
Loss of Myostatin Affects m6A Modification but Not Semen Characteristics in Bull Spermatozoa

 , and
, and
Abstract
1. Introduction
2. Results
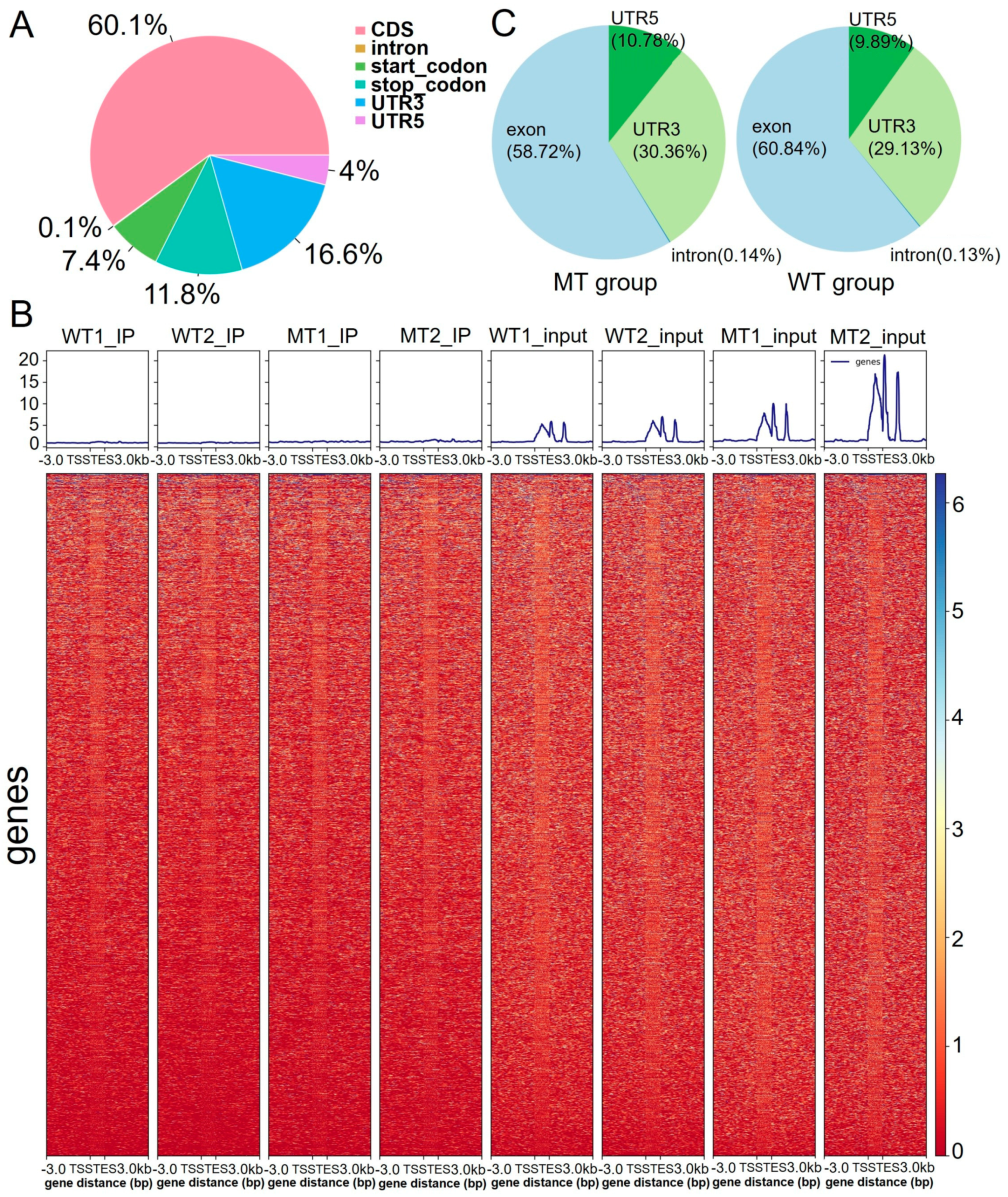
2.1. Overall Analysis of the m6A Modification Pattern
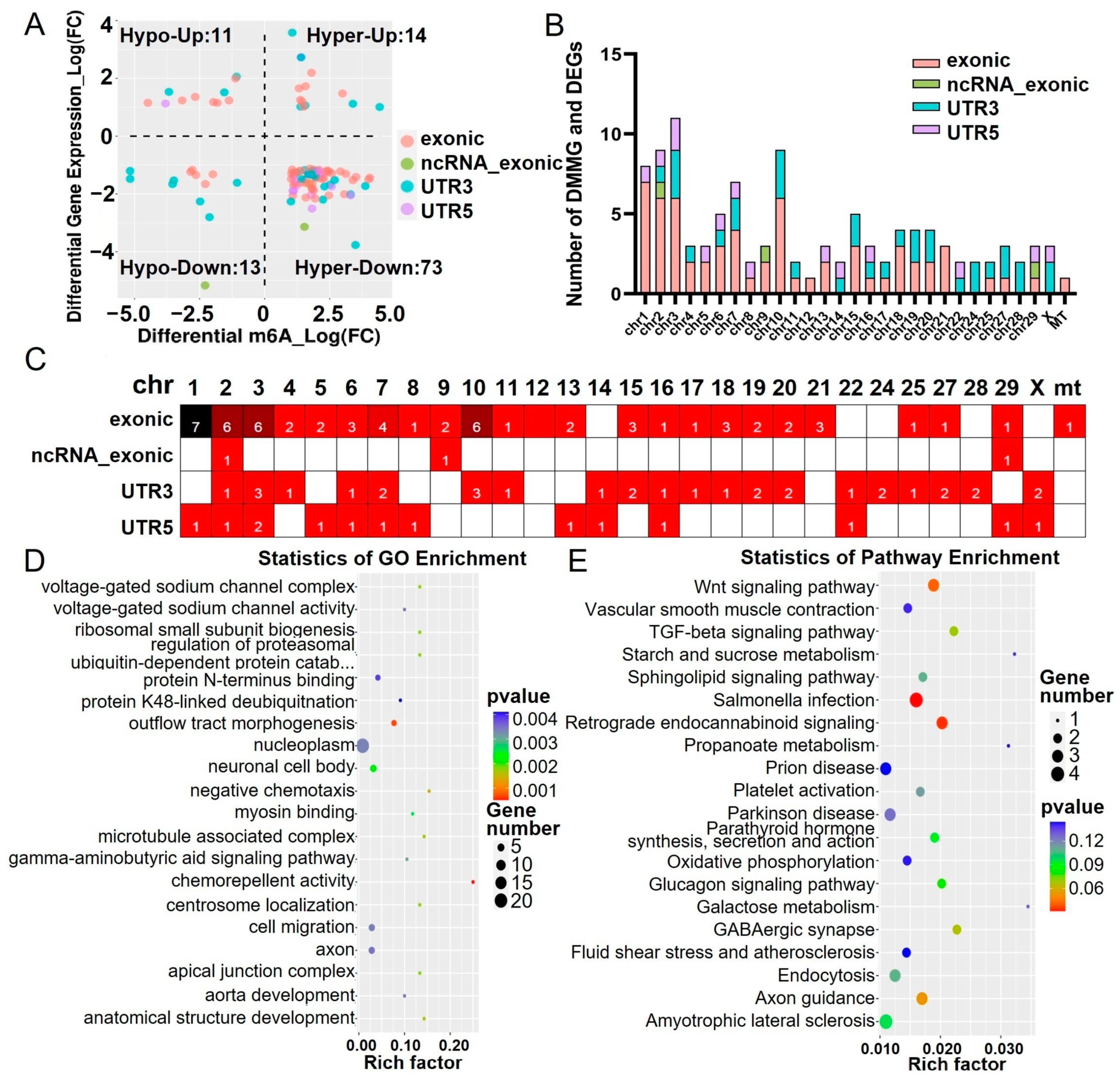
2.2. Distribution of Differentially m6A-Methylated Sites
2.3. Functional Analysis of m6A Methylated Genes
2.4. Combined Analysis of Differentially m6A-Methylated and Expressed Genes
