434. [Google Scholar] [CrossRef] [PubMed]
- Kamran, M.; Long, Z.J.; Xu, D.; Lv, S.; Liu, B.; Wang, C.L.; Xu, J.; Lam, E.W.F.; Liu, Q. Aurora Kinase a Regulates Survivin Stability Through Targeting FBXL7 in Gastric Cancer Drug Resistance and Prognosis. Oncogenesis 2017, 6, e298. [Google Scholar] [CrossRef] [PubMed]
- Wijnen, R.; Pecoraro, C.; Carbone, D.; Fiuji, H.; Rezayi, M.; Peters, G.J.; Giovannetti, E.; Diana, P. Cyclin Dependent Kinase-1 (CDK-1) Inhibition as a Novel Therapeutic Strategy Against Pancreatic Ductal Adenocarcinoma (PDAC). Cancers 2021, 13, 4389. [Google Scholar] [CrossRef]
- Pezeshki, S.; Hashemi, P.; Salimi, A.; Ebrahimi, S.; Javanzad, M.; Amir, M. Evaluation of NUF2 and GMNN Expression in Prostate Cancer: Potential Biomarkers for Prostate Cancer Screening. Rep. Biochem. Mol. Biol. 2021, 10, 224–232. [Google Scholar] [CrossRef]
- Rocca, M.S.; Benna, C.; Mocellin, S.; Rossi, C.R.; Msaki, A.; Nisio, A.D.; Opocher, G. E2F1 Germline Copy Number Variations and Melanoma Susceptibility. J. Transl. Med. 2019, 17, 181. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.-H.; Kim, K.P. E2F1 Facilitates DNA Break Repair by Localizing to Break Sites and Enhancing the Expression of Homologous Recombination Factors. Exp. Mol. Med. 2019, 51, 1–12. [Google Scholar] [CrossRef]
- Ning, J.; Sun, K.; Fan, X.; Jia, K.; Meng, L.; Wang, X.; Li, H.; Ma, R.; Liu, S.; Li, F. Use of Machine Learning-Based Integration to Develop an Immune-Related Signature for Improving Prognosis in Patients With Gastric Cancer. Sci. Rep. 2023, 13, 7019. [Google Scholar] [CrossRef] [PubMed]
- Mostavi, M.; Chiu, Y.-C.; Huang, Y.; Chen, Y. Convolutional Neural Network Models for Cancer Type Prediction Based on Gene Expression. BMC Med. Genom. 2020, 13, 44. [Google Scholar] [CrossRef]
- Yin, H.; Que, R.; Liu, C.; Ji, W.; Sun, B.; Lin, X.; Zhang, Q.; Zhao, X.; Peng, Z.; Zhang, X.; et al. Survivin-targeted drug screening platform identifies a matrine derivative WM-127 as a potential therapeutics against hepatocellular carcinoma. Cancer Lett. 2018, 425, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.M.; Zhang, D.; Xiao, P.; Qu, B.; Sun, Y.F. E2F1-mediated KDM4A-AS1 up-regulation promotes EMT of hepatocellular carcinoma cells by recruiting ILF3 to stabilize AURKA mRNA. Cancer Gene Ther. 2023, 30, 1007–1017. [Google Scholar] [CrossRef]
- Lei, Y.; Xu, X.; Liu, H.; Chen, L.; Zhou, H.; Jiang, J.; Yang, Y.; Wu, B. HBx induces hepatocellular carcinogenesis through ARRB1-mediated autophagy to drive the G(1)/S cycle. Autophagy 2021, 17, 4423–4441. [Google Scholar] [CrossRef] [PubMed]
- Li, T.F.; Zeng, H.J.; Shan, Z.; Ye, R.Y.; Cheang, T.Y.; Zhang, Y.J.; Lu, S.H.; Zhang, Q.; Shao, N.; Lin, Y. Overexpression of kinesin superfamily members as prognostic biomarkers of breast cancer. Cancer Cell Int. 2020, 20, 123. [Google Scholar] [CrossRef] [PubMed]
- Ling, B.; Liao, X.; Huang, Y.; Liang, L.; Jiang, Y.; Pang, Y.; Qi, G. Identification of prognostic markers of lung cancer through bioinformatics analysis and in vitro experiments. Int. J. Oncol. 2020, 56, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.; Zhan, Y.; Chen, X.; Wu, B.; Liu, B. Identification of Biomarkers for Controlling Cancer Stem Cell Characteristics in Bladder Cancer by Network Analysis of Transcriptome Data Stemness Indices. Front. Oncol. 2019, 9, 613. [Google Scholar] [CrossRef]
