Abstract
Several mutations of the uppermost arginine, R219, in the voltage-sensing sliding helix S4I of cardiac sodium channel Nav1.5 are reported in the ClinVar databases, but the clinical significance of the respective variants is unknown (VUSs). AlphaFold 3 models predicted a significant downshift of S4I in the R219C VUS. Analogous downshift S4I, upon its in silico deactivation, resulted in a salt bridge between R219 and the uppermost glutamate, E161, in helix S2I. To understand how salt bridge elimination affects biophysical characteristics, we generated mutant channel R219E, expressed it in the HEK293-T cells, and employed the patch-clamp method in a whole-cell configuration. Mutation R219E did not change the peak current density but shortened time to the peak current at several potentials, significantly enhanced activation, enhanced steady-state inactivation and steady-state fast inactivation, and slowed recovery from inactivation. Taken together, these data suggest that mutation R219E destabilized the resting state of Nav1.5. Cardiac syndromes associated with mutations R219P/H/C/P or E161Q/K are consistent with the observed changes of biophysical characteristics of mutant channel R219E suggesting pathogenicity of the respective VUSs, as well as ClinVar-reported VUSs involving arginine or glutamate in homologous positions of several Nav1.5 paralogs.
1. Introduction
Cardiac voltage-gated sodium channels (Nav1.5) are responsible for the initiation and propagation of action potentials (AP) in cardiomyocytes by allowing the rapid entry of sodium ions into the cell during phase 0 [1]. The Nav1.5 channel has a pore-forming α subunit of 220 kDa [2] and one or more regulatory β subunits of 30–40 kDa [3]. The α subunit, which is encoded by the SCN5A gene localized in chromosome 3p21 [4], is a pseudo hetero-tetramer of four homologous repeats (I–IV). Each repeat contains six transmembrane α-helical segments, S1–S6. Helices S1 to S4 form a voltage-sensing domain (VSD). Helices S5, S6, and a large extracellular membrane re-entering P-loop between S5 and S6 contribute a quarter to the pore domain (PD) [5,6,7]. Each P-loop contains ascending (P1) and descending (P2) helices with a selectivity filter residue between them [8]. A voltage-sensing siding helix, S4, features positively charged Arg or Lys residues, which collectively move the helix across the membrane in response to changes of the membrane voltage. Navs exist in several states. At the resting membrane potential (−90 to −80 mV), the channels are in the resting state [9]. Membrane depolarization activates VSDI, VSDII, and VSDIII, and the activation gate in the pore domain opens up. Subsequent activation of VSDIV leads to fast inactivation [10]. With prolonged or repeated depolarization, the channels enter slow inactivated state(s). Upon recovery from inactivation, the channel transits to deactivated and then to the resting state; see [11] for a review.
According to the World Health Organization, by 2030, the number of diagnosed cases of cardiovascular diseases may increase by 40% relative to 2022 [12]. Thus, the pathology of the cardiovascular system, in particular arrhythmias, is an urgent problem in modern healthcare. Pathogenic variants of the SCN5A gene are associated with the development of arrhythmogenic syndromes. Missense variants are predominantly present in the heterozygous state. SCN5A-associated syndromes include Brugada syndrome type 1 (BrS1), long QT syndrome type 3 (LQT3), sick sinus syndrome, progressive cardiac conduction disorder (Lev–Lenegre syndrome), and other diseases [13]. ClinVar [14] and other databases accumulate data on inherited variants of the SCN5A gene. Most of the identified missense variants are observed in single patients and, according to the American College of Medical Genetics classification, they are variants of unknown significance, VUSs [15]. Information about the clinical significance of a specific genetic variant is essential for proper diagnosis, risk stratification, and patient management. Functional studies allow for the determination of biophysical mechanisms of arrhythmias. The patch-clamp technique allows the biophysical properties of Nav channels to be studied and their functional activity to be evaluated.
Pathogenic genetic variants can stabilize or destabilize various channel states. Respective changes in the equilibrium between different Nav1.5 states may affect channel conductance, duration and amplitude of the peak and late sodium currents, or disrupt inactivation, resulting in prolonged sodium currents during the AP plateau phase [1]. These changes can affect the normal heart physiology and lead to formation of arrhythmogenic foci. Moreover, disturbances in the structure of Nav1.5 can lead to both gain-of-function (GoF) and loss-of-function (LoF) phenotypes [16]. The GoF syndromes are associated with delayed inactivation, accelerated activation, or accelerated recovery from the inactivation. As a result, later repolarization increases the duration of the ventricular AP and could increase the QT interval [9]. A decreased channel activity can cause stabilization of the inactivated or resting state and destabilization of the activated state. As a result, myocardial excitability decreases and conduction slows down [9].
The vast majority of mutations, which are localized in VSDs, are described in the ClinVar database as VUSs. Functional studies of mutant channels are necessary to understand molecular mechanisms of possible channel dysfunction associated with such mutations. If a functional study demonstrates significant changes in the biophysical characteristics of a mutant channel, VUSs in the respective position may be reclassified as likely pathogenic (LP) variants.
In this study, we focus on the uppermost arginine in the voltage-sensing helix S4 of VSDI (R1_S4I or R1). Various mutations of this arginine in the Nav1.5 channel and its paralogues are reported in the ClinVar database as VUSs. We first analyzed state-dependent contacts of R1 in helix S4I in experimental structures of Navs, in AlphaFold 3 (AF3) models of VSDI variants, and in in silico deactivated S4I in the WT and R219E mutant channels. These structural data strongly suggest a state-dependent salt bridge, R219:E161. We further generated mutant channel R219E, expressed it in the HEK293-T cells, and demonstrated significant changes in various biophysical characteristics of the mutant channel versus the WT channel. Based on these data, we proposed an atomic mechanism of the observed dysfunction of the Nav1.5-R219E mutant channel. In view of significant changes in the biophysical characteristics of the mutant channel R219E, we propose that fifteen VUSs in respective positions of Nav1.5 and its sodium channel paralogs may be reclassified as P/LP variants.
