
Mitochondrial microRNAs: Key Drivers in Unraveling Neurodegenerative Diseases

 ,
,  , , , and
, , , and 
Abstract
1. Introduction
2. MitomiRs in Neurodegenerative Diseases
2.1. Biogenesis, Regulation, and Function of MitomiRs
2.2. Dysregulated MitomiR in Neurodegenerative Disorders
2.2.1. MitomiR in Alzheimer’s Disease
2.2.2. MitomiR in Parkinson’s Disease
2.2.3. MitomiR in Amyotrophic Lateral Sclerosis
3. Limitations and Challenges for MitomiR-Based Therapeutic Strategies
4. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dugger, B.N.; Dickson, D.W. Pathology of Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a028035. [Google Scholar] [CrossRef] [PubMed]
- Duranti, E.; Villa, C. From Brain to Muscle: The Role of Muscle Tissue in Neurodegenerative Disorders. Biology 2024, 13, 719. [Google Scholar] [CrossRef] [PubMed]
- Duranti, E.; Villa, C. Molecular Investigations of Protein Aggregation in the Pathogenesis of Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2022, 24, 704. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yang, J. Mitochondria-associated membranes: A hub for neurodegenerative diseases. Biomed. Pharmacother. 2022, 149, 112890. [Google Scholar] [CrossRef]
- Trigo, D.; Avelar, C.; Fernandes, M.; Sá, J.; da Cruz, E.S.O. Mitochondria, energy, and metabolism in neuronal health and disease. FEBS Lett. 2022, 596, 1095–1110. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Durães, F.; Pinto, M.; Sousa, E. Old Drugs as New Treatments for Neurodegenerative Diseases. Pharmaceuticals 2018, 11, 44. [Google Scholar] [CrossRef]
- Catanesi, M.; d’Angelo, M.; Tupone, M.G.; Benedetti, E.; Giordano, A.; Castelli, V.; Cimini, A. MicroRNAs Dysregulation and Mitochondrial Dysfunction in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 5986. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Goel, H.; Goel, A. MicroRNA and Rare Human Diseases. Genes 2024, 15, 1243. [Google Scholar] [CrossRef] [PubMed]
- Galimberti, D.; Villa, C.; Fenoglio, C.; Serpente, M.; Ghezzi, L.; Cioffi, S.M.; Arighi, A.; Fumagalli, G.; Scarpini, E. Circulating miRNAs as potential biomarkers in Alzheimer’s disease. J. Alzheimer’s Dis. 2014, 42, 1261–1267. [Google Scholar] [CrossRef] [PubMed]
- Ridolfi, E.; Fenoglio, C.; Cantoni, C.; Calvi, A.; De Riz, M.; Pietroboni, A.; Villa, C.; Serpente, M.; Bonsi, R.; Vercellino, M.; et al. Expression and Genetic Analysis of MicroRNAs Involved in Multiple Sclerosis. Int. J. Mol. Sci. 2013, 14, 4375–4384. [Google Scholar] [CrossRef] [PubMed]
- Fenoglio, C.; Cantoni, C.; De Riz, M.; Ridolfi, E.; Cortini, F.; Serpente, M.; Villa, C.; Comi, C.; Monaco, F.; Mellesi, L.; et al. Expression and genetic analysis of miRNAs involved in CD4+ cell activation in patients with multiple sclerosis. Neurosci. Lett. 2011, 504, 9–12. [Google Scholar] [CrossRef]
- Barrey, E.; Saint-Auret, G.; Bonnamy, B.; Damas, D.; Boyer, O.; Gidrol, X. Pre-microRNA and mature microRNA in human mitochondria. PLoS ONE 2011, 6, e20220. [Google Scholar] [CrossRef]
- Mercer, T.R.; Neph, S.; Dinger, M.E.; Crawford, J.; Smith, M.A.; Shearwood, A.M.; Haugen, E.; Bracken, C.P.; Rackham, O.; Stamatoyannopoulos, J.A.; et al. The human mitochondrial transcriptome. Cell 2011, 146, 645–658. [Google Scholar] [CrossRef]
- Ro, S.; Ma, H.Y.; Park, C.; Ortogero, N.; Song, R.; Hennig, G.W.; Zheng, H.; Lin, Y.M.; Moro, L.; Hsieh, J.T.; et al. The mitochondrial genome encodes abundant small noncoding RNAs. Cell Res. 2013, 23, 759–774. [Google Scholar] [CrossRef]
- Fan, S.; Tian, T.; Chen, W.; Lv, X.; Lei, X.; Zhang, H.; Sun, S.; Cai, L.; Pan, G.; He, L.; et al. Mitochondrial miRNA Determines Chemoresistance by Reprogramming Metabolism and Regulating Mitochondrial Transcription. Cancer Res. 2019, 79, 1069–1084. [Google Scholar] [CrossRef]
- Li, P.; Jiao, J.; Gao, G.; Prabhakar, B.S. Control of mitochondrial activity by miRNAs. J. Cell. Biochem. 2012, 113, 1104–1110. [Google Scholar] [CrossRef]
- Bartel, D.P. Metazoan MicroRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef]
- Lee, Y.; Kim, M.; Han, J.; Yeom, K.H.; Lee, S.; Baek, S.H.; Kim, V.N. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004, 23, 4051–4060. [Google Scholar] [CrossRef] [PubMed]
- Denli, A.M.; Tops, B.B.; Plasterk, R.H.; Ketting, R.F.; Hannon, G.J. Processing of primary microRNAs by the Microprocessor complex. Nature 2004, 432, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Hutvágner, G.; McLachlan, J.; Pasquinelli, A.E.; Bálint, E.; Tuschl, T.; Zamore, P.D. A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science 2001, 293, 834–838. [Google Scholar] [CrossRef]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef] [PubMed]
- Khvorova, A.; Reynolds, A.; Jayasena, S.D. Functional siRNAs and miRNAs exhibit strand bias. Cell 2003, 115, 209–216. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef]
- Forman, J.J.; Coller, H.A. The code within the code: microRNAs target coding regions. Cell Cycle 2010, 9, 1533–1541. [Google Scholar] [CrossRef]
- Meijer, H.A.; Kong, Y.W.; Lu, W.T.; Wilczynska, A.; Spriggs, R.V.; Robinson, S.W.; Godfrey, J.D.; Willis, A.E.; Bushell, M. Translational repression and eIF4A2 activity are critical for microRNA-mediated gene regulation. Science 2013, 340, 82–85. [Google Scholar] [CrossRef]