- Mo, S.; Fang, D.; Zhao, S.; Thai Hoa, P.T.; Zhou, C.; Liang, T.; He, Y.; Yu, T.; Chen, Y.; Qin, W.; et al. Down regulated oncogene KIF2C inhibits growth, invasion, and metastasis of hepatocellular carcinoma through the Ras/MAPK signaling pathway and epithelial-to-mesenchymal transition. Ann. Transl. Med. 2022, 10, 151. [Google Scholar] [CrossRef] [PubMed]
- Sung, W.W.; Lin, Y.M.; Wu, P.R.; Yen, H.H.; Lai, H.W.; Su, T.C.; Huang, R.H.; Wen, C.K.; Chen, C.Y.; Chen, C.J.; et al. High nuclear/cytoplasmic ratio of Cdk1 expression predicts poor prognosis in colorectal cancer patients. BMC Cancer 2014, 14, 951. [Google Scholar] [CrossRef]
- Willder, J.M.; Heng, S.J.; McCall, P.; Adams, C.E.; Tannahill, C.; Fyffe, G.; Seywright, M.; Horgan, P.G.; Leung, H.Y.; Underwood, M.A.; et al. Androgen receptor phosphorylation at serine 515 by Cdk1 predicts biochemical relapse in prostate cancer patients. Br. J. Cancer 2013, 108, 139–148. [Google Scholar] [CrossRef]
- Fu, Z.; Yang, G.; Wang, T.; Gao, D.; Lin, D.; Liu, X. Comprehensive Analysis of the Sorafenib-Associated Druggable Targets on Differential Gene Expression and ceRNA Network in Hepatocellular Carcinoma. J. Environ. Pathol. Toxicol. Oncol. Off. Organ Int. Soc. Environ. Toxicol. Cancer 2023, 42, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Depamphilis, M.L. Selective killing of cancer cells by suppression of geminin activity. Cancer Res. 2009, 69, 4870–4877. [Google Scholar] [CrossRef] [PubMed]
- Ananthula, S.; Sinha, A.; El Gassim, M.; Batth, S.; Marshall, G.D., Jr.; Gardner, L.H.; Shimizu, Y.; ElShamy, W.M. Geminin overexpression-dependent recruitment and crosstalk with mesenchymal stem cells enhance aggressiveness in triple negative breast cancers. Oncotarget 2016, 7, 20869–20889. [Google Scholar] [CrossRef] [PubMed]
- Kushwaha, P.P.; Rapalli, K.C.; Kumar, S. Geminin a multi task protein involved in cancer pathophysiology and developmental process: A review. Biochimie 2016, 131, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Salabat, M.R.; Melstrom, L.G.; Strouch, M.J.; Ding, X.Z.; Milam, B.M.; Ujiki, M.B.; Chen, C.; Pelling, J.C.; Rao, S.; Grippo, P.J.; et al. Geminin is overexpressed in human pancreatic cancer and downregulated by the bioflavanoid apigenin in pancreatic cancer cell lines. Mol. Carcinog. 2008, 47, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Montanari, M.; Boninsegna, A.; Faraglia, B.; Coco, C.; Giordano, A.; Cittadini, A.; Sgambato, A. Increased expression of geminin stimulates the growth of mammary epithelial cells and is a frequent event in human tumors. J. Cell. Physiol. 2005, 202, 215–222. [Google Scholar] [CrossRef]
- Du, R.; Li, K.; Zhou, Z.; Huang, Y.; Guo, K.; Zhang, H.; Chen, Z.; Han, L.; Bian, H. Bioinformatics and Experimental Validation of an AURKA/TPX2 Axis as a Potential Target in Esophageal Squamous Cell Carcinoma. Oncol. Rep. 2023, 49, 116. [Google Scholar] [CrossRef] [PubMed]
- Cui, D.J.; Wu, Y.; Wen, D.H. CD34, PCNA and CK19 expressions in AFP- hepatocellular carcinoma. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 5200–5205. [Google Scholar] [CrossRef] [PubMed]
- Sohn, E.H.; Flamme-Wiese, M.J.; Whitmore, S.S.; Wang, K.; Tucker, B.A.; Mullins, R.F. Loss of CD34 Expression in Aging Human Choriocapillaris Endothelial Cells. PLoS ONE 2014, 9, e86538. [Google Scholar] [CrossRef] [PubMed]