2. Results
2.1. Arginine R1 in Helix S4I (R1_S4I) and Glutamate E1 in Helix S2I (E1_S2I) of VSDI in Experimental and Computed Structures of Nav1.x Channels
In the cryoEM structure of channel hNav1.5 (PDB ID: 6lqa), R1_S4I (R219) forms a polar contact with Q859 in helix S5II of the pore domain, whereas E1_S2I (E161) forms a salt bridge with R2_S4I (R222) (Figure 1a). Among other cryoEM structures of Nav1.x channels with activated VSDs, R1_S4I interacts with E1_S2I only in the Nav1.8 channel (PDB ID: 7wel, 7we4, 7wfr). Thus, available cryoEM structures of eukaryotic sodium channels with activated VSDs do not provide a consensus view on contacts of ionizable residues in the extracellular half of VSDI.
Earlier, we used crystal structures of the prokaryotic sodium channel NavAb with the sliding helices S4 in the activated (PDB ID: 6p6x) and resting (PDB ID: 6p6w) states [17] to build homology models of the hNav1.5 channel with resting and activated VSDs [18]. In our resting-state model of hNav1.5, R1_S4I is salt-bridged to E1_S2I (Figure 1b). Respective arginine and glutamate residues are present in VSDI of all Nav1.x channels, indicating their functional importance.

Figure 1.
(a) Membrane view of the cryoEM structure with activated VSDs (PDB ID: 6lqa). Repeats I, II, III, and IV are shown in pink, yellow, green, and gray, respectively. R219 forms a polar contact with glutamine Q859 in helix S5I of the pore domain, while glutamate E161 forms a salt bridge with arginine R222. (b) Deactivated VSDI visualized using coordinates from [18]. R219 forms a salt bridge with glutamate E161. Mutation R219E would cause electrostatic repulsion between the two glutamates.
An important piece of evidence regarding state-dependent contacts of voltage-sensing arginines R1 in Nav1.5 channels was provided by William Catterall and colleagues [19]. The authors obtained cryoEM structures of the rat channel Nav1.5 in the apo-state and in complex with the deathstalker scorpion α-toxin LqhIII that traps VSDIV in the resting state. A comparison of the 3D aligned structures of rNav1.5 channel in the apo and toxin-bound states showed that the toxin binding downshifted the backbone atoms of R1_S4IV by two helical turns as compared with the apo channel (Figure 2a,c). In the toxin-bound structure, the side chain of R1_S4IV was unresolved, implying its high flexibility. In Monte Carlo (MC) minimized models of this structure, R1_S4IV adopted two energetically optimal orientations, forming salt bridges either with E1_S3IV or E2_S2IV.
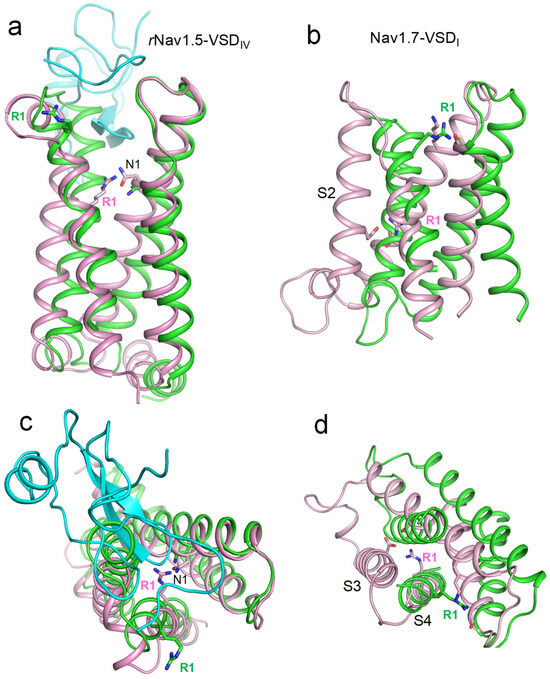
Figure 2.
Activated and deactivated VSDs in cryoEM structures of toxin-bound rNav1.5 and mutated hNav1.7. Shown are membrane (a,b) and extracellular (c,d) views of 3D-aligned cryo-EM structures with activated (green) and deactivated (magenta) VSDs. Upon VSD deactivation, state-dependent contacts of the uppermost arginines (R1) contribute to VSD stabilization. (a,c) rNav1.5 in the apo state (PDB ID: 6uz3) and in complex with gating-modifying toxin LQHIII (PDB ID: 7xsu). The toxin (cyan) downshifts S4IV by two helical turns (a). It also twists S4IV anticlockwise (c) and somewhat shifts helices in the membrane plane (c). In the toxin-bound deactivated state, R1 interacts with the uppermost D1_S2, which sequentially aligns with Nav1.5 glutamate E1_S2I. (c,d) WT channel hNav1.7 (PDB ID: 7xvf) and mutant channel with deactivated VSDI (PDB ID: 7xve). In the deactivated VSDI, R1 forms salt bridges with an asparagine in helix S3 and a glutamate in helix S2.
The CryoEM structure of the hNav1.7 channel with 11 engineered point mutations (PDB ID: 7xve) [20] shows VSDI in a completely deactivated, down state (Figure 2b,d). In this structure, the side chain of R1_S4I points in the cytoplasmic direction, forming state-dependent salt bridges with glutamate residues in the cytoplasmic halves of helices S2I and S3I. However, this structure does not rule out the possibility of a salt bridge between R1_S4I and E1_S2I in the hNav1.5 channel. Indeed, in the mutated channel hNav1.7, E1_S2I is substituted with lysine that repels rather than attracts R1_S4I. Furthermore, the hNav1.5 channel lacks an acidic residue in the cytoplasmic half of S3I, which, together with E2_S2I, attracts R1_S4I in the cryoEM structure of mutant channel hNav1.7.
2.2. AlphaFold 3 Structures of VSDI in WT Nav1.5 and ClinVar-Reported Variants of R1_S4I and E1_S2I
We generated AF3 models of VSDI in the WT channel hNav1.5 and in four mutant channels where R1_S4I is substituted with Glu, Cys, His, or Pro and 3D-aligned the models with PyMol. Glutamate or proline substitutions of R1_S4I resulted in only minimal changes in the VSDI folding (Figure 3b). In contrast, the cysteine substitution of R1_S4I down-shifted the cysteine by two helical turns, and caused a disulfide bond with a cysteine in the middle of helix S1I. Helix S4I adopted a deactivated position (Figure 3b), which was seen in the cryoEM structure hNav1.7 with eleven engineered residues in VSDI (Figure 2b). A significant downshift was also predicted for the histidine substitution of R1_S4I, likely due to attraction of the protonated histidine to the uppermost glutamate E1_S2I (Figure 3b). Thus, AF3 models do predict significant downshifts of S4I in mutant channels where R1_S4I is substituted by cysteine or histidine, suggesting dysfunction of these ClinVar-reported variants of hNav1.5.