- Rencelj, A.; Gvozdenovic, N.; Cemazar, M. MitomiRs: Their roles in mitochondria and importance in cancer cell metabolism. Radiol. Oncol. 2021, 55, 379–392. [Google Scholar] [CrossRef]
- Macgregor-Das, A.M.; Das, S. A microRNA’s journey to the center of the mitochondria. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H206–H215. [Google Scholar] [CrossRef]
- Wang, G.; Chen, H.W.; Oktay, Y.; Zhang, J.; Allen, E.L.; Smith, G.M.; Fan, K.C.; Hong, J.S.; French, S.W.; McCaffery, J.M.; et al. PNPASE regulates RNA import into mitochondria. Cell 2010, 142, 456–467. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, D.L.; Hathaway, Q.A.; Pinti, M.V.; Nichols, C.E.; Durr, A.J.; Sreekumar, S.; Hughes, K.M.; Stine, S.M.; Martinez, I.; Hollander, J.M. Exploring the mitochondrial microRNA import pathway through Polynucleotide Phosphorylase (PNPase). J. Mol. Cell. Cardiol. 2017, 110, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.G.; McBurney, K.L.; Thompson, K.J.; Stickney, L.M.; Mackie, G.A. S1 and KH domains of polynucleotide phosphorylase determine the efficiency of RNA binding and autoregulation. J. Bacteriol. 2013, 195, 2021–2031. [Google Scholar] [CrossRef]
- Zeng, Y.; Sankala, H.; Zhang, X.; Graves, P.R. Phosphorylation of Argonaute 2 at serine-387 facilitates its localization to processing bodies. Biochem. J. 2008, 413, 429–436. [Google Scholar] [CrossRef]
- Giordani, C.; Silvestrini, A.; Giuliani, A.; Olivieri, F.; Rippo, M.R. MicroRNAs as Factors in Bidirectional Crosstalk Between Mitochondria and the Nucleus During Cellular Senescence. Front. Physiol. 2021, 12, 734976. [Google Scholar] [CrossRef]
- Bandiera, S.; Matégot, R.; Girard, M.; Demongeot, J.; Henrion-Caude, A. MitomiRs delineating the intracellular localization of microRNAs at mitochondria. Free Radic. Biol. Med. 2013, 64, 12–19. [Google Scholar] [CrossRef]
- Colombini, M. Structure and mode of action of a voltage dependent anion-selective channel (VDAC) located in the outer mitochondrial membrane. Ann. N. Y. Acad. Sci. 1980, 341, 552–563. [Google Scholar] [CrossRef]
- Das, S.; Wong, R.; Rajapakse, N.; Murphy, E.; Steenbergen, C. Glycogen synthase kinase 3 inhibition slows mitochondrial adenine nucleotide transport and regulates voltage-dependent anion channel phosphorylation. Circ. Res. 2008, 103, 983–991. [Google Scholar] [CrossRef]
- Salinas, T.; Duchêne, A.M.; Delage, L.; Nilsson, S.; Glaser, E.; Zaepfel, M.; Maréchal-Drouard, L. The voltage-dependent anion channel, a major component of the tRNA import machinery in plant mitochondria. Proc. Natl. Acad. Sci. USA 2006, 103, 18362–18367. [Google Scholar] [CrossRef]
- Murri, M.; El Azzouzi, H. MicroRNAs as regulators of mitochondrial dysfunction and obesity. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H291–H302. [Google Scholar] [CrossRef]
- Kussainova, A.; Bulgakova, O.; Aripova, A.; Khalid, Z.; Bersimbaev, R.; Izzotti, A. The Role of Mitochondrial miRNAs in the Development of Radon-Induced Lung Cancer. Biomedicines 2022, 10, 428. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Ferlito, M.; Kent, O.A.; Fox-Talbot, K.; Wang, R.; Liu, D.; Raghavachari, N.; Yang, Y.; Wheelan, S.J.; Murphy, E.; et al. Nuclear miRNA regulates the mitochondrial genome in the heart. Circ. Res. 2012, 110, 1596–1603. [Google Scholar] [CrossRef] [PubMed]
- Geiger, J.; Dalgaard, L.T. Interplay of mitochondrial metabolism and microRNAs. Cell. Mol. Life Sci. 2017, 74, 631–646. [Google Scholar] [CrossRef]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef]
- Raya, K.Y.; Ari, Q.N. The miR-146a-5p and miR-125b-5p levels as biomarkers for early prediction of Alzheimer’s disease. Hum. Gene 2022, 34, 201129. [Google Scholar] [CrossRef]
- Yashooa, R.K.; Nabi, A.Q. Telomerase Reverse Transcriptase and Telomerase RNA Component Gene Expression as a Novel Biomarkers for Alzheimer’s disease. Cell. Mol. Biol. 2022, 68, 14–20. [Google Scholar] [CrossRef]
- Buée, L.; Bussière, T.; Buée-Scherrer, V.; Delacourte, A.; Hof, P.R. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res. Brain Res. Rev. 2000, 33, 95–130. [Google Scholar] [CrossRef]
- Michalska, P.; León, R. When It Comes to an End: Oxidative Stress Crosstalk with Protein Aggregation and Neuroinflammation Induce Neurodegeneration. Antioxidants 2020, 9, 740. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef]
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef]
- Reddy, P.H.; Beal, M.F. Amyloid beta, mitochondrial dysfunction and synaptic damage: Implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol. Med. 2008, 14, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Blass, J.P. Brain metabolism and brain disease: Is metabolic deficiency the proximate cause of Alzheimer dementia? J. Neurosci. Res. 2001, 66, 851–856. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Choi, K.Y.; Park, Y.; McLean, C.; Park, J.; Lee, J.H.; Lee, K.H.; Kim, B.C.; Huh, Y.H.; Song, W.K. Enhanced Expression of microRNA-1273g-3p Contributes to Alzheimer’s Disease Pathogenesis by Regulating the Expression of Mitochondrial Genes. Cells 2021, 10, 2697. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Zhou, H.; Jiang, L.; Mao, Y.; Cui, X.; Xie, B.; Cui, D.; Wang, H.; Zhang, Q.; Xu, S. MiR-195 dependent roles of mitofusin2 in the mitochondrial dysfunction of hippocampal neurons in SAMP8 mice. Brain Res. 2016, 1652, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.C.; Wang, L.M.; Wang, M.; Song, B.; Tan, S.; Teng, J.F.; Duan, D.X. MicroRNA-195 downregulates Alzheimer’s disease amyloid-β production by targeting BACE1. Brain Res. Bull. 2012, 88, 596–601. [Google Scholar] [CrossRef]
