
Deciphering the Proteome and Phosphoproteome of Peanut (Arachis hypogaea L.) Pegs Penetrating into the Soil

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
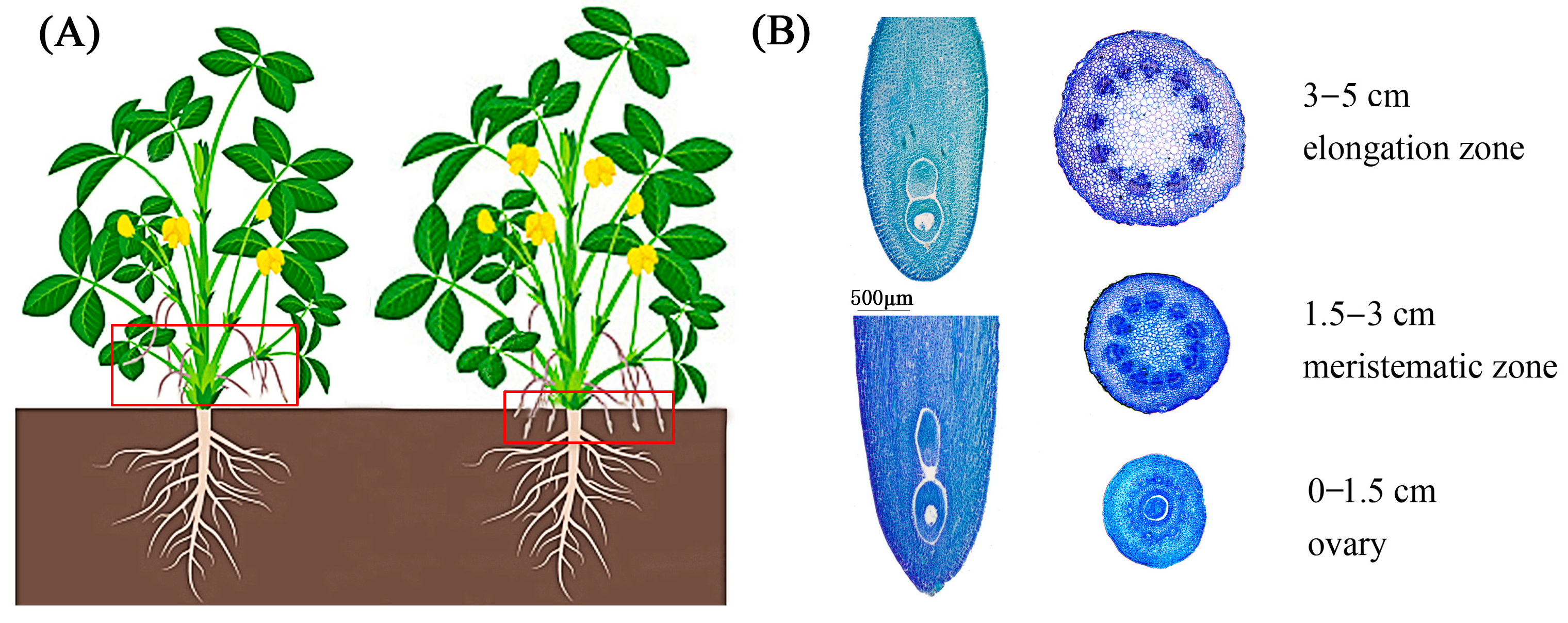
2.1. Phenotype Characteristics of Aerial and Underground Pegs
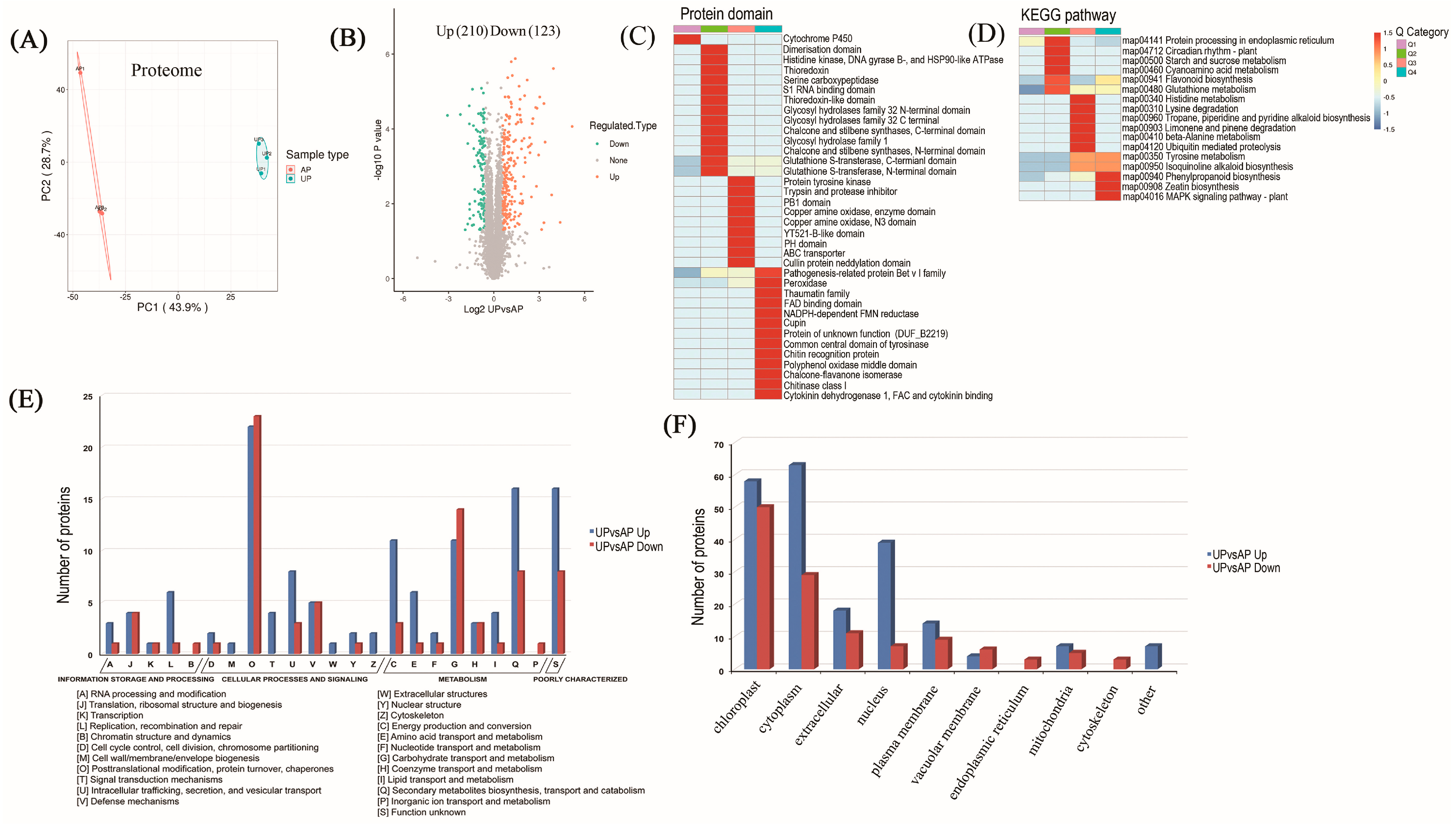
2.2. Proteomic Identification of Underground Pegs
2.3. Functional Classification and Verification of DAPs
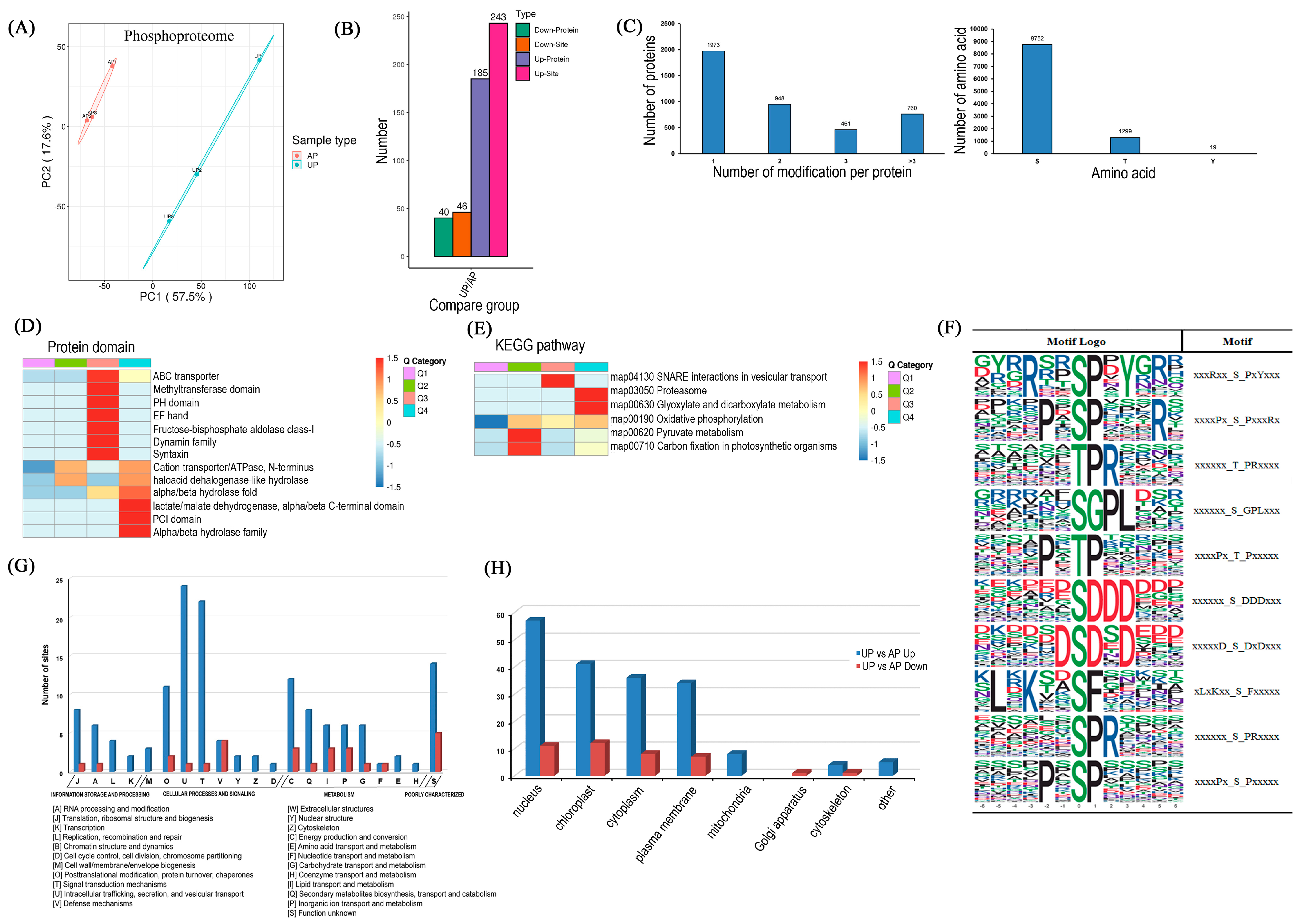
2.4. Functional Classification and Verification of DRPPs
2.5. Significantly Differentiated Kinase Phosphorylation Sites
2.6. Detection of Phosphorylated Sucrose-Phosphate Synthase and Verification of Omics Data
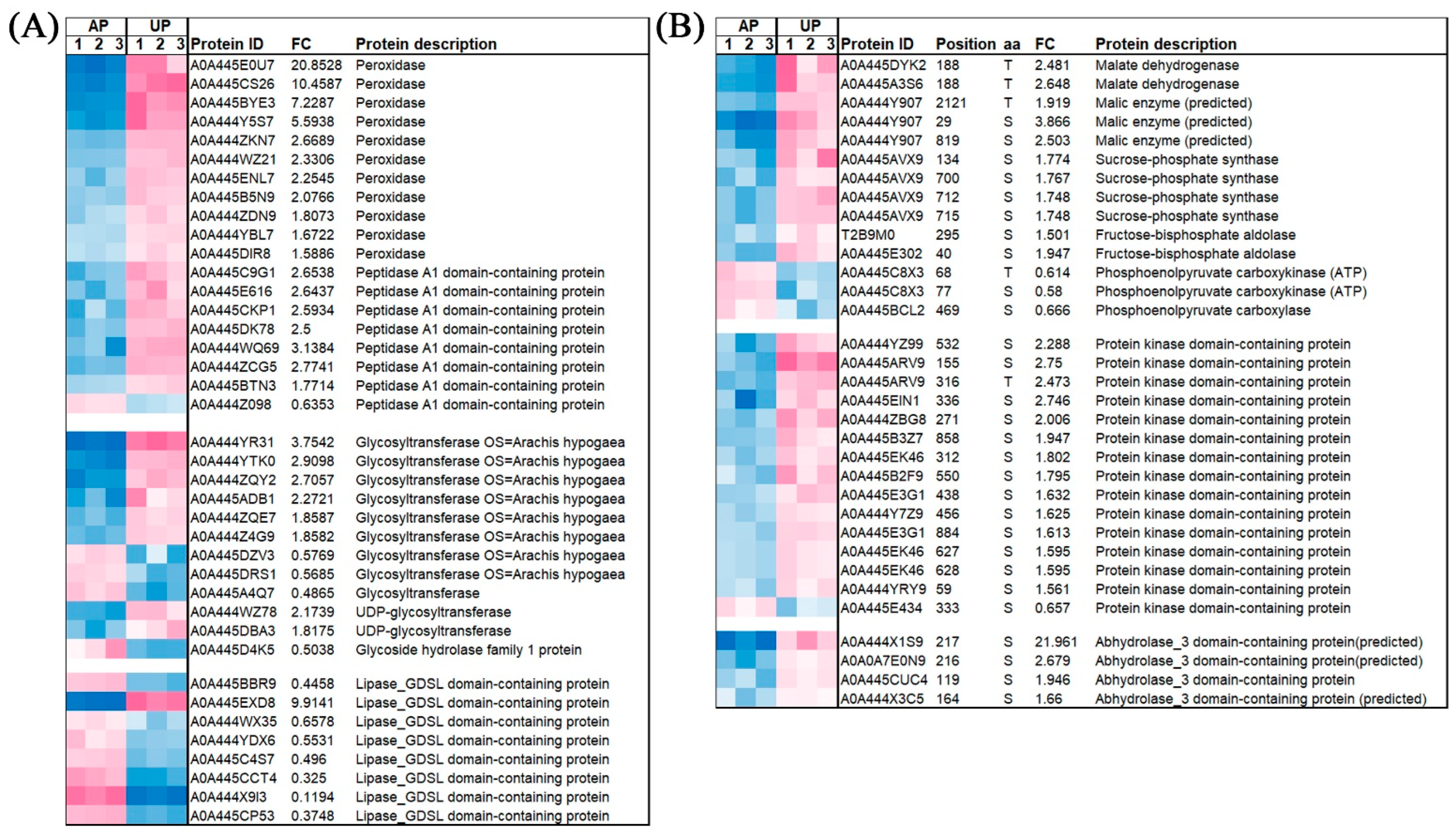
2.7. The Expression of Some Significantly Enriched DAPs and Phosphorylation Level of DRPPs in APs and UPs
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Paraffin Section
4.2. Protein Preparation and Mass Spectrometry Analysis
4.3. Phosphoproteomic Analysis
4.4. Bioinformatics Analysis
4.5. Quantification of Protein Abundance and Phosphorylation Level of Selected Phosphosites by PRM
4.6. Phos-Tag SDS-PAGE
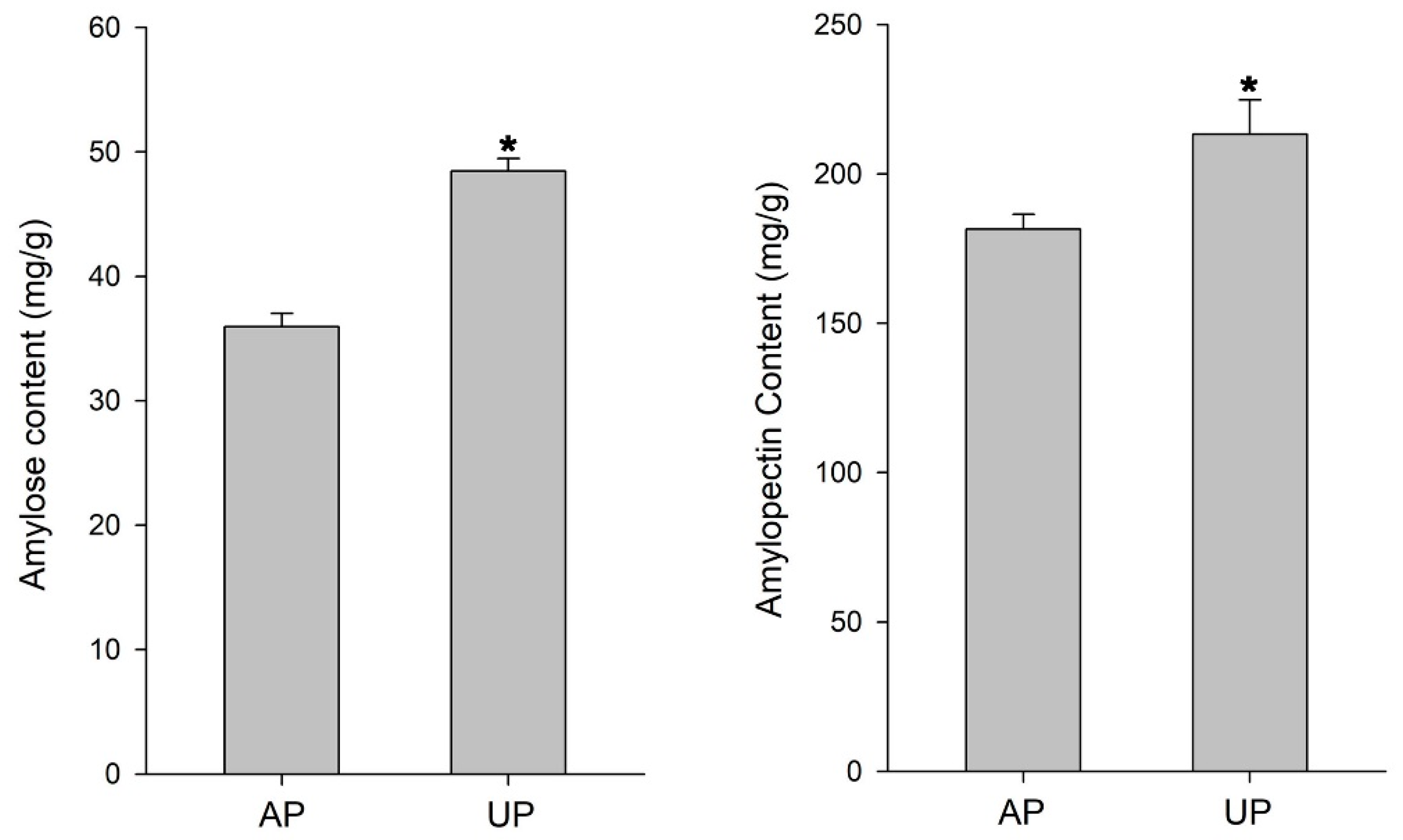
4.7. Detection of Starch
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, J.; Qi, F.; Zheng, Z.; Sun, Z.; Tian, M.; Wang, X.; Huang, B.; Dong, W.; Zhang, X. Global Transcriptome Analyses Provide into Several Fatty Acid Biosynthesis-related Genes in Peanut (Arachis hypogaea L.). Trop. Plant Biol. 2021, 14, 267–282. [Google Scholar] [CrossRef]