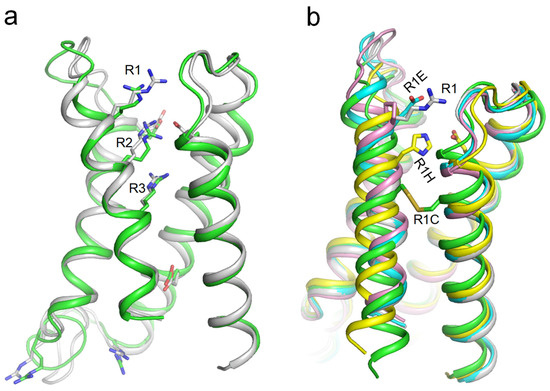
Figure 3.
(a) VSDI in cryo-EM structure of hNav1.5 (PDB ID: 6lqa, green) and AF3 model of lone VSDI from hNav1.5 (gray). Arginine and glutamate residues are shown as sticks. (b) 3D aligned VSDI models generated by AF3: WT (gray), R1E (cyan), R1H (yellow), R1C (green), and R1P (magenta). R1 in the WT model and Glu in the R1E model do not make intersegment contacts. Cys in the R1C model formed a disulfide bond with a cysteine in helix S1, causing a downshift of S4I by two helical turns.
2.3. In Silico Deactivating Lone VSDI of the WT Channel Nav1.5 and Mutant Channel R219E
To model deactivation of VSDI in the WT and R219E channels, we in silico downshifted S4I as described in the methods section. Downshifting of S4I in the WT channel yielded 21 MC-minimized structures. The strongest attraction between R219 and E161 was found at the final step of the trajectory, where these residues were salt-bridged (Figure 4a). At the extracellular view of the 21 structures, S4I turned anticlockwise (Figure 4b), resembling the anticlockwise turns in the cryoEM structures of toxin-deactivated S4IV of rNav1.5 (Figure 2c) and deactivated VSDI in the mutant channel hNav1.7 (Figure 2d).
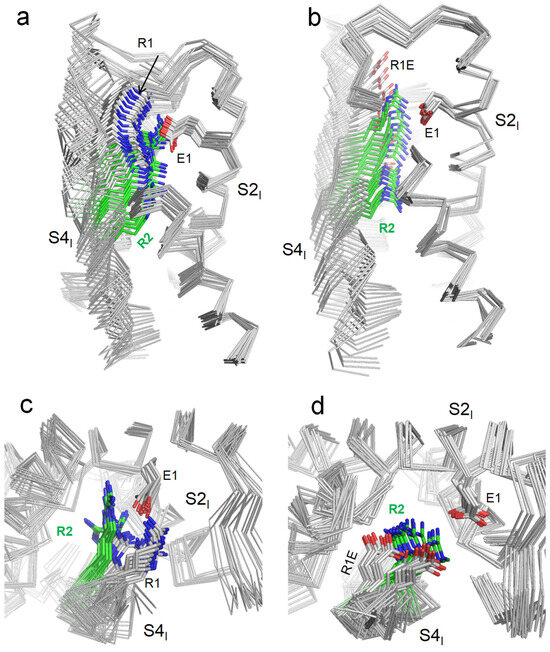
Figure 4.
In silico deactivation of VSDI in the WT channel hNav1.5 (a,c) and mutant channel R219E (b,d). The backbones are shown by ribbons, with some parts removed for clarity. (a,b) membrane views. (c,d) Extracellular views. Note green color of sidechain carbon atoms in R2.
Upon in silico deactivation of VSDI in the mutant channel R219E (Figure 4b,d), carbon atoms of the COO groups in R219E and E161 were 9.7 Å apart in the first step of the S4I downshifting trajectory. The distance decreased to 5.1 Å in step 14 and increased to 8.1 Å in the final step, 21. In step 14, the strong repulsion between the two carboxylates was compensated by their electrostatic attraction to R222. As in the WT VSDI, S4I turned anticlockwise (when viewed from the extracellular space).
The above analysis of cryoEM structures and our molecular models suggests that, in the resting state of hNav1.5_VSDI, R219 was salt-bridged with glutamate E161. Mutations of R219 in ClinVar-reported variants eliminated this salt bridge and may have affected the channel gating. The strongest impact on the gating was expected for mutant channel R219E where the electrostatic repulsion between the engineered and native glutamates would destabilize the resting state of VSDI. With this idea in mind, we generated mutant channel R219E and explored its biophysical properties.
2.4. Biophysical Characteristics of the WT and R219E Channels
We generated mutant construction R219E-pcDNA3.1 using site-directed mutagenesis via PCR amplification (Figure 5a). The substitution was verified with Sanger sequencing (Figure 5b). The plasmid vector with the substitution was expressed in the HEK293-T cell culture using GFP as a marker of transfection efficiency.
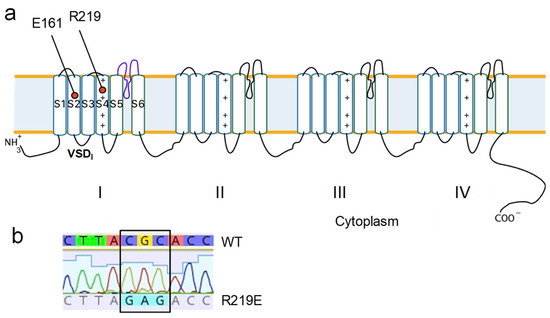
Figure 5.
Generating mutant construct R219E-Nav1.5. (a) E161 and R219 in the topological model of the Nav1.5 channel. (b) Results of site-specific mutagenesis verified using the Sanger sequencing method.