- Liang, C.; Mu, Y.; Tian, H.; Wang, D.; Zhang, S.; Wang, H.; Liu, Y.; Di, C. MicroRNA-140 silencing represses the incidence of Alzheimer’s disease. Neurosci. Lett. 2021, 758, 135674. [Google Scholar] [CrossRef]
- Nelson, P.T.; Wang, W.X. MiR-107 is reduced in Alzheimer’s disease brain neocortex: Validation study. J. Alzheimer’s Dis. 2010, 21, 75–79. [Google Scholar] [CrossRef]
- Wang, W.X.; Rajeev, B.W.; Stromberg, A.J.; Ren, N.; Tang, G.; Huang, Q.; Rigoutsos, I.; Nelson, P.T. The expression of microRNA miR-107 decreases early in Alzheimer’s disease and may accelerate disease progression through regulation of beta-site amyloid precursor protein-cleaving enzyme 1. J. Neurosci. 2008, 28, 1213–1223. [Google Scholar] [CrossRef]
- Rech, M.; Kuhn, A.R.; Lumens, J.; Carai, P.; van Leeuwen, R.; Verhesen, W.; Verjans, R.; Lecomte, J.; Liu, Y.; Luiken, J.; et al. AntagomiR-103 and -107 Treatment Affects Cardiac Function and Metabolism. Mol. Ther. Nucleic Acids 2019, 14, 424–437. [Google Scholar] [CrossRef]
- Shu, B.; Zhang, X.; Du, G.; Fu, Q.; Huang, L. MicroRNA-107 prevents amyloid-β-induced neurotoxicity and memory impairment in mice. Int. J. Mol. Med. 2018, 41, 1665–1672. [Google Scholar] [CrossRef]
- Wan, X.; Garg, N.J. Sirtuin Control of Mitochondrial Dysfunction, Oxidative Stress, and Inflammation in Chagas Disease Models. Front. Cell. Infect. Microbiol. 2021, 11, 693051. [Google Scholar] [CrossRef] [PubMed]
- Cieślik, M.; Czapski, G.A.; Wójtowicz, S.; Wieczorek, I.; Wencel, P.L.; Strosznajder, R.P.; Jaber, V.; Lukiw, W.J.; Strosznajder, J.B. Alterations of Transcription of Genes Coding Anti-oxidative and Mitochondria-Related Proteins in Amyloid β Toxicity: Relevance to Alzheimer’s Disease. Mol. Neurobiol. 2020, 57, 1374–1388. [Google Scholar] [CrossRef] [PubMed]
- Jaber, V.; Zhao, Y.; Lukiw, W.J. Alterations in micro RNA-messenger RNA (miRNA-mRNA) Coupled Signaling Networks in Sporadic Alzheimer’s Disease (AD) Hippocampal CA1. J. Alzheimer’s Dis. Park. 2017, 7, 312. [Google Scholar] [CrossRef]
- Weinberg, R.B.; Mufson, E.J.; Counts, S.E. Evidence for a neuroprotective microRNA pathway in amnestic mild cognitive impairment. Front. Neurosci. 2015, 9, 430. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, Z.; Tan, L.; Wang, Y.; Lu, C.; Chen, R.; Zhang, S.; Gao, Y.; Liu, Y.; Yin, Y.; et al. Correcting miR92a-vGAT-Mediated GABAergic Dysfunctions Rescues Human Tau-Induced Anxiety in Mice. Mol. Ther. 2017, 25, 140–152. [Google Scholar] [CrossRef]
- Sempere, L.F.; Freemantle, S.; Pitha-Rowe, I.; Moss, E.; Dmitrovsky, E.; Ambros, V. Expression profiling of mammalian microRNAs uncovers a subset of brain-expressed microRNAs with possible roles in murine and human neuronal differentiation. Genome Biol. 2004, 5, R13. [Google Scholar] [CrossRef]
- Cogswell, J.P.; Ward, J.; Taylor, I.A.; Waters, M.; Shi, Y.; Cannon, B.; Kelnar, K.; Kemppainen, J.; Brown, D.; Chen, C.; et al. Identification of miRNA changes in Alzheimer’s disease brain and CSF yields putative biomarkers and insights into disease pathways. J. Alzheimer’s Dis. 2008, 14, 27–41. [Google Scholar] [CrossRef]
- Zhuang, J.; Chen, Z.; Cai, P.; Wang, R.; Yang, Q.; Li, L.; Yang, H.; Zhu, R. Targeting MicroRNA-125b Promotes Neurite Outgrowth but Represses Cell Apoptosis and Inflammation via Blocking PTGS2 and CDK5 in a FOXQ1-Dependent Way in Alzheimer Disease. Front. Cell. Neurosci. 2020, 14, 587747. [Google Scholar] [CrossRef]
- Banzhaf-Strathmann, J.; Benito, E.; May, S.; Arzberger, T.; Tahirovic, S.; Kretzschmar, H.; Fischer, A.; Edbauer, D. MicroRNA-125b induces tau hyperphosphorylation and cognitive deficits in Alzheimer’s disease. EMBO J. 2014, 33, 1667–1680. [Google Scholar] [CrossRef]
- Zhao, Y.; Lang, Y.; Zhang, M.; Liang, S.; Zhu, X.; Liu, Z. miR-125b Disrupts Mitochondrial Dynamics via Targeting Mitofusin 1 in Cisplatin-Induced Acute Kidney Injury. Kidney Dis. 2022, 8, 137–147. [Google Scholar] [CrossRef]
- Cheng, L.; Doecke, J.D.; Sharples, R.A.; Villemagne, V.L.; Fowler, C.J.; Rembach, A.; Martins, R.N.; Rowe, C.C.; Macaulay, S.L.; Masters, C.L.; et al. Prognostic serum miRNA biomarkers associated with Alzheimer’s disease shows concordance with neuropsychological and neuroimaging assessment. Mol. Psychiatry 2015, 20, 1188–1196. [Google Scholar] [CrossRef]
- Sun, C.; Jia, N.; Li, R.; Zhang, Z.; Zhong, Y.; Han, K. miR-143-3p inhibition promotes neuronal survival in an Alzheimer’s disease cell model by targeting neuregulin-1. Folia Neuropathol. 2020, 58, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Ennequin, G.; Capel, F.; Caillaud, K.; Chavanelle, V.; Etienne, M.; Teixeira, A.; Li, X.; Boisseau, N.; Sirvent, P. Neuregulin 1 improves complex 2-mediated mitochondrial respiration in skeletal muscle of healthy and diabetic mice. Sci. Rep. 2017, 7, 1742. [Google Scholar] [CrossRef] [PubMed]