- Cui, Y.Y.; Bian, J.X.; Lv, Y.Y.; Li, J.H.; Deng, X.W.; Liu, X.Q. Analysis of the transcriptional dynamics of regulatory genes during peanut pod development caused by darkness and mechanical stress. Front. Plant Sci. 2022, 13, 904162. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, P.F.; Xia, H.; Zhao, C.Z.; Hou, L.; Li, C.; Gao, C.; Zhao, S.Z.; Wang, X.J. Comparative transcriptome analysis of basal and zygote-located tip regions of peanut ovaries provides insight into the mechanism of light regulation in peanut embryo and pod development. BMC Genom. 2016, 17, 606. [Google Scholar]
- Zamski, E.; Ziv, M. Pod formation and its geotropic orientation in the peanut, Arachis hypogaea L., in relation to light and mechanical stress. Ann. Bot. 1976, 40, 631–636. [Google Scholar] [CrossRef]
- Puppala, N.; Nayak, S.N.; Sanz-Saez, A.; Chen, C.; Devi, M.J.; Nivedita, N.; Bao, Y.; He, G.; Traore, S.M.; Wright, D.A. Sustaining yield and nutritional quality of peanuts in harsh environments: Physiological and molecular basis of drought and heat stress tolerance. Front. Genet. 2023, 8, 1121462. [Google Scholar] [CrossRef] [PubMed]
- Isaias, L.; Susana, F.; Milagros, M. Understanding the FMN cofactor chemistry within the Anabaena Flavodoxin environment. Biochim. Biophys. Acta. 2012, 1817, 2118–2127. [Google Scholar]
- Tan, H.L.; Yang, X.H.; Zhang, F.X.; Zheng, X.; Qu, C.M.; Mu, J.Y.; Fu, F.Y.; Li, J.N.; Guan, R.Z.; Zhang, H.S.; et al. Enhanced Seed Oil Production in Canola by Conditional Expression of Brassica napus LEAFY COTYLEDON1 and LEC1-LIKE in Developing Seeds. Plant Physiol. 2011, 156, 1577–1588. [Google Scholar] [CrossRef]
- Kaczmarzyk, D.; Fulda, M. Fatty acid activation in cyanobacteria mediated by acyl-acyl carrier protein synthetase enables fatty acid recycling. Plant Physiol. 2010, 152, 1598–1610. [Google Scholar] [CrossRef] [PubMed]