Mutation R219E led to functionally active sodium channels with typical currents (Figure 6a). The capacitance of cells expressing Nav1.5-R219E did not significantly differ from that of Nav1.5-WT (WT: 8.6 ± 0.5 pF, n = 32; R219E: 9.2 ± 0.8 pF, n = 21), indicating that the cell size was similar in the control and experimental groups. R219E did not significantly change the peak sodium current density (Figure 6b; Table 1), but the current density significantly decreased at potentials from −40 and +40 mV (Figure 6c). The time to peak of the R219E channel was significantly lower at potentials from −50 to −40 mV (Figure 6d), indicating the enhanced activation. Mutation R219E did not affect the current decay to 50% at potentials between −50 and −45 mV (Figure 6e).
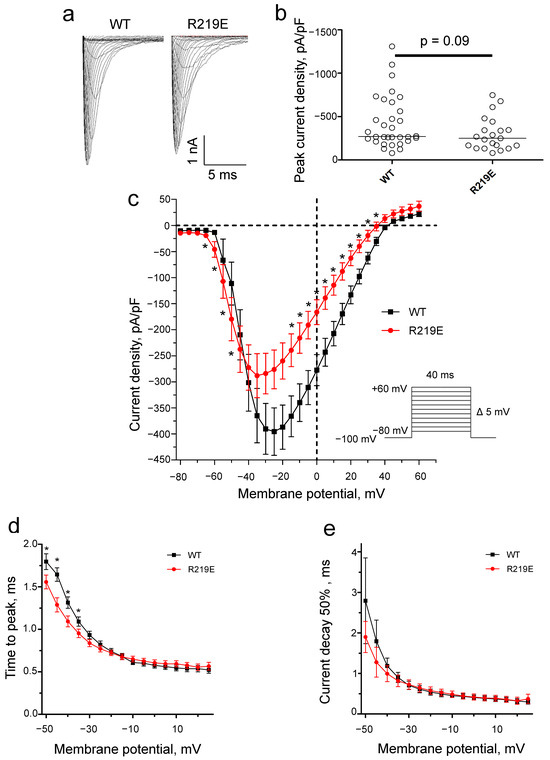
Figure 6.
Changes in Na+ current due to mutation of R219E. (a) Representative recordings of Na+ current for the WT and mutant channel R219E. (b) Distribution of Na+ current density at peak values. There was no significant difference for the WT and R219E channels. Each circle corresponds to an experiment with one cell (WT: n = 32; R219E: n = 21). (c) Volt-ampere characteristics of Na+ currents. Mutation R219E caused significant changes in the current density at some voltages. Asterisks (*) indicate significantly different values. (d) The voltage dependence of time to peak Na+ current for the WT and R219E channels. Mutation R219E caused a marked reduction in time to peak in the range from −50 to −35 mV. (e) Time of 50% decay for WT (black squares) and R219E (red circles) channels. Mutation R219E did not significantly influence the time of 50% decay.

Table 1.
Biophysical characteristics of Nav1.5 wildtype and R219E mutant channels a.
Mutation R219E caused a significant hyperpolarization shift (9.6 mV) of the steady-state activation curve (Figure 7a; Table 1), indicating an accelerated activation that is consistent with observed reduction in time to peak. Mutation R219E also statistically significantly shifted the curve of the steady-state inactivation by −5.6 mV (Figure 7b, Table 1), indicating accelerated inactivation.
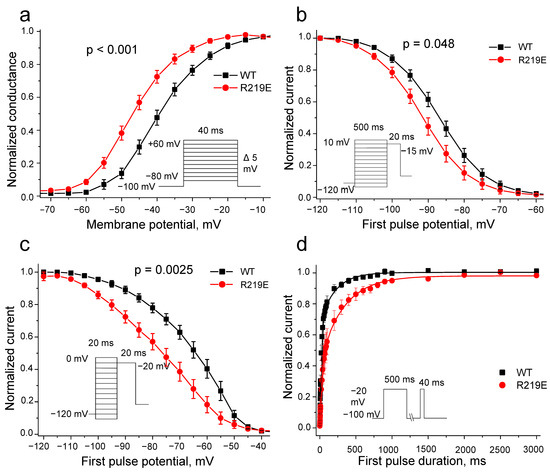
Figure 7.
Biophysical characteristics of wild-type WT (black squares) and R219E (red circles) channels. (a) Steady-state activation. Mutation R219E caused a significant shift (−9.6 mV) of the steady-state activation (WT: n = 32; R219E: n = 21). (b) Steady-state inactivation for the WT (black squares) and R219E (red circles) channels. Mutation caused a negative shift of steady-state inactivation (WT: n = 24; R219E: n = 15). (c) Steady-state fast inactivation for the WT (black squares) and R219E (red circles) channels. Mutation R219E cased a significant shift of (15 mV) of the fast inactivation curve towards hyperpolarized potentials (WT: n = 14; R219E: n = 14). (d) Time course of recovery from inactivation. Mutation R219E markedly decelerated recovery from inactivation.
Next, we explored the fast inactivation of mutant channels to determine the role of this process in the changes in steady-state inactivation and observed a significant negative shift of −15.1 mV (Figure 7c; Table 1). The changed kinetics of the steady-state inactivation may be associated with the enhanced activation (“easy on—easy off”). Thus, the observed alterations in inactivation could be explained as the consequences of the enhanced activation. The R219E channels also demonstrated slower recovery from inactivation (Figure 7d, Table 1), supporting the resting-state destabilization.
3. Discussion
Earlier, we proposed that R219 in the hNav1.5 channel is salt-bridged with E219 in the resting but not activated state of VSDI [18]. Here, we analyzed cryoEM structures of Nav1.x channels, including two channels in which VSDs are captured in both the resting and activated states. We further deactivated VSDI in silico in the WT hNav1.5 channel and R219E channel. Our analyses of cryoEM structures and computational results strongly suggest that R219 forms a salt bridge with E219 in the resting but not activated VSDI. To experimentally test this hypothesis, we generated mutant channel hNav1.5-R219E where the electrostatic repulsion between R219E and E161 would be strong, and explored the biophysical properties of the WT and mutant channels. We found that mutation R219E enhanced activation, inactivation, and fast inactivation. It also slowed recovery from inactivation, but did not change the peak current density.