- Nong, W.; Wei, Z.-q.; Mo, X.-n.; Wu, L.; Tang, N. miR-137 overexpression protects neurons from Aβ-induced neurotoxicity via ERK1/2. All Life 2021, 14, 522–529. [Google Scholar] [CrossRef]
- Channakkar, A.S.; Singh, T.; Pattnaik, B.; Gupta, K.; Seth, P.; Adlakha, Y.K. MiRNA-137-mediated modulation of mitochondrial dynamics regulates human neural stem cell fate. Stem Cells 2020, 38, 683–697. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Reddy, A.P.; Yin, X.; Reddy, P.H. Novel MicroRNA-455-3p and its protective effects against abnormal APP processing and amyloid beta toxicity in Alzheimer’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2428–2440. [Google Scholar] [CrossRef]
- Parkinson, J. An essay on the shaking palsy, 1817. J. Neuropsychiatry Clin. Neurosci. 2002, 14, 223–236, discussion 222. [Google Scholar] [CrossRef]
- Lees, A.J.; Hardy, J.; Revesz, T. Parkinson’s disease. Lancet 2009, 373, 2055–2066. [Google Scholar] [CrossRef]
- Xu, L.; Pu, J. Alpha-Synuclein in Parkinson’s Disease: From Pathogenetic Dysfunction to Potential Clinical Application. Park. Dis. 2016, 2016, 1720621. [Google Scholar] [CrossRef]
- Duranti, E.; Villa, C. Insights into Dysregulated Neurological Biomarkers in Cancer. Cancers 2024, 16, 2680. [Google Scholar] [CrossRef]
- Pang, S.Y.; Ho, P.W.; Liu, H.F.; Leung, C.T.; Li, L.; Chang, E.E.S.; Ramsden, D.B.; Ho, S.L. The interplay of aging, genetics and environmental factors in the pathogenesis of Parkinson’s disease. Transl. Neurodegener. 2019, 8, 23. [Google Scholar] [CrossRef] [PubMed]
- Davie, C.A. A review of Parkinson’s disease. Br. Med. Bull. 2008, 86, 109–127. [Google Scholar] [CrossRef] [PubMed]
- Langston, J.W.; Irwin, I.; Langston, E.B.; Forno, L.S. 1-Methyl-4-phenylpyridinium ion (MPP+): Identification of a metabolite of MPTP, a toxin selective to the substantia nigra. Neurosci. Lett. 1984, 48, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, R.R.; Salach, J.I.; Singer, T.P. Uptake of the neurotoxin 1-methyl-4-phenylpyridine (MPP+) by mitochondria and its relation to the inhibition of the mitochondrial oxidation of NAD+-linked substrates by MPP+. Biochem. Biophys. Res. Commun. 1986, 134, 743–748. [Google Scholar] [CrossRef]
- Borsche, M.; Pereira, S.L.; Klein, C.; Grünewald, A. Mitochondria and Parkinson’s Disease: Clinical, Molecular, and Translational Aspects. J. Park. Dis. 2021, 11, 45–60. [Google Scholar] [CrossRef]
- Castelli, V.; Benedetti, E.; Antonosante, A.; Catanesi, M.; Pitari, G.; Ippoliti, R.; Cimini, A.; d’Angelo, M. Neuronal Cells Rearrangement During Aging and Neurodegenerative Disease: Metabolism, Oxidative Stress and Organelles Dynamic. Front. Mol. Neurosci. 2019, 12, 132. [Google Scholar] [CrossRef]
- Sanders, L.H.; Timothy Greenamyre, J. Oxidative damage to macromolecules in human Parkinson disease and the rotenone model. Free Radic. Biol. Med. 2013, 62, 111–120. [Google Scholar] [CrossRef]
- Subramaniam, S.R.; Chesselet, M.F. Mitochondrial dysfunction and oxidative stress in Parkinson’s disease. Prog. Neurobiol. 2013, 106–107, 17–32. [Google Scholar] [CrossRef]
- Hsu, L.J.; Sagara, Y.; Arroyo, A.; Rockenstein, E.; Sisk, A.; Mallory, M.; Wong, J.; Takenouchi, T.; Hashimoto, M.; Masliah, E. alpha-synuclein promotes mitochondrial deficit and oxidative stress. Am. J. Pathol. 2000, 157, 401–410. [Google Scholar] [CrossRef]
- Tansey, M.G.; McCoy, M.K.; Frank-Cannon, T.C. Neuroinflammatory mechanisms in Parkinson’s disease: Potential environmental triggers, pathways, and targets for early therapeutic intervention. Exp. Neurol. 2007, 208, 1–25. [Google Scholar] [CrossRef]
- Buendia, I.; Michalska, P.; Navarro, E.; Gameiro, I.; Egea, J.; León, R. Nrf2-ARE pathway: An emerging target against oxidative stress and neuroinflammation in neurodegenerative diseases. Pharmacol. Ther. 2016, 157, 84–104. [Google Scholar] [CrossRef] [PubMed]
- Junn, E.; Lee, K.W.; Jeong, B.S.; Chan, T.W.; Im, J.Y.; Mouradian, M.M. Repression of alpha-synuclein expression and toxicity by microRNA-7. Proc. Natl. Acad. Sci. USA 2009, 106, 13052–13057. [Google Scholar] [CrossRef] [PubMed]
- McMillan, K.J.; Murray, T.K.; Bengoa-Vergniory, N.; Cordero-Llana, O.; Cooper, J.; Buckley, A.; Wade-Martins, R.; Uney, J.B.; O’Neill, M.J.; Wong, L.F.; et al. Loss of MicroRNA-7 Regulation Leads to α-Synuclein Accumulation and Dopaminergic Neuronal Loss In Vivo. Mol. Ther. 2017, 25, 2404–2414. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, A.D.; Choi, D.C.; Kabaria, S.; Tran, A.; Junn, E. MicroRNA-7 Regulates the Function of Mitochondrial Permeability Transition Pore by Targeting VDAC1 Expression. J. Biol. Chem. 2016, 291, 6483–6493. [Google Scholar] [CrossRef]
- Li, S.; Lv, X.; Zhai, K.; Xu, R.; Zhang, Y.; Zhao, S.; Qin, X.; Yin, L.; Lou, J. MicroRNA-7 inhibits neuronal apoptosis in a cellular Parkinson’s disease model by targeting Bax and Sirt2. Am. J. Transl. Res. 2016, 8, 993–1004. [Google Scholar]
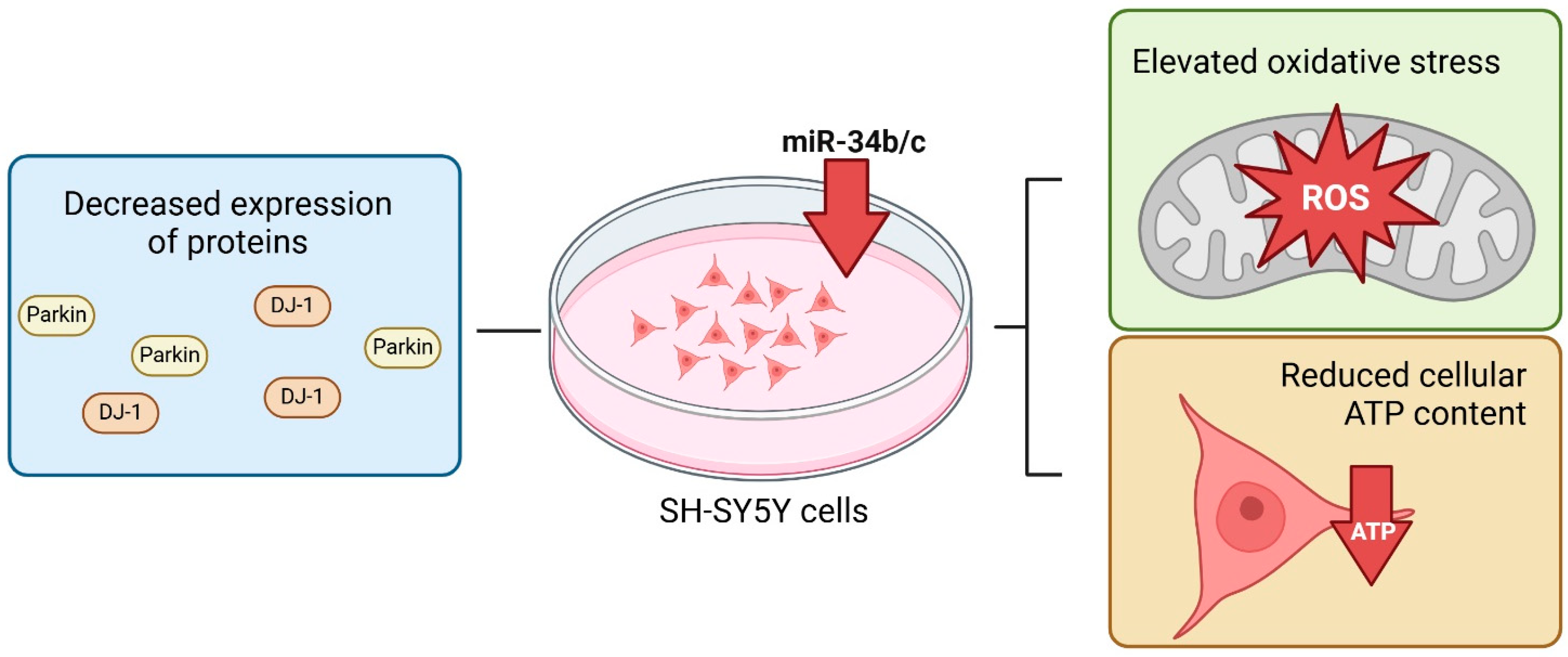