- Rawsthorne, S. Carbon flux and fatty acid synthesis in plants. Prog. Lipid Res. 2002, 41, 182–196. [Google Scholar] [CrossRef] [PubMed]
- Schwender, J.; Ohlrogge, J.B.; Shachar-Hill, Y. A flux model of glycolysis and the oxidative pentosephosphate pathway in developing Brassica napus embryos. J. Biol. Chem. 2003, 278, 29442–29453. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Yan, F.; Zong, Y.; Li, J.W.; Gao, H.; Liu, Y.J.; Wang, Y.; Zhu, Y.C.; Wang, Q.Y. Proteomic and lipidomics analyses of high fatty acid AhDGAT3 transgenic soybean reveals the key lipase gene associated with the lipid internal mechanism. Genome 2022, 65, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Kimbrough, J.M.; Salinas-Mondragon, R.; Boss, W.F.; Brown, C.S.; Sederoff, H.W. The fast and transient transcriptional network of gravity and mechanical stimulation in the Arabidopsis root apex. Plant Physiol. 2004, 136, 2790–2805. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.Z.; Zhao, S.Z.; Hou, L.; Xia, H.; Wang, J.S.; Li, C.S.; Li, A.Q.; Li, T.T.; Zhang, X.Y.; Wang, X.J. Proteomics analysis reveals differentially activated pathways that operate in peanut pegs at different developmental stages. BMC Plant Biol. 2015, 15, 188. [Google Scholar] [CrossRef] [PubMed]
- Sacco, F.; Perfetto, L.; Castagnoli, L.; Cesareni, G. The human phosphatase interactome: An intricate family portrait. FEBS Lett. 2012, 586, 2732–2739. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wilmanns, M.; Thornton, J.; Köhn, M. Elucidating human phosphatase-substrate networks. Sci. Signal. 2013, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Qi, R.Z.; Paudel, H.; Zhu, H. Regulation and function of protein kinases and phosphatases. Enzym. Res. 2011, 2011, 794089. [Google Scholar] [CrossRef] [PubMed]