Several Nav1.5 genetic variants involving E161 or R219 have previously been functionally characterized. Thus, mutation E161K caused a positive shift of the steady-state activation, but did not change the kinetics of recovery from the inactivation, which can decrease the cardiac excitability [13]. Other functional studies of the E161K mutant channel using the tsA201 expression model demonstrated similar changes in electrophysiological characteristics [21]. The above studies show that mutation E161K, which eliminates the salt bridge between R1_S4I and E1_S2I, impairs activation. In contrast, in our experiments, mutation R219E accelerated the channel activation. A likely cause for this discrepancy is that E161 may form state-dependent salt bridges with two voltage sensing arginines, R219 and R222 (Figure 4a). In the mutant channel E161K, repulsion between the engineered lysine K161 and arginine R222 would obstruct the voltage-dependent shift of S4I in the extracellular direction, thus retarding activation of VSDI. In contrast, the major effect of mutation of R219E is destabilization of the resting-state salt bridge R219:E161 in the WT channel, which facilitates channel activation. Destabilization of the resting state could also explain the observed impairment of recovery from inactivation. The curve of recovery from inactivation reflects the transition between inactivated and resting states. The channel inactivation involves activation of VSDIV [11], whereas channel transition to the resting state involves deactivation of all VSDs. Therefore, the slower recovery from inactivation in the R219E mutant channel could be due to destabilization of the VSDI resting (deactivated) state because of repulsion between R219E and E161.
We observed the enhanced steady state and fast inactivation of the mutant channel R219E (Figure 7b,c). The facilitated activation may accelerate inactivation due to a reduced duration of the channel activated state (“easy on—easy off”). This suggestion is supported by the shorter time to the peak current without changes in the inactivation decay characteristics (Figure 6d,e). Mutation R222Q, associated with dilated cardiomyopathy, caused enhanced activation and inactivation [22], suggesting that dysfunctions of mutant channels R219E and R222Q have similar mechanisms. Our data are consistent with a recent study in which charge-neutralizing mutations (R1Q, R2Q, and R3Q) were generated for each VSD in several Navs, and movement VSDI was proposed to be rate limiting for the Nav1.5 pore opening [23]. Earlier, a similar conclusion was made for the Nav1.4 channel [10].
Some mutations beyond VSDI also cause both gain-of-function (GoF) and lost-of-function (LoF) effects. Mutation T1620K in the S3-S4IV extracellular loop accelerates steady-state activation, increases the slope of the steady-state inactivation, and causes a late current, i.e., both LoF and GoF [24]. Mutation R340Q in the P-loop S5-S6I caused a negative shift in the steady-state activation and inactivation, as well as facilitating fast inactivation [25]. Mutation V411M accelerated activation and inactivation and caused late currents, but no changes in current density were recorded [26]. Mutations V1777M and S1904L in the C-terminal region caused a negative shift in the steady-state activation and inactivation and increased the sodium current [27,28]. Most of the above substitutions lead to complex changes in biophysical characteristics and are associated with the development of various syndromes and mixed phenotypes: BrS1, LQT3, dilated cardiomyopathy, sick sinus syndrome, etc.
Some Nav 1.5 variants involving R219 or E161 were explored in earlier biophysical studies. Thus, mutation E161K impaired activation [13,21]. Mutation R219H is proposed to cause a proton leakage current [29,30]. Five of six variants of R219 or E161, which are reported in the ClinVar database, have been identified in patients with syndromes associated with decreased Nav1.5 activity and are classified as VUSs (Table 2). An exception is genetic variant R219Q, which is listed in the ClinVar database as likely benign (Table 2). We propose that the long side chain of glutamine R219Q would form an H-bond with E161 in the resting VSDI. And although an H-bond is usually weaker than a salt bridge, the contact would still provide a stabilizing contribution to the resting-state VSDI.
Table 2.
Nav1.5 genetic missense variants of E161 and R219 reported to the ClinVar database.
We also found in the ClinVar database eleven mutations of R1_S4I or E1_S2I in five paralogs of the Nav1.5 channel (Nav1.1, Nav1.6, Nav1.7, Nav1.8, and Nav1.9). Two of these variants are reported as pathogenic, and nine variants as VUSs (Table 3). We suggest that salt bridge elimination in these variants is a possible mechanism of the channel dysfunction. Functional studies are necessary to estimate the likely pathogenicity of the VUSs. Our data suggest that such functional studies are promising and would allow the VUSs to be reclassified as pathogenic/likely pathogenic variants. Such a reclassification would be important for diagnostics of arrhythmias and the choice of treatment.
Table 3.
ClinVar-reported missense variants of R1_S4I or E1_S2I in paralogs of Nav1.5.
4. Materials and Methods
4.1. Structural Analyses and Modeling
The AlphaFold 3 (AF3) server [31] (https://golgi.sandbox.google.com/ was used to generate models of VSDI in the WT channel Nav1.5 and mutant channels R219E/C/H/P. Structures were visualized and 3D aligned using PyMol (Schrödinger, New York, NY, USA, v.099). Molecular modeling was performed with the ZMM program (www.zmmsoft.ca) as described, e.g., in [32]. Briefly, energy calculations were performed using the AMBER force field [33] with environment and distance-dependent dielectric function [34]. The energetically optimal structures were predicted with the method of Monte Carlo (MC) energy minimization [35]. MC trajectories were terminated when the last 100th energy minimization did not improve the channel energy.
In silico deactivation of VSDI was performed as described in [36,37]. Four CA atoms of voltage-sensing basic residues in helix S4I (R1, R2, R3, and K4) were progressively downshifted with steps of 0.5 Å. At each step, the energy was MC-minimized with the CA atoms permitted to move freely within the plane normal to the pore axis, but not to leave the plane. To prevent refolding of VSDI, at each step of the downshifting trajectory, CA atoms in helices S1, S2, and S3 were allowed to deviate penalty-free from the starting position up to 4 Å, but at larger deviations, the energy penalty of 10 kcal mol−1 Å−2 was imposed.
CryoEM structures and AF3 models of sodium channels were 3D aligned by minimizing the root mean square deviations of Cα atoms in P1 helices from matching atoms in the Kv1.2-Kv2.1 channel (PDB code: 2R9R), the first eukaryotic P-loop potassium channels whose crystal structure was obtained with the resolution below 2.5 Å [38]. Such alignment shows not only deformations within VSDs, but also shifts of VSDs over the pore domain.