- Miñones-Moyano, E.; Porta, S.; Escaramís, G.; Rabionet, R.; Iraola, S.; Kagerbauer, B.; Espinosa-Parrilla, Y.; Ferrer, I.; Estivill, X.; Martí, E. MicroRNA profiling of Parkinson’s disease brains identifies early downregulation of miR-34b/c which modulate mitochondrial function. Hum. Mol. Genet. 2011, 20, 3067–3078. [Google Scholar] [CrossRef]
- Kabaria, S.; Choi, D.C.; Chaudhuri, A.D.; Mouradian, M.M.; Junn, E. Inhibition of miR-34b and miR-34c enhances α-synuclein expression in Parkinson’s disease. FEBS Lett. 2015, 589, 319–325. [Google Scholar] [CrossRef]
- Shendelman, S.; Jonason, A.; Martinat, C.; Leete, T.; Abeliovich, A. DJ-1 is a redox-dependent molecular chaperone that inhibits alpha-synuclein aggregate formation. PLoS Biol. 2004, 2, e362. [Google Scholar] [CrossRef]
- Chen, Y.; Gao, C.; Sun, Q.; Pan, H.; Huang, P.; Ding, J.; Chen, S. MicroRNA-4639 Is a Regulator of DJ-1 Expression and a Potential Early Diagnostic Marker for Parkinson’s Disease. Front. Aging Neurosci. 2017, 9, 232. [Google Scholar] [CrossRef]
- Xiong, R.; Wang, Z.; Zhao, Z.; Li, H.; Chen, W.; Zhang, B.; Wang, L.; Wu, L.; Li, W.; Ding, J.; et al. MicroRNA-494 reduces DJ-1 expression and exacerbates neurodegeneration. Neurobiol. Aging 2014, 35, 705–714. [Google Scholar] [CrossRef]
- McCoy, M.K.; Cookson, M.R. DJ-1 regulation of mitochondrial function and autophagy through oxidative stress. Autophagy 2011, 7, 531–532. [Google Scholar] [CrossRef] [PubMed]
- Thomas, K.J.; McCoy, M.K.; Blackinton, J.; Beilina, A.; van der Brug, M.; Sandebring, A.; Miller, D.; Maric, D.; Cedazo-Minguez, A.; Cookson, M.R. DJ-1 acts in parallel to the PINK1/parkin pathway to control mitochondrial function and autophagy. Hum. Mol. Genet. 2011, 20, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Yang, X.; Lou, J. Geniposide reduces α-synuclein by blocking microRNA-21/lysosome-associated membrane protein 2A interaction in Parkinson disease models. Brain Res. 2016, 1644, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Ghavami, S.; Shojaei, S.; Yeganeh, B.; Ande, S.R.; Jangamreddy, J.R.; Mehrpour, M.; Christoffersson, J.; Chaabane, W.; Moghadam, A.R.; Kashani, H.H.; et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog. Neurobiol. 2014, 112, 24–49. [Google Scholar] [CrossRef]
- Wang, H.; Ye, Y.; Zhu, Z.; Mo, L.; Lin, C.; Wang, Q.; Gong, X.; He, X.; Lu, G.; Lu, F.; et al. MiR-124 Regulates Apoptosis and Autophagy Process in MPTP Model of Parkinson’s Disease by Targeting to Bim. Brain Pathol. 2016, 26, 167–176. [Google Scholar] [CrossRef]
- Gong, X.; Wang, H.; Ye, Y.; Shu, Y.; Deng, Y.; He, X.; Lu, G.; Zhang, S. miR-124 regulates cell apoptosis and autophagy in dopaminergic neurons and protects them by regulating AMPK/mTOR pathway in Parkinson’s disease. Am. J. Transl. Res. 2016, 8, 2127–2137. [Google Scholar]
- Yamano, K.; Matsuda, N.; Tanaka, K. The ubiquitin signal and autophagy: An orchestrated dance leading to mitochondrial degradation. EMBO Rep. 2016, 17, 300–316. [Google Scholar] [CrossRef]
- Kane, L.A.; Lazarou, M.; Fogel, A.I.; Li, Y.; Yamano, K.; Sarraf, S.A.; Banerjee, S.; Youle, R.J. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 2014, 205, 143–153. [Google Scholar] [CrossRef]
- Geisler, S.; Holmström, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef]
- Wang, L.; Cho, Y.L.; Tang, Y.; Wang, J.; Park, J.E.; Wu, Y.; Wang, C.; Tong, Y.; Chawla, R.; Zhang, J.; et al. PTEN-L is a novel protein phosphatase for ubiquitin dephosphorylation to inhibit PINK1-Parkin-mediated mitophagy. Cell Res. 2018, 28, 787–802. [Google Scholar] [CrossRef]
- Kim, J.; Fiesel, F.C.; Belmonte, K.C.; Hudec, R.; Wang, W.X.; Kim, C.; Nelson, P.T.; Springer, W. miR-27a and miR-27b regulate autophagic clearance of damaged mitochondria by targeting PTEN-induced putative kinase 1 (PINK1). Mol. Neurodegener. 2016, 11, 55. [Google Scholar] [CrossRef] [PubMed]
- Jauhari, A.; Singh, T.; Mishra, S.; Shankar, J.; Yadav, S. Coordinated Action of miR-146a and Parkin Gene Regulate Rotenone-induced Neurodegeneration. Toxicol. Sci. 2020, 176, 433–445. [Google Scholar] [CrossRef] [PubMed]
- Di Rita, A.; Maiorino, T.; Bruqi, K.; Volpicelli, F.; Bellenchi, G.C.; Strappazzon, F. miR-218 Inhibits Mitochondrial Clearance by Targeting PRKN E3 Ubiquitin Ligase. Int. J. Mol. Sci. 2020, 21, 355. [Google Scholar] [CrossRef] [PubMed]
- Xing, R.X.; Li, L.G.; Liu, X.W.; Tian, B.X.; Cheng, Y. Down regulation of miR-218, miR-124, and miR-144 relates to Parkinson’s disease via activating NF-κB signaling. Kaohsiung J. Med. Sci. 2020, 36, 786–792. [Google Scholar] [CrossRef]
- Mortiboys, H.; Johansen, K.K.; Aasly, J.O.; Bandmann, O. Mitochondrial impairment in patients with Parkinson disease with the G2019S mutation in LRRK2. Neurology 2010, 75, 2017–2020. [Google Scholar] [CrossRef]
- Wang, X.; Yan, M.H.; Fujioka, H.; Liu, J.; Wilson-Delfosse, A.; Chen, S.G.; Perry, G.; Casadesus, G.; Zhu, X. LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum. Mol. Genet. 2012, 21, 1931–1944. [Google Scholar] [CrossRef]