- Bao, J.; He, M.; Liu, Y.; Lu, Y.; Hong, Y.; Luo, H.; Ren, Z.; Zhao, S.; Jiang, Y. AGE/ RAGE/Akt pathway contributes to prostate cancer cell proliferation by promoting Rb phosphorylation and degradation. Am. J. Cancer Res. 2015, 5, 1741–1750. [Google Scholar] [CrossRef]
- Lim, S.; Smith, K.R.; Lim, S.T.S.; Tian, R.; Lu, J.; Tan, M. Regulation of mitochondrial functions by protein phosphorylation and dephosphorylation. Cell Biosci. 2016, 6, 25. [Google Scholar] [CrossRef]
- Wang, X.; Sun, S.; Cao, X.; Gao, J. Quantitative phosphoproteomic analysis reveals the regulatory networks of Elovl6 on lipid and glucose metabolism in zebrafish. Int. J. Mol. Sci. 2020, 21, 2860. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.C.; Sun, Z.D.; Ji, C.L.; Li, F.; Wu, H.F. Toxicological effects of tris (1,3-dichloro-2-propyl) phosphate in oyster Crassostrea gigas using proteomic and phosphoproteomic analyses. J. Hazard. Mater. 2022, 434, 128824. [Google Scholar] [CrossRef] [PubMed]
- Longsaward, R.; Pengnoo, A.; Kongsawadworakul, P.; Viboonjun, U. A novel rubber tree PR-10 protein involved in host-defense response against the white root rot fungus Rigidoporus microporus. BMC Plant Biol. 2023, 23, 157. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.L.; Li, C.; Chen, X.F.; Cheng, J.S.; Xie, J.T.; Lin, Y.; Fu, Y.P.; Jiang, D.H.; Chen, T. Overexpression of chitinase PbChia1 from Plasmodiophora brassicae improves broad-spectrum disease resistance of Arabidopsis. Virulence 2023, 14, 2233147. [Google Scholar] [CrossRef] [PubMed]