4.2. Site-Directed Mutagenesis and Heterologous Expression System Analyses and Modeling
Vector pcDNA3.1 with the WT hNav1.5 and GFP (hH1-pcDNA3.1) was kindly provided by Prof. Hugues Abriel (Institute of Biochemistry and Molecular Medicine, University of Bern, Bern, Switzerland). Site-directed mutagenesis was carried out by PCR amplification with overlapping primers complementary to the gene region containing the nucleotide substitution leading to R219E. Then, we eliminated the template WT cDNA via DpnI (New England Biolabs, Ipswich, MA, USA) restriction (overnight) and performed the chemical bacterial transformation using XL1-blue strain (Eurogene, Moscow, Russia). Next, we extracted mutated cDNA and verified mutagenesis via Sanger sequencing.
hH1-pcDNA3.1 (1 μg) or R219E-pcDNA3.1 (1 μg) were transfected into HEK293-T cells growing on 3 cm dishes using 1 mg/mL aqueous solution of linear polyethylenimine hydrochloride (PEI, molecular weight 40,000, Polysciences, Warrington, PA, USA) at a 2:1 v/m ratio with pDNA. On the first day, cells were seeded in 3 cm dishes, and on the second day, transfection was performed. The transfection mix (1 μg hH1-pcDNA3.1 (WT or R219E), 0.1 μg GFP, 2.2 μL PEI, and 1 mL DMEM medium without supplements) was incubated for 15 min at room temperature and then added to the cell culture. GFP was used as a marker of transfection efficiency and analyzed using fluorescent microscopy (Supplemental Figure S1). Cells were maintained in DMEM supplemented with 2 mM glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin (Thermo Fisher Scientific, Waltham, MA, USA) in a CO2 incubator at +37 °C for 24 h, and then seeded onto glass slides coated with poly-lysine (Sigma Aldrich, Burlington, MA, USA) for subsequent electrophysiological experiments.
4.3. Electrophysiology
The sodium current (INa) was recorded using the patch-clamp method in the whole-cell configuration. All measurements were carried out at room temperature. The extracellular solution contained (mmol/L): 140 NaCl, 1 MgCl2, 1.8 CaCl2, 10 HEPES, and 10 glucose (pH 7.4 CsOH). The intracellular solution contained (mmol/L): 130 CsCl, 10 NaCl, 10 EGTA, and 10 HEPES (pH 7.3 CsOH). Glass microelectrodes were made from borosilicate glass using a P-1000 puller (Sutter Instrument, Novato, CA, USA). The electrode resistance varied from 2 to 3.5 MΩ. Series resistance was compensated by 75–80%. Data acquisition was carried out using an Axopatch 200B amplifier and Clampfit 10.3 software (Molecular Devices, San Jose, CA, USA). Currents were recorded at a frequency of 20 kHz, and low-pass filtering was carried out at a frequency of 5 kHz using an analog-to-digital interface (Digidata 1440A data acquisition system, Molecular Devices, San Jose, CA, USA). At least 3 independent transfections were used for electrophysiological recordings.
4.4. Data Analysis
The holding potential was −100 mV. The protocols used in the experiments are schematically represented in the figures. To record the current–voltage characteristics, we used the following protocol, with depolarizing pulses from −80 to 60 mV for 40 ms and a step of 5 mV at a frequency of 1 Hz. The current density at each test potential was estimated by normalizing INa to the cell capacitance.
To access the voltage dependency of the peak current advent, we analyzed current traces at potentials from −50 to +25 mV. The pulse start was considered as the zero time. We normalized each current trace to its maximal value and estimated time to the peak current. The latter was used as the initial point to analyze the inactivation decay. To simplify the analysis, we used 50% of the decay time instead of mono-exponential fitting. The decay time constant in the case of mono-exponential or bi-exponential fitting is highly variable and depends on the start and end points in the analyzed region, making difficult to standardize this parameter. We suggest using the time when the current returns to the level of 50% of maximum current (current decay 50%).
To access the steady-state activation, the maximal INa at each voltage value was recorded, and the corresponding conductance was calculated using the following equation:
where G is the conductance (Sm), Gmax is the maximum conductivity for Na+ ions, INa is the sodium current (pA), V is the membrane potential (mV), and Vrev is the reversal potential.
Next, we plotted the curve of steady-state activation of Nav1.5 (the voltage-dependence of normalized conductivity). The curves were approximated using the Boltzmann function:
where G is the conductance for Na+, Gmax is the maximum conductivity for Na+ ions, V is the membrane potential, V1/2 is the potential at which 50% of channels are activated, and k is the slope factor.
The voltage dependence of steady-state inactivation was measured using a two-step protocol with a 500 ms first pulse varying from −120 to 10 mV in 5 mV steps and a −15 mV test pulse lasting 20 ms. A curve of steady-state inactivation was plotted as the dependence of normalized INa in response to the first testing voltage pulse and approximated using the Boltzmann function:
where INa is the sodium current, Imax is the peak current, V is the membrane potential, V1/2 is the potential at which 50% of channels are inactivated, and k is the slope factor. The voltage dependence of fast inactivation was assessed in a similar way, but a protocol with a first-pulse duration of 20 ms was used. All results are presented as mean ± standard error (SEM). Statistical comparisons were performed using the unpaired Student test, with p < 0.05 considered statistically significant.
5. Conclusions
Glutamate substitution of the of the uppermost arginine R219 (R1) in helix S4 of the first voltage-sensing domain (VSDI) of the cardiac sodium channel hNav1.5 (R219E) caused negative shifts of the steady-state activation and inactivation and decelerated the recovery from inactivation. The enhanced activation of the mutant channel and its decelerated recovery from inactivation are consistent with the molecular modeling prediction on the salt bridge E161:R219 in the activated VSDI and destabilization of the resting VSDI due to electrostatic repulsion between glutamates E161 and R219E in the mutant channel. Elimination of the salt bridge is likely the atomic mechanism of dysfunction of ClinVar-reported disease variants of E161 or R219 for the hNav1.5 channel and mutations in analogous positions of sodium channel paralogs, suggesting that respective variants of unknown clinical significance are pathogenic/likely pathogenic variants.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms26020712/s1.