- Cho, H.J.; Liu, G.; Jin, S.M.; Parisiadou, L.; Xie, C.; Yu, J.; Sun, L.; Ma, B.; Ding, J.; Vancraenenbroeck, R.; et al. MicroRNA-205 regulates the expression of Parkinson’s disease-related leucine-rich repeat kinase 2 protein. Hum. Mol. Genet. 2013, 22, 608–620. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, Z.; Le, W. Tiny But Mighty: Promising Roles of MicroRNAs in the Diagnosis and Treatment of Parkinson’s Disease. Neurosci. Bull. 2017, 33, 543–551. [Google Scholar] [CrossRef]
- Valdés, P.; Schneider, B.L. Gene Therapy: A Promising Approach for Neuroprotection in Parkinson’s Disease? Front. Neuroanat. 2016, 10, 123. [Google Scholar] [CrossRef]
- Duranti, E.; Villa, C. Muscle Involvement in Amyotrophic Lateral Sclerosis: Understanding the Pathogenesis and Advancing Therapeutics. Biomolecules 2023, 13, 1582. [Google Scholar] [CrossRef]
- Duranti, E.; Villa, C. Influence of DUX4 Expression in Facioscapulohumeral Muscular Dystrophy and Possible Treatments. Int. J. Mol. Sci. 2023, 24, 9503. [Google Scholar] [CrossRef] [PubMed]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef] [PubMed]
- Akçimen, F.; Lopez, E.R.; Landers, J.E.; Nath, A.; Chiò, A.; Chia, R.; Traynor, B.J. Amyotrophic lateral sclerosis: Translating genetic discoveries into therapies. Nat. Rev. Genet. 2023, 24, 642–658. [Google Scholar] [CrossRef]
- Nakaya, T.; Maragkakis, M. Amyotrophic Lateral Sclerosis associated FUS mutation shortens mitochondria and induces neurotoxicity. Sci. Rep. 2018, 8, 15575. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, X.; Huo, Z.; Chen, Y.; Liu, J.; Zhao, Z.; Meng, F.; Su, Q.; Bao, W.; Zhang, L.; et al. The Impact of Mitochondrial Dysfunction in Amyotrophic Lateral Sclerosis. Cells 2022, 11, 2049. [Google Scholar] [CrossRef]
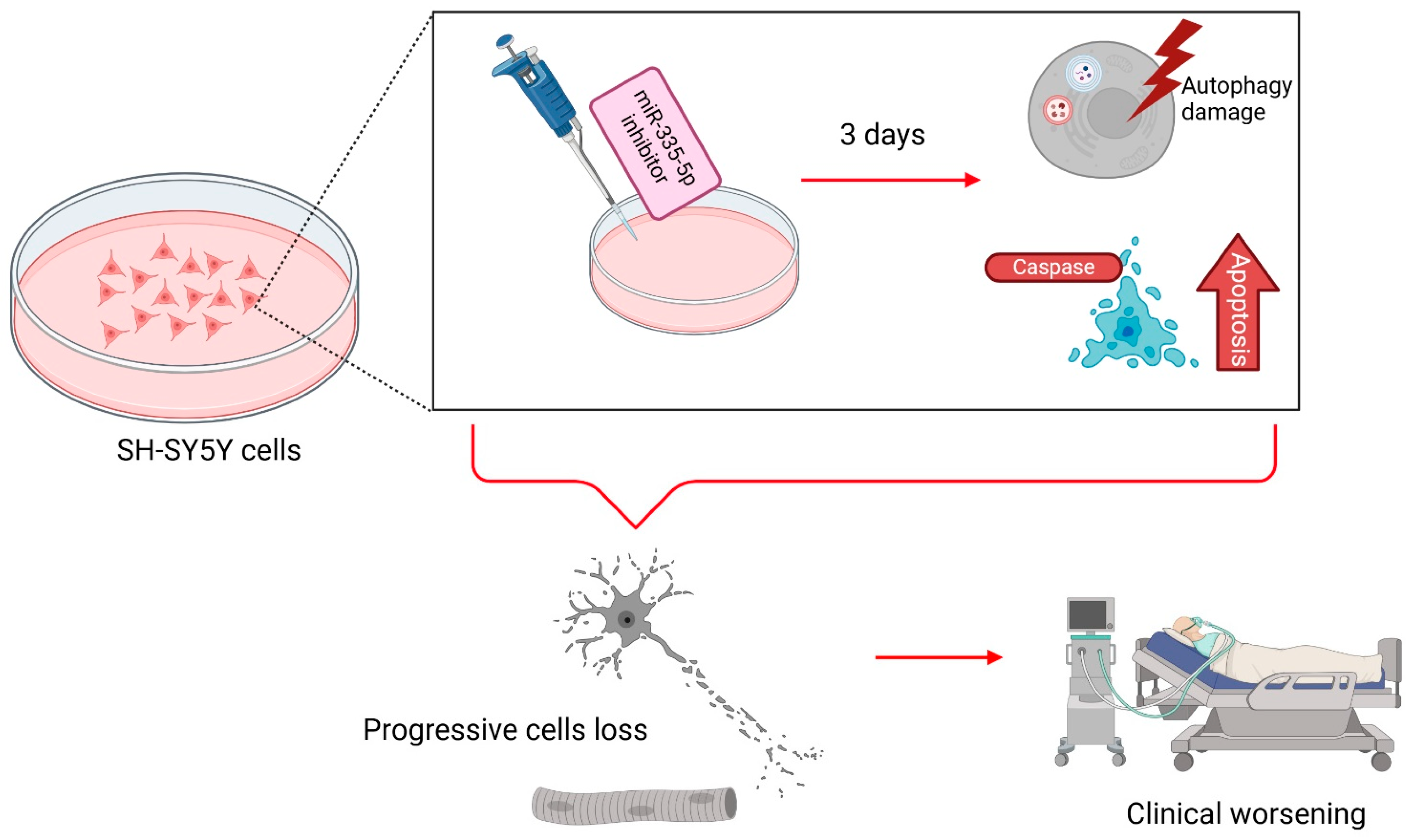
- De Luna, N.; Turon-Sans, J.; Cortes-Vicente, E.; Carrasco-Rozas, A.; Illán-Gala, I.; Dols-Icardo, O.; Clarimón, J.; Lleó, A.; Gallardo, E.; Illa, I.; et al. Downregulation of miR-335-5P in Amyotrophic Lateral Sclerosis Can Contribute to Neuronal Mitochondrial Dysfunction and Apoptosis. Sci. Rep. 2020, 10, 4308. [Google Scholar] [CrossRef]
- Jhanji, R.; Behl, T.; Sehgal, A.; Bungau, S. Mitochondrial dysfunction and traffic jams in amyotrophic lateral sclerosis. Mitochondrion 2021, 58, 102–110. [Google Scholar] [CrossRef]
- Nguyen, T.P.N.; Kumar, M.; Fedele, E.; Bonanno, G.; Bonifacino, T. MicroRNA Alteration, Application as Biomarkers, and Therapeutic Approaches in Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 4718. [Google Scholar] [CrossRef]
- Waller, R.; Wyles, M.; Heath, P.R.; Kazoka, M.; Wollff, H.; Shaw, P.J.; Kirby, J. Small RNA Sequencing of Sporadic Amyotrophic Lateral Sclerosis Cerebrospinal Fluid Reveals Differentially Expressed miRNAs Related to Neural and Glial Activity. Front. Neurosci. 2017, 11, 731. [Google Scholar] [CrossRef]
- Kmetzsch, V.; Anquetil, V.; Saracino, D.; Rinaldi, D.; Camuzat, A.; Gareau, T.; Jornea, L.; Forlani, S.; Couratier, P.; Wallon, D.; et al. Plasma microRNA signature in presymptomatic and symptomatic subjects with C9orf72-associated frontotemporal dementia and amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2021, 92, 485–493. [Google Scholar] [CrossRef]
- Tai, Y.; Pu, M.; Yuan, L.; Guo, H.; Qiao, J.; Lu, H.; Wang, G.; Chen, J.; Qi, X.; Tao, Z.; et al. miR-34a-5p regulates PINK1-mediated mitophagy via multiple modes. Life Sci. 2021, 276, 119415. [Google Scholar] [CrossRef]
- Loffreda, A.; Nizzardo, M.; Arosio, A.; Ruepp, M.D.; Calogero, R.A.; Volinia, S.; Galasso, M.; Bendotti, C.; Ferrarese, C.; Lunetta, C.; et al. miR-129-5p: A key factor and therapeutic target in amyotrophic lateral sclerosis. Prog. Neurobiol. 2020, 190, 101803. [Google Scholar] [CrossRef] [PubMed]
- Bronicki, L.M.; Jasmin, B.J. Emerging complexity of the HuD/ELAVl4 gene; implications for neuronal development, function, and dysfunction. RNA 2013, 19, 1019–1037. [Google Scholar] [CrossRef] [PubMed]
- Martinez, B.; Peplow, P.V. MicroRNA expression in animal models of amyotrophic lateral sclerosis and potential therapeutic approaches. Neural Regen. Res. 2022, 17, 728–740. [Google Scholar] [CrossRef]
- De Felice, B.; Annunziata, A.; Fiorentino, G.; Borra, M.; Biffali, E.; Coppola, C.; Cotrufo, R.; Brettschneider, J.; Giordana, M.L.; Dalmay, T.; et al. miR-338-3p is over-expressed in blood, CFS, serum and spinal cord from sporadic amyotrophic lateral sclerosis patients. Neurogenetics 2014, 15, 243–253. [Google Scholar] [CrossRef]
- Aschrafi, A.; Schwechter, A.D.; Mameza, M.G.; Natera-Naranjo, O.; Gioio, A.E.; Kaplan, B.B. MicroRNA-338 regulates local cytochrome c oxidase IV mRNA levels and oxidative phosphorylation in the axons of sympathetic neurons. J. Neurosci. 2008, 28, 12581–12590. [Google Scholar] [CrossRef]
- Russell, A.P.; Wada, S.; Vergani, L.; Hock, M.B.; Lamon, S.; Léger, B.; Ushida, T.; Cartoni, R.; Wadley, G.D.; Hespel, P.; et al. Disruption of skeletal muscle mitochondrial network genes and miRNAs in amyotrophic lateral sclerosis. Neurobiol. Dis. 2013, 49, 107–117. [Google Scholar] [CrossRef]
- Mthembu, S.X.H.; Mazibuko-Mbeje, S.E.; Ziqubu, K.; Muvhulawa, N.; Marcheggiani, F.; Cirilli, I.; Nkambule, B.B.; Muller, C.J.F.; Basson, A.K.; Tiano, L.; et al. Potential regulatory role of PGC-1α within the skeletal muscle during metabolic adaptations in response to high-fat diet feeding in animal models. Pflug. Arch. 2024, 476, 283–293. [Google Scholar] [CrossRef]