- Eschenbrenner, M.; Covès, J.; Fontecave, M. NADPH-sulfite reductase flavoprotein from Escherichia coli: Contribution to the flavin content and subunit interaction. FEBS Lett. 1995, 374, 82–84. [Google Scholar] [CrossRef] [PubMed]
- Masuda, S. Light detection and signal transduction in the BLUF photoreceptors. Plant Cell Physiol. 2014, 55, 1858. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhao, J.; Song, J.C.; Jameson, P.E. Cytokinin dehydrogenase: A genetic target for yield improvement in wheat. Plant Biotechnol. J. 2020, 18, 614–630. [Google Scholar] [CrossRef]
- Zhong, M.; Li, S.; Huang, F.; Qiu, J.; Zhang, J.; Sheng, Z. The Phosphoproteomic Response of Rice Seedlings to Cadmium Stress. Int. J. Mol. Sci. 2017, 18, 2055. [Google Scholar] [CrossRef]
- Liu, H.; Wang, F.F.; Peng, X.J.; Huang, J.H.; Shen, S.H. Global Phosphoproteomic Analysis Reveals the Defense and Response Mechanisms of Jatropha Curcas Seedling under Chilling Stress. Int. J. Mol. Sci. 2019, 20, 208. [Google Scholar] [CrossRef]
- Kato, Y.; Sakamoto, W. Phosphorylation of the Chloroplastic Metalloprotease FtsH in Arabidopsis Characterized by Phos-Tag SDS-PAGE. Front. Plant Sci. 2019, 10, 1080. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Chen, X.; Li, H.; Zhu, F.; Hong, Y.; Varshney, R.K.; Liang, X.Q. Comparative transcriptome analysis of aerial and subterranean pods development provides insights into seed abortion in peanut. Plant Mol. Biol. 2014, 85, 395–409. [Google Scholar] [CrossRef]
- Chen, X.; Yang, Q.; Li, H.; Hong, Y.; Pan, L.; Chen, N.; Zhu, F.; Chi, X.; Zhu, W. Transcriptome-wide sequencing provides insights into geocarpy in peanut (Arachis hypogaea L.). Plant Biotechnol. J. 2016, 14, 1215–1224. [Google Scholar] [CrossRef]