- Ершoва, Е.С.; Shmarina, G.; Martynov, A.V.; Zakharova, N.; Veiko, R.V.; Umriukhin, P.E.; Kostyuk, G.P.; Kutsev, S.I.; Veiko, N.N.; Kostyuk, S.V. NADPH-oxidase 4 Gene Over-Expression in Peripheral Blood Lymphocytes of the Schizophrenia Patients. PLoS ONE 2022, 17, e0269130. [Google Scholar] [CrossRef]
- Jourde-Chiche, N.; Dou, L.; Cérini, C.; Dignat-George, F.; Brunet, P. Vascular Incompetence in Dialysis Patients—Protein-Bound Uremic Toxins and Endothelial Dysfunction. Semin. Dial. 2011, 24, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Peñuelas-Haro, I.; Espinosa-Sotelo, R.; Crosas-Molist, E.; Herranz-Itúrbide, M.; Caballero-Díaz, D.; Alay, A.; Solé, X.; Ramos, E.; Serrano, T.; Martínez-Chantar, M.L.; et al. The NADPH oxidase NOX4 regulates redox and metabolic homeostasis preventing HCC progression. Hepatology 2023, 78, 416–433. [Google Scholar] [CrossRef] [PubMed]
- Arneth, B. Tumor Microenvironment. Medicina 2019, 56, 15. [Google Scholar] [CrossRef]
- Liu, X.; Si, F.; Bagley, D.M.; Ma, F.; Zhang, Y.; Tao, Y.; Shaw, E.; Peng, G. Blockades of Effector T Cell Senescence and Exhaustion Synergistically Enhance Antitumor Immunity and Immunotherapy. J. Immunother. Cancer 2022, 10, e005020. [Google Scholar] [CrossRef]
- Etxebeste-Mitxeltorena, M.; Rincón-Loza, I.d.; Martín-Antonio, B. Tumor Secretome to Adoptive Cellular Immunotherapy: Reduce Me Before I Make You My Partner. Front. Immunol. 2021, 12, 717850. [Google Scholar] [CrossRef]
- Battram, A.M.; Bachiller, M.; Martín-Antonio, B. Senescence in the Development and Response to Cancer with Immunotherapy: A Double-Edged Sword. Int. J. Mol. Sci. 2020, 21, 4346. [Google Scholar] [CrossRef] [PubMed]
- Forner, A.; Reig, M.; Bruix, J. Hepatocellular carcinoma. Lancet 2018, 391, 1301–1314. [Google Scholar] [CrossRef] [PubMed]
- Ainiwaer, A.; Chen, Y.; Lu, Y. Precise staging of advanced HCC promotes higher quality of personalized treatment management: Chinese experts consensus on precision diagnosis and management of advanced hepatocellular carcinoma (2023). Hepatoma Res. 2024, 10, 4. [Google Scholar] [CrossRef]
- Yang, X.; Yang, C.; Zhang, S.; Geng, H.; Zhu, A.X.; Bernards, R.; Qin, W.; Fan, J.; Wang, C.; Gao, Q. Precision treatment in advanced hepatocellular carcinoma. Cancer Cell 2024, 42, 180–197. [Google Scholar] [CrossRef] [PubMed]
- Liang, R.; Zhang, J.; Liu, Z.; Liu, Z.; Li, Q.; Luo, X.; Li, Y.; Ye, J.; Lin, Y. Mechanism and Molecular Network of RBM8A-Mediated Regulation of Oxaliplatin Resistance in Hepatocellular Carcinoma. Front. Oncol. 2020, 10, 585452. [Google Scholar] [CrossRef]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Long, M.; Zhou, Z.; Wei, X.; Lin, Q.; Qiu, M.; Zhou, Y.; Chen, P.; Jiang, Y.; Wen, Q.; Liu, Y.; et al. A novel risk score based on immune-related genes for hepatocellular carcinoma as a reliable prognostic biomarker and correlated with immune infiltration. Front. Immunol. 2022, 13, 1023349. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Singh, D.; Zeng, Z.; Coleman, S.J.; Huang, Y.; Savich, G.L.; He, X.; Mieczkowski, P.; Grimm, S.A.; Perou, C.M.; et al. MapSplice: Accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res. 2010, 38, e178. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Tacutu, R.; Thornton, D.; Johnson, E.; Budovsky, A.; Barardo, D.; Craig, T.; Diana, E.; Lehmann, G.; Toren, D.; Wang, J.; et al. Human Ageing Genomic Resources: New and updated databases. Nucleic Acids Res. 2018, 46, D1083–D1090. [Google Scholar] [CrossRef] [PubMed]