- Hawley, Z.C.E.; Campos-Melo, D.; Strong, M.J. MiR-105 and miR-9 regulate the mRNA stability of neuronal intermediate filaments. Implications for the pathogenesis of amyotrophic lateral sclerosis (ALS). Brain Res. 2019, 1706, 93–100. [Google Scholar] [CrossRef]
- Hu, J.H.; Zhang, H.; Wagey, R.; Krieger, C.; Pelech, S.L. Protein kinase and protein phosphatase expression in amyotrophic lateral sclerosis spinal cord. J. Neurochem. 2003, 85, 432–442. [Google Scholar] [CrossRef]
- Liu, J.; Zuo, X.; Han, J.; Dai, Q.; Xu, H.; Liu, Y.; Cui, S. MiR-9-5p inhibits mitochondrial damage and oxidative stress in AD cell models by targeting GSK-3β. Biosci. Biotechnol. Biochem. 2020, 84, 2273–2280. [Google Scholar] [CrossRef] [PubMed]
- Liguori, M.; Nuzziello, N.; Introna, A.; Consiglio, A.; Licciulli, F.; D’Errico, E.; Scarafino, A.; Distaso, E.; Simone, I.L. Dysregulation of MicroRNAs and Target Genes Networks in Peripheral Blood of Patients With Sporadic Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2018, 11, 288. [Google Scholar] [CrossRef] [PubMed]
- Magen, I.; Yacovzada, N.S.; Yanowski, E.; Coenen-Stass, A.; Grosskreutz, J.; Lu, C.H.; Greensmith, L.; Malaspina, A.; Fratta, P.; Hornstein, E. Circulating miR-181 is a prognostic biomarker for amyotrophic lateral sclerosis. Nat. Neurosci. 2021, 24, 1534–1541. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.; Liu, L.; Lao, Y.; Liao, W.; Liao, M.; Luo, X.; Wu, J.; Xie, W.; Zhang, Y.; Xu, N. MicroRNA-181a suppresses parkin-mediated mitophagy and sensitizes neuroblastoma cells to mitochondrial uncoupler-induced apoptosis. Oncotarget 2016, 7, 42274–42287. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, Y.B.; Lu, Y.; Yue, S.; Giffard, R.G. miR-181 targets multiple Bcl-2 family members and influences apoptosis and mitochondrial function in astrocytes. Mitochondrion 2012, 12, 213–219. [Google Scholar] [CrossRef]
- Lang, A.; Grether-Beck, S.; Singh, M.; Kuck, F.; Jakob, S.; Kefalas, A.; Altinoluk-Hambüchen, S.; Graffmann, N.; Schneider, M.; Lindecke, A.; et al. MicroRNA-15b regulates mitochondrial ROS production and the senescence-associated secretory phenotype through sirtuin 4/SIRT4. Aging 2016, 8, 484–505. [Google Scholar] [CrossRef]
- Toivonen, J.M.; Manzano, R.; Oliván, S.; Zaragoza, P.; García-Redondo, A.; Osta, R. MicroRNA-206: A potential circulating biomarker candidate for amyotrophic lateral sclerosis. PLoS ONE 2014, 9, e89065. [Google Scholar] [CrossRef]
- Liu, H.; Lan, S.; Shi, X.J.; Fan, F.C.; Liu, Q.S.; Cong, L.; Cheng, Y. Systematic review and meta-analysis on microRNAs in amyotrophic lateral sclerosis. Brain Res. Bull. 2023, 194, 82–89. [Google Scholar] [CrossRef]
- Novák, J.; Kružliak, P.; Bienertová-Vašků, J.; Slabý, O.; Novák, M. MicroRNA-206: A promising theranostic marker. Theranostics 2014, 4, 119–133. [Google Scholar] [CrossRef]
- Przanowska, R.K.; Sobierajska, E.; Su, Z.; Jensen, K.; Przanowski, P.; Nagdas, S.; Kashatus, J.A.; Kashatus, D.F.; Bhatnagar, S.; Lukens, J.R.; et al. miR-206 family is important for mitochondrial and muscle function, but not essential for myogenesis in vitro. FASEB J. 2020, 34, 7687–7702. [Google Scholar] [CrossRef]
- Williams, A.H.; Valdez, G.; Moresi, V.; Qi, X.; McAnally, J.; Elliott, J.L.; Bassel-Duby, R.; Sanes, J.R.; Olson, E.N. MicroRNA-206 delays ALS progression and promotes regeneration of neuromuscular synapses in mice. Science 2009, 326, 1549–1554. [Google Scholar] [CrossRef] [PubMed]
- Marcuzzo, S.; Bonanno, S.; Kapetis, D.; Barzago, C.; Cavalcante, P.; D’Alessandro, S.; Mantegazza, R.; Bernasconi, P. Up-regulation of neural and cell cycle-related microRNAs in brain of amyotrophic lateral sclerosis mice at late disease stage. Mol. Brain 2015, 8, 5. [Google Scholar] [CrossRef] [PubMed]
- Yardeni, T.; Fine, R.; Joshi, Y.; Gradus-Pery, T.; Kozer, N.; Reichenstein, I.; Yanowski, E.; Nevo, S.; Weiss-Tishler, H.; Eisenberg-Bord, M.; et al. High content image analysis reveals function of miR-124 upstream of Vimentin in regulating motor neuron mitochondria. Sci. Rep. 2018, 8, 59. [Google Scholar] [CrossRef] [PubMed]
- Vaz, A.R.; Vizinha, D.; Morais, H.; Colaço, A.R.; Loch-Neckel, G.; Barbosa, M.; Brites, D. Overexpression of miR-124 in Motor Neurons Plays a Key Role in ALS Pathological Processes. Int. J. Mol. Sci. 2021, 22, 6128. [Google Scholar] [CrossRef]
- Paul, S.; Bravo Vázquez, L.A.; Pérez Uribe, S.; Roxana Reyes-Pérez, P.; Sharma, A. Current Status of microRNA-Based Therapeutic Approaches in Neurodegenerative Disorders. Cells 2020, 9, 1698. [Google Scholar] [CrossRef]
- Seyhan, A.A. Trials and Tribulations of MicroRNA Therapeutics. Int. J. Mol. Sci. 2024, 25, 1469. [Google Scholar] [CrossRef]
- Yan, Y.; Liu, X.Y.; Lu, A.; Wang, X.Y.; Jiang, L.X.; Wang, J.C. Non-viral vectors for RNA delivery. J. Control. Release 2022, 342, 241–279. [Google Scholar] [CrossRef]
- Yang, N. An overview of viral and nonviral delivery systems for microRNA. Int. J. Pharm. Investig. 2015, 5, 179–181. [Google Scholar] [CrossRef]
- Kulkarni, J.A.; Cullis, P.R.; van der Meel, R. Lipid Nanoparticles Enabling Gene Therapies: From Concepts to Clinical Utility. Nucleic Acid Ther. 2018, 28, 146–157. [Google Scholar] [CrossRef]