- Chen, X.; Li, H.; Pandey, M.K.; Yang, Q.; Yu, S. Draft genome of the peanut A-genome progenitor (Arachis duranensis) provides insights into geocarpy, oil biosynthesis, and allergens. Proc. Natl. Acad. Sci. USA. 2016, 113, 6785–6790. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.C.; Fan, K.; Shen, J.Z.; Wang, Y.; Jeyaraj, A.; Hu, S.K.; Chen, X.; Ding, Z.T.; Li, X.H. Glycine-Induced Phosphorylation Plays a Pivotal Role in Energy Metabolism in Roots and Amino Acid Metabolism in Leaves of Tea Plant. Foods 2023, 12, 334. [Google Scholar] [CrossRef] [PubMed]
- Sharkey, T.D. The end game(s) of photosynthetic carbon metabolism. Plant Physiol. 2023, 195, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Krasuska, U.; Andrzejczak, O.; Staszek, P.; Bogatek, R.; Gniazdowska, A. Canavanine Alters ROS/RNS Level and Leads to Post-translational Modification of Proteins in Roots of Tomato Seedlings. Front. Plant Sci. 2016, 7, 840. [Google Scholar] [CrossRef]
- Matamoros, M.A.; Becana, M. Molecular responses of legumes to abiotic stress: Protein post-translational modifications and redox signaling. J. Exp. Bot. 2021, 72, 5876–5892. [Google Scholar] [CrossRef] [PubMed]
- Huber, S.C.; Huber, J.L. Role of sucrose-phosphate synthase in sucrose metabolism in leaves. Plant Physiol. 1992, 99, 1275–1278. [Google Scholar] [CrossRef] [PubMed]
- Partida, V.G.S.; Dias, H.M.; Corcino, D.S.M.; Sluys, M.A.V. Sucrose-phosphate phosphatase from sugarcane reveals an ancestral tandem duplication. BMC Plant Biol. 2021, 21, 23. [Google Scholar] [CrossRef] [PubMed]
- Le, X.H.; Lee, C.P.; Millar, A.H. The mitochondrial pyruvate carrier (MPC) complex mediates one of three pyruvate-supplying pathways that sustain Arabidopsis respiratory metabolism. Plant Cell. 2021, 33, 2776–2793. [Google Scholar] [CrossRef]