Author Contributions
Conceptualization, B.S.Z. and A.K.Z.; methodology, B.S.Z. and A.K.Z.; software, B.S.Z.; validation, B.S.Z. and A.K.Z.; formal analysis, B.S.Z. and A.K.Z.; investigation, O.E.K., B.S.Z. and A.K.Z.; resources, A.A.K. and B.S.Z.; data curation, A.K.Z.; writing—original draft preparation, O.E.K., A.K.Z. and B.S.Z.; writing—review and editing, A.A.K. and B.S.Z.; visualization, O.E.K. and B.S.Z.; supervision, B.S.Z.; project administration, A.A.K. and B.S.Z.; funding acquisition, B.S.Z. All authors have read and agreed to the published version of the manuscript.
Funding
The work is supported by the Sechenov Institute of Evolutionary Physiology and Biochemistry Research Program. B.S.Z. acknowledges support from the Natural Sciences and Engineering Research Council of Canada (RGPIN-2020-07100).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in this study are included in the article/Supplementary Materials. Further inquiries can be directed to the corresponding author.
Acknowledgments
We thank Denis Tikhonov for helpful discussions. Computations were performed, in part, using facilities provided by Compute Ontario (https://www.computeontario.ca) and the Digital Research Alliance of Canada (https://www.alliancecan.ca).
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Abbreviations
AP: action potential; BrS1, Brugada syndrome type 1; Cryo-EM, Cryo-electron microscopy; INa, sodium current; LQT3, long QT syndrome 3 type; MC, Monte Carlo; Nav, voltage-gated sodium channel; Nav1.5, cardiac voltage-gated sodium channel; PDB, Protein Data Bank; PCR, polymerase chain reaction.
References
- Amin, A.S.; Asghari-Roodsari, A.; Tan, H.L. Cardiac sodium channelopathies. Pflug. Arch. 2010, 460, 223–237. [Google Scholar] [CrossRef]
- Abriel, H.; Rougier, J.S.; Jalife, J. Ion channel macromolecular complexes in cardiomyocytes: Roles in sudden cardiac death. Circ. Res. 2015, 116, 1971–1988. [Google Scholar] [CrossRef]
- Remme, C.A. Cardiac sodium channelopathy associated with SCN5A mutations: Electrophysiological, molecular and genetic aspects. J. Physiol. 2013, 591, 4099–4116. [Google Scholar] [CrossRef] [PubMed]
- Veerman, C.C.; Wilde, A.A.; Lodder, E.M. The cardiac sodium channel gene SCN5A and its gene product NaV1.5: Role in physiology and pathophysiology. Gene 2015, 573, 177–187. [Google Scholar] [CrossRef]
- Detta, N.; Frisso, G.; Salvatore, F. The multi-faceted aspects of the complex cardiac Nav1.5 protein in membrane function and pathophysiology. Biochim. Biophys. Acta 2015, 1854, 1502–1509. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A. Voltage-gated sodium channels at 60: Structure, function and pathophysiology. J. Physiol. 2012, 590, 2577–2589. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A. Structure and function of voltage-gated sodium channels at atomic resolution. Exp. Physiol. 2014, 99, 35–51. [Google Scholar] [CrossRef] [PubMed]
- Payandeh, J.; Scheuer, T.; Zheng, N.; Catterall, W.A. The crystal structure of a voltage-gated sodium channel. Nature 2011, 475, 353–358. [Google Scholar] [CrossRef]
- Han, D.; Tan, H.; Sun, C.; Li, G. Dysfunctional Nav1.5 channels due to SCN5A mutations. Exp. Biol. Med. 2018, 243, 852–863. [Google Scholar] [CrossRef]
- Capes, D.L.; Goldschen-Ohm, M.P.; Arcisio-Miranda, M.; Bezanilla, F.; Chanda, B. Domain IV voltage-sensor movement is both sufficient and rate limiting for fast inactivation in sodium channels. J. Gen. Physiol. 2013, 142, 101–112. [Google Scholar] [CrossRef]
- Ahern, C.A.; Payandeh, J.; Bosmans, F.; Chanda, B. The hitchhiker’s guide to the voltage-gated sodium channel galaxy. J. Gen. Physiol. 2016, 147, 1–24. [Google Scholar] [CrossRef]
- Piot, O.; Boveda, S.; Defaye, P.; Klug, D.; Lacotte, J.; Marijon, E. Prospective evolution of cardiac arrhythmia care: 2030 vision. Arch. Cardiovasc. Dis. 2022, 115, 179–189. [Google Scholar] [CrossRef]
- Gui, J.; Wang, T.; Jones, R.P.; Trump, D.; Zimmer, T.; Lei, M. Multiple loss-of-function mechanisms contribute to SCN5A-related familial sick sinus syndrome. PLoS ONE 2010, 5, e10985. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Steinberg, C.; Pilote, S.; Philippon, F.; Laksman, Z.W.; Champagne, J.; Simard, C.; Krahn, A.D.; Drolet, B. SCN5A-C683R exhibits combined gain-of-function and loss-of-function properties related to adrenaline-triggered ventricular arrhythmia. Exp. Physiol. 2021, 106, 683–699. [Google Scholar] [CrossRef]
- Wisedchaisri, G.; Tonggu, L.; McCord, E.; Gamal El-Din, T.M.; Wang, L.; Zheng, N.; Catterall, W.A. Resting-State Structure and Gating Mechanism of a Voltage-Gated Sodium Channel. Cell 2019, 178, 993–1003.E12. [Google Scholar] [CrossRef]
- Korkosh, V.S.; Zaytseva, A.K.; Kostareva, A.A.; Zhorov, B.S. Intersegment Contacts of Potentially Damaging Variants of Cardiac Sodium Channel. Front. Pharmacol. 2021, 12, 756415. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Tonggu, L.; Gamal El-Din, T.M.; Banh, R.; Pomes, R.; Zheng, N.; Catterall, W.A. Structural basis for voltage-sensor trapping of the cardiac sodium channel by a deathstalker scorpion toxin. Nat. Commun. 2021, 12, 128. [Google Scholar] [CrossRef]