- Patra, J.K.; Das, G.; Fraceto, L.F.; Campos, E.V.R.; Rodriguez-Torres, M.D.P.; Acosta-Torres, L.S.; Diaz-Torres, L.A.; Grillo, R.; Swamy, M.K.; Sharma, S.; et al. Nano based drug delivery systems: Recent developments and future prospects. J. Nanobiotechnol. 2018, 16, 71. [Google Scholar] [CrossRef]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Tung, C.W.; Huang, P.Y.; Chan, S.C.; Cheng, P.H.; Yang, S.H. The regulatory roles of microRNAs toward pathogenesis and treatments in Huntington’s disease. J. Biomed. Sci. 2021, 28, 59. [Google Scholar] [CrossRef] [PubMed]
- Gareev, I.; Beylerli, O.; Tamrazov, R.; Ilyasova, T.; Shumadalova, A.; Du, W.; Yang, B. Methods of miRNA delivery and possibilities of their application in neuro-oncology. Noncoding RNA Res. 2023, 8, 661–674. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Lee, H.; Zhu, Z.; Minhas, J.K.; Jin, Y. Enrichment of selective miRNAs in exosomes and delivery of exosomal miRNAs in vitro and in vivo. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 312, L110–L121. [Google Scholar] [CrossRef]
- Diener, C.; Keller, A.; Meese, E. Emerging concepts of miRNA therapeutics: From cells to clinic. Trends Genet. 2022, 38, 613–626. [Google Scholar] [CrossRef]
- Segal, M.; Slack, F.J. Challenges identifying efficacious miRNA therapeutics for cancer. Expert Opin. Drug Discov. 2020, 15, 987–992. [Google Scholar] [CrossRef]
- Weaver, D.T.; Pishas, K.I.; Williamson, D.; Scarborough, J.; Lessnick, S.L.; Dhawan, A.; Scott, J.G. Network potential identifies therapeutic miRNA cocktails in Ewing sarcoma. PLoS Comput. Biol. 2021, 17, e1008755. [Google Scholar] [CrossRef]
- Duarte, F.V.; Palmeira, C.M.; Rolo, A.P. The Emerging Role of MitomiRs in the Pathophysiology of Human Disease. Adv. Exp. Med. Biol. 2015, 888, 123–154. [Google Scholar] [CrossRef]
- Saikia, B.J.; Bhardwaj, J.; Paul, S.; Sharma, S.; Neog, A.; Paul, S.R.; Bk, B. Understanding the roles and regulation of mitochondrial microRNAs (MitomiRs) in neurodegenerative diseases: Current status and advances. Mech. Ageing Dev. 2023, 213, 111838. [Google Scholar] [CrossRef]
- Hu, X.; Go, Y.M.; Jones, D.P. Omics Integration for Mitochondria Systems Biology. Antioxid. Redox Signal. 2020, 32, 853–872. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MitomiRs | Expression | Disorders | Key Target(s) | References |
|---|---|---|---|---|
| miR-7 | ↓ | PD | SNCA | [92] |
| ↓ | PD | VDAC1 | [94] | |
| ↓ | PD | SIRT2 | [95] | |
| ↓ | PD | BAX | [95] | |
| miR-9 | ↑ | AD | SIRT1 | [62] |
| ↓ | ALS | GSK-3β | [139,141] | |
| miR-15b | ↓ | ALS | SIRT4 | [142,146] |
| miR-21 | ↑ | PD | LAMP2A | [103] |
| miR-23a | ↑ | ALS | PGC-1α | [137] |
| miR-23a/23b | ↓ | AD | SIRT1 | [64] |
| miR-27a/b | Unknown | PD | PINK1 | [111] |
| miR-27b-3p | ↑ | ALS | PINK1 | [111,129] |
| miR-34a | ↑ | AD | SIRT1 | [62,63] |
| miR-34a-5p | ↑ | ALS | PINK1 | [130,131] |
| miR-34b/c | ↓ | PD | SNCA | [97] |
| miR-92a | ↑ | AD | cytochrome b | [65] |
| miR-107 | ↓ | AD | ETC complexes I, III, IV and V | [59] |
| miR-124 | ↓ | PD | BIM | [105] |
| ↑ | ALS | VIM | [153,154] | |
| miR-125b | ↑ | AD | DUSP6, PPP1CA | [69] |
| ↑ | AD | MFN1 | [70] | |
| miR-129-5p | ↑ | ALS | ELAVL4 | [132] |
| miR-137 | ↓ | AD | MEF2A, NRF2, TFAM | [75] |
| miR-140 | ↑ | AD | PINK1 | [56] |
| miR-143-3p | ↑ | AD | NRG1 | [72] |
| miR-146a | ↑ | AD | SIRT1 | [62,63] |
| ↑ | PD | PRKN | [112] | |
| miR-155 | ↑ | AD | SIRT1 | [62] |
| miR-181 | ↑ | ALS | PRKN | [144] |
| ↑ | ALS | BCL2, MCL1 | [145] | |
| miR-195 | ↑ | AD | MFN2 | [54] |
| ↑ | AD | BACE1 | [55] | |
| miR-205 | ↓ | PD | LRRK2 | [117] |
| miR-206 | ↑ | ALS | HDAC4 | [147,151] |
| miR-212/132 | ↓ | AD | SIRT1 | [64] |
| miR-218 | Unknown | PD | PRKN | [113] |
| Unknown | PD | KPNA4 | [114] | |
| miR-335-5p | ↓ | ALS | CASP7 | [126] |
| miR-338-3p | ↑ | ALS | cytochrome C oxidase | [135,136] |
| miR-455-3p | ↓ | AD | DRP1, FIS1 | [76] |
| miR-494 | ↑ | PD | DJ-1 | [100] |
| miR-1273g-3p | ↑ | AD | TIMM13 | [53] |
| miR-4639–5p | ↑ | PD | DJ-1 | [99] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yashooa, R.K.; Duranti, E.; Conconi, D.; Lavitrano, M.; Mustafa, S.A.; Villa, C. Mitochondrial microRNAs: Key Drivers in Unraveling Neurodegenerative Diseases. Int. J. Mol. Sci. 2025, 26, 626. https://doi.org/10.3390/ijms26020626
Yashooa RK, Duranti E, Conconi D, Lavitrano M, Mustafa SA, Villa C. Mitochondrial microRNAs: Key Drivers in Unraveling Neurodegenerative Diseases. International Journal of Molecular Sciences. 2025; 26(2):626. https://doi.org/10.3390/ijms26020626
Chicago/Turabian StyleYashooa, Raya Kh., Elisa Duranti, Donatella Conconi, Marialuisa Lavitrano, Suhad A. Mustafa, and Chiara Villa. 2025. "Mitochondrial microRNAs: Key Drivers in Unraveling Neurodegenerative Diseases" International Journal of Molecular Sciences 26, no. 2: 626. https://doi.org/10.3390/ijms26020626
APA StyleYashooa, R. K., Duranti, E., Conconi, D., Lavitrano, M., Mustafa, S. A., & Villa, C. (2025). Mitochondrial microRNAs: Key Drivers in Unraveling Neurodegenerative Diseases. International Journal of Molecular Sciences, 26(2), 626. https://doi.org/10.3390/ijms26020626