- Khan, M.M.; Jan, A.; Karibe, H.; Komatsu, S. Identification of phosphoproteins regulated by gibberellin in rice leaf sheath. Plant Mol. Biol. 2005, 58, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Rojas, B.E.; Hartman, M.D.; Figueroa, C.M.; Leaden, L.; Podestá, F.E.; Iglesias, A.A. Biochemical characterization of phosphoenolpyruvate carboxykinases from Arabidopsis thaliana. Biochem. J. 2019, 476, 2939–2952. [Google Scholar] [CrossRef] [PubMed]
- Cerný, M.; Doubnerová, V.; Müller, K.; Ryslavá, H. Characterization of phosphoenolpyruvate carboxylase from mature maize seeds: Properties of phosphorylated and dephosphorylated forms. Biochimie 2010, 92, 1362–1370. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Su, L.; Wang, R.; Dai, X.; Li, X.; Chang, Y.; Zhao, S.; Chen, H.; Yin, Z.; Wu, G. Comparative 4D Label-Free Quantitative Proteomic Analysis of Bombus terrestris Provides Insights into Proteins and Processes Associated with Diapause. Int. J. Mol. Sci. 2023, 25, 326. [Google Scholar] [CrossRef]
- Bonhomme, L.; Valot, B.; Tardieu, F.; Zivy, M. Phosphoproteome dynamics upon changes in plant water status reveal early events associated with rapid growth adjustment in maize leaves. Mol. Cell Proteom. 2012, 11, 957–972. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.S.; Chen, Q.S.; Zheng, Q.X.; Shen, J.J.; Luo, Z.P.; Fan, K.; Xu, S.H.; Shen, Q.; Liu, P.P. Proteomic and Phosphoproteomic Analysis in Tobacco Mosaic Virus-Infected Tobacco (Nicotiana tabacum). Biomolecules 2019, 9, 39. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Z.; Li, X.R.; Zhang, J.; Li, D.X.; Yan, L.J.; You, M.H.; Zhang, J.B.; Lei, X.; Chang, D.; Ji, X.F.; et al. Physiological and Proteomic Responses of Contrasting Alfalfa (Medicago sativa L.) Varieties to High Temperature Stress. Front. Plant Sci. 2021, 12, 753011. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.C. Testing Theory and Method of Cereal Quality; Science Press: Beijing, China, 2006. (In Chinese) [Google Scholar]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, S.; He, M.; Tang, Z.; Liu, K.; Wang, J.; Cui, L.; Guo, F.; Liu, P.; Zhang, J.; Wan, S. Deciphering the Proteome and Phosphoproteome of Peanut (Arachis hypogaea L.) Pegs Penetrating into the Soil. Int. J. Mol. Sci. 2025, 26, 634. https://doi.org/10.3390/ijms26020634
Yang S, He M, Tang Z, Liu K, Wang J, Cui L, Guo F, Liu P, Zhang J, Wan S. Deciphering the Proteome and Phosphoproteome of Peanut (Arachis hypogaea L.) Pegs Penetrating into the Soil. International Journal of Molecular Sciences. 2025; 26(2):634. https://doi.org/10.3390/ijms26020634
Chicago/Turabian StyleYang, Sha, Mei He, Zhaohui Tang, Keke Liu, Jianguo Wang, Li Cui, Feng Guo, Ping Liu, Jialei Zhang, and Shubo Wan. 2025. "Deciphering the Proteome and Phosphoproteome of Peanut (Arachis hypogaea L.) Pegs Penetrating into the Soil" International Journal of Molecular Sciences 26, no. 2: 634. https://doi.org/10.3390/ijms26020634
APA StyleYang, S., He, M., Tang, Z., Liu, K., Wang, J., Cui, L., Guo, F., Liu, P., Zhang, J., & Wan, S. (2025). Deciphering the Proteome and Phosphoproteome of Peanut (Arachis hypogaea L.) Pegs Penetrating into the Soil. International Journal of Molecular Sciences, 26(2), 634. https://doi.org/10.3390/ijms26020634