- Huang, G.; Wu, Q.; Li, Z.; Jin, X.; Huang, X.; Wu, T.; Pan, X.; Yan, N. Unwinding and spiral sliding of S4 and domain rotation of VSD during the electromechanical coupling in Na(v)1.7. Proc. Natl. Acad. Sci. USA 2022, 119, e2209164119. [Google Scholar] [CrossRef]
- Smits, J.P.; Koopmann, T.T.; Wilders, R.; Veldkamp, M.W.; Opthof, T.; Bhuiyan, Z.A.; Mannens, M.M.; Balser, J.R.; Tan, H.L.; Bezzina, C.R.; et al. A mutation in the human cardiac sodium channel (E161K) contributes to sick sinus syndrome, conduction disease and Brugada syndrome in two families. J. Mol. Cell. Cardiol. 2005, 38, 969–981. [Google Scholar] [CrossRef]
- Mann, S.A.; Castro, M.L.; Ohanian, M.; Guo, G.; Zodgekar, P.; Sheu, A.; Stockhammer, K.; Thompson, T.; Playford, D.; Subbiah, R.; et al. R222Q SCN5A mutation is associated with reversible ventricular ectopy and dilated cardiomyopathy. J. Am. Coll. Cardiol. 2012, 60, 1566–1573. [Google Scholar] [CrossRef] [PubMed]
- Brake, N.; Mancino, A.S.; Yan, Y.; Shimomura, T.; Kubo, Y.; Khadra, A.; Bowie, D. Closed-state inactivation of cardiac, skeletal, and neuronal sodium channels is isoform specific. J. Gen. Physiol. 2022, 154, e202112921. [Google Scholar] [CrossRef] [PubMed]
- Surber, R.; Hensellek, S.; Prochnau, D.; Werner, G.S.; Benndorf, K.; Figulla, H.R.; Zimmer, T. Combination of cardiac conduction disease and long QT syndrome caused by mutation T1620K in the cardiac sodium channel. Cardiovasc. Res. 2008, 77, 740–748. [Google Scholar] [CrossRef] [PubMed]
- Olesen, M.S.; Yuan, L.; Liang, B.; Holst, A.G.; Nielsen, N.; Nielsen, J.B.; Hedley, P.L.; Christiansen, M.; Olesen, S.P.; Haunsø, S.; et al. High prevalence of long QT syndrome-associated SCN5A variants in patients with early-onset lone atrial fibrillation. Circ. Cardiovasc. Genet. 2012, 5, 450–459. [Google Scholar] [CrossRef]
- Horne, A.J.; Eldstrom, J.; Sanatani, S.; Fedida, D. A novel mechanism for LQT3 with 2:1 block: A pore-lining mutation in Nav1.5 significantly affects voltage-dependence of activation. Heart Rhythm. 2011, 8, 770–777. [Google Scholar] [CrossRef]
- Bankston, J.R.; Sampson, K.J.; Kateriya, S.; Glaaser, I.W.; Malito, D.L.; Chung, W.K.; Kass, R.S. A novel LQT-3 mutation disrupts an inactivation gate complex with distinct rate-dependent phenotypic consequences. Channels 2007, 1, 273–280. [Google Scholar] [CrossRef][Green Version]
- Lupoglazoff, J.M.; Cheav, T.; Baroudi, G.; Berthet, M.; Denjoy, I.; Cauchemez, B.; Extramiana, F.; Chahine, M.; Guicheney, P. Homozygous SCN5A mutation in long-QT syndrome with functional two-to-one atrioventricular block. Circ. Res. 2001, 89, E16–E21. [Google Scholar] [CrossRef] [PubMed]
- Gosselin-Badaroudine, P.; Keller, D.I.; Huang, H.; Pouliot, V.; Chatelier, A.; Osswald, S.; Brink, M.; Chahine, M. A proton leak current through the cardiac sodium channel is linked to mixed arrhythmia and the dilated cardiomyopathy phenotype. PLoS ONE 2012, 7, e38331. [Google Scholar] [CrossRef]
- Moreau, A.; Gosselin-Badaroudine, P.; Mercier, A.; Burger, B.; Keller, D.I.; Chahine, M. A leaky voltage sensor domain of cardiac sodium channels causes arrhythmias associated with dilated cardiomyopathy. Sci. Rep. 2018, 8, 13804. [Google Scholar] [CrossRef]
- Varadi, M.; Bertoni, D.; Magana, P.; Paramval, U.; Pidruchna, I.; Radhakrishnan, M.; Tsenkov, M.; Nair, S.; Mirdita, M.; Yeo, J.; et al. AlphaFold Protein Structure Database in 2024: Providing structure coverage for over 214 million protein sequences. Nucleic Acids Res. 2024, 52, D368–D375. [Google Scholar] [CrossRef]
- Zhorov, B.S. Possible Mechanism of Ion Selectivity in Eukaryotic Voltage-Gated Sodium Channels. J. Phys. Chem. B 2021, 125, 2074–2088. [Google Scholar] [CrossRef]
- Weiner, S.J.; Kollman, P.A.; Nguyen, D.T.; Case, D.A. An all atom force field for simulations of proteins and nucleic acids. J. Comput. Chem. 1986, 7, 230–252. [Google Scholar] [CrossRef]
- Garden, D.P.; Zhorov, B.S. Docking flexible ligands in proteins with a solvent exposure- and distance-dependent dielectric function. J. Comput. Aided Mol. Des. 2010, 24, 91–105. [Google Scholar] [CrossRef]
- Li, Z.; Scheraga, H.A. Monte Carlo-minimization approach to the multiple-minima problem in protein folding. Proc. Natl. Acad. Sci. USA 1987, 84, 6611–6615. [Google Scholar] [CrossRef] [PubMed]
- Zhorov, B.S.; Du, Y.; Song, W.; Luo, N.; Gordon, D.; Gurevitz, M.; Dong, K. Mapping the interaction surface of scorpion beta-toxins with an insect sodium channel. Biochem. J. 2021, 478, 2843–2869. [Google Scholar] [CrossRef] [PubMed]
- Zaytseva, A.K.; Boitsov, A.S.; Kostareva, A.A.; Zhorov, B.S. Possible Interactions of Extracellular Loop IVP2-S6 With Voltage-Sensing Domain III in Cardiac Sodium Channel. Front. Pharmacol. 2021, 12, 742508. [Google Scholar] [CrossRef]
- Long, S.B.; Tao, X.; Campbell, E.B.; MacKinnon, R. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature 2007, 450, 376–382. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).