- Heagerty, P.J.; Lumley, T.; Pepe, M.S. Time-dependent ROC curves for censored survival data and a diagnostic marker. Biometrics 2000, 56, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Meyer, D.; Dimitriadou, E.; Hornik, K.; Weingessel, A.; Leisch, F. Misc Functions of the Department of Statistics, Probability Theory Group (Formerly: E1071), TU Wien [R Package e1071 Version 1.7-4]. 2020. Available online: https://www.semanticscholar.org/paper/Misc-Functions-of-the-Department-of-Statistics%2C-TU-Meyer-Dimitriadou/571964e71aa822b950c6c6dfed52fa63694c05a9 (accessed on 24 April 2024).
- Karatzoglou, A.; Smola, A.; Hornik, K. kernlab: Kernel-Based Machine Learning Lab. 2018. Available online: https://CRAN.R-project.org/package=kernlab (accessed on 24 April 2024).
- Kuhn, M. Building Predictive Models in R Using the caret Package. J. Stat. Softw. 2008, 28, 1–26. [Google Scholar] [CrossRef]
- Wilkerson, M.D.; Hayes, D.N. ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking. Bioinformatics 2010, 26, 1572–1573. [Google Scholar] [CrossRef] [PubMed]
- Dayimu, A. forestploter: Create a Flexible Forest Plot. 2024. Available online: https://CRAN.R-project.org/package=forestploter (accessed on 24 April 2024).
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Mayakonda, A.; Lin, D.C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef]
- Salomonis, N. Investigating Cell Fate Decisions with ICGS Analysis of Single Cells. Methods Mol. Biol. 2019, 1975, 251–275. [Google Scholar] [CrossRef] [PubMed]
- Korsunsky, I.M.H.; Patikas, N.; Yao, H.; Millard, N.; Fan, J.; Slowikowski, K.; Raychaudhuri, S. harmony: Fast, Sensitive, and Accurate Integration of Single Cell Data. 2023. Available online: https://CRAN.R-project.org/package=harmony (accessed on 24 April 2024).
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M., 3rd; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587.e3529. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, X.; Luo, Y.; Huang, Y.; Zhu, Z.; Yin, H.; Xu, S. Cellular Senescence in Hepatocellular Carcinoma: Immune Microenvironment Insights via Machine Learning and In Vitro Experiments. Int. J. Mol. Sci. 2025, 26, 773. https://doi.org/10.3390/ijms26020773
Lu X, Luo Y, Huang Y, Zhu Z, Yin H, Xu S. Cellular Senescence in Hepatocellular Carcinoma: Immune Microenvironment Insights via Machine Learning and In Vitro Experiments. International Journal of Molecular Sciences. 2025; 26(2):773. https://doi.org/10.3390/ijms26020773
Chicago/Turabian StyleLu, Xinhe, Yuhang Luo, Yun Huang, Zhiqiang Zhu, Hongyan Yin, and Shunqing Xu. 2025. "Cellular Senescence in Hepatocellular Carcinoma: Immune Microenvironment Insights via Machine Learning and In Vitro Experiments" International Journal of Molecular Sciences 26, no. 2: 773. https://doi.org/10.3390/ijms26020773
APA StyleLu, X., Luo, Y., Huang, Y., Zhu, Z., Yin, H., & Xu, S. (2025). Cellular Senescence in Hepatocellular Carcinoma: Immune Microenvironment Insights via Machine Learning and In Vitro Experiments. International Journal of Molecular Sciences, 26(2), 773. https://doi.org/10.3390/ijms26020773