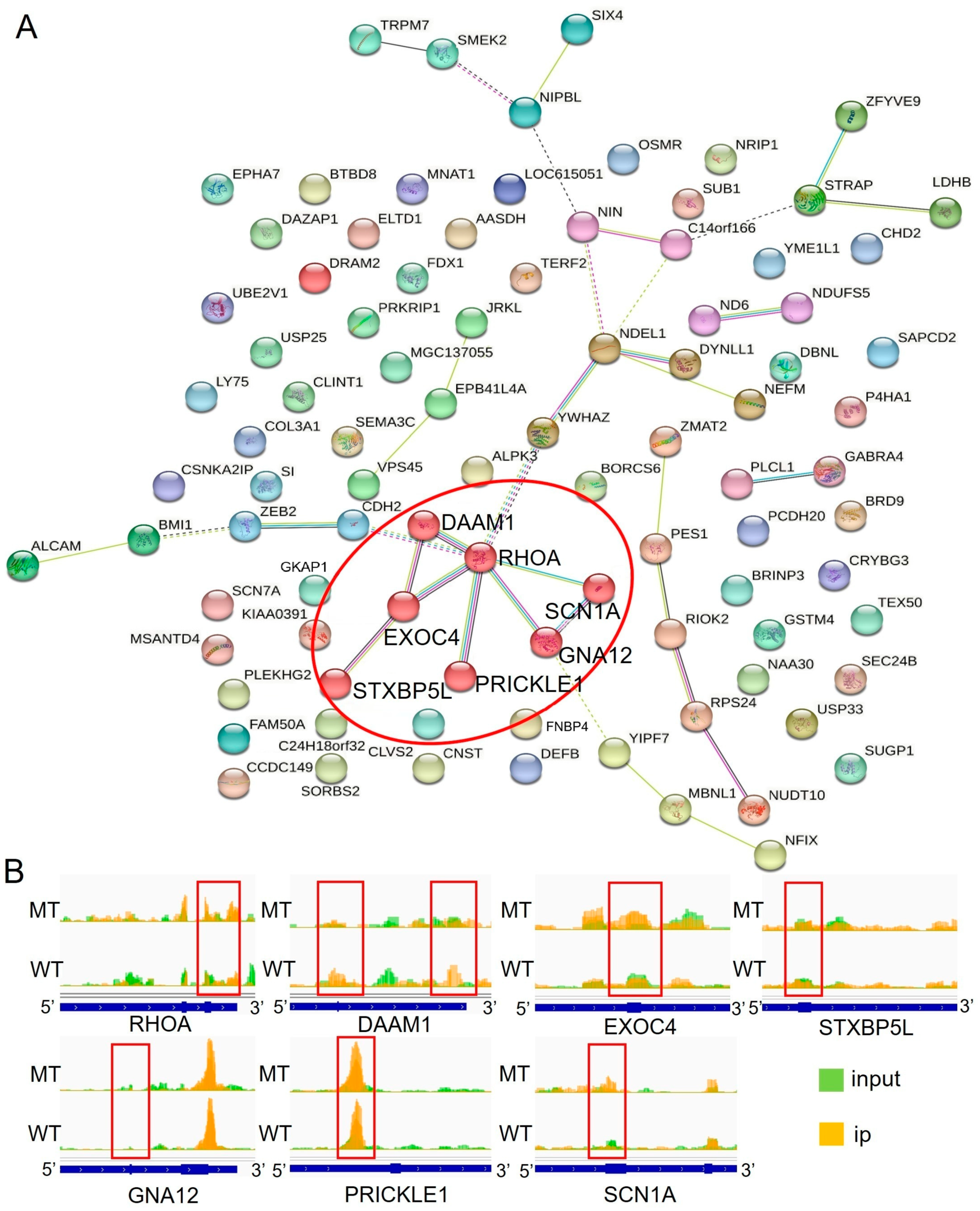
2.5. Network Analysis of Differentially m6A-Methylated and Expressed Genes
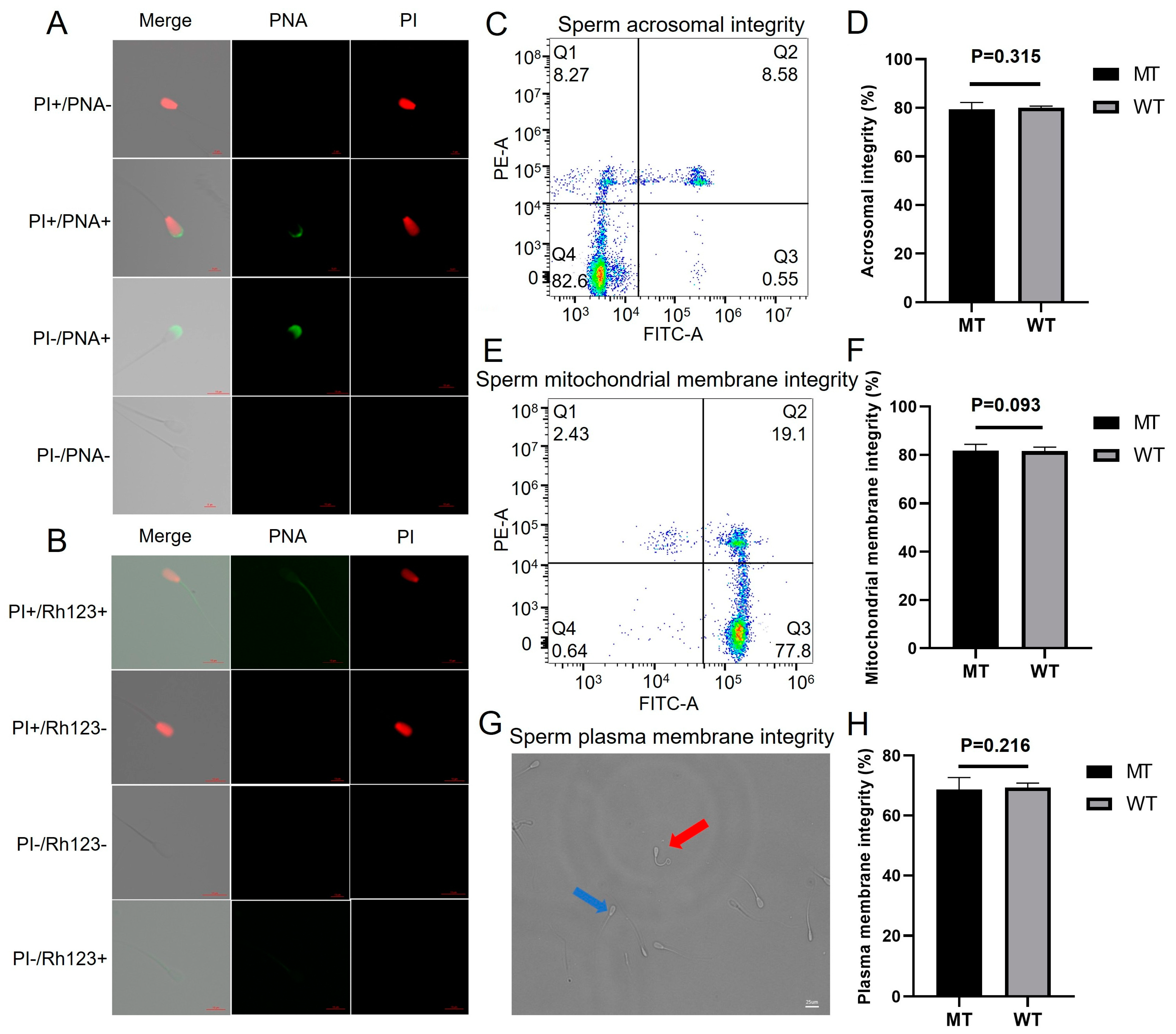
2.6. Semen Characteristics, Sperm Motility, and Kinetic Parameters of Movement
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Semen Cryopreservation and Sperm Sample Collection
4.3. Sperm Quality Analysis
4.4. RNA Isolation, Library Construction, and Sequencing
4.5. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lee, S.J. Myostatin: A Skeletal Muscle Chalone. Annu. Rev. Physiol. 2023, 85, 269–291. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yang, L.; Su, G.; Wei, Z.; Liu, X.; Song, L.; Hai, C.; Wu, D.; Hao, Z.; Wu, Y.; et al. Growth Traits and Sperm Proteomics Analyses of Myostatin Gene-Edited Chinese Yellow Cattle. Life 2022, 12, 627. [Google Scholar] [CrossRef] [PubMed]
- Grobet, L.; Martin, L.J.; Poncelet, D.; Pirottin, D.; Brouwers, B.; Riquet, J.; Schoeberlein, A.; Dunner, S.; Ménissier, F.; Massabanda, J.; et al. A deletion in the bovine myostatin gene causes the double-muscled phenotype in cattle. Nat. Genet. 1997, 17, 71–74. [Google Scholar] [CrossRef]
- McPherron, A.C.; Lee, S.J. Double muscling in cattle due to mutations in the myostatin gene. Proc. Natl. Acad. Sci. USA 1997, 94, 12457–12461. [Google Scholar] [CrossRef]
- Han, S.Z.; Jin, S.S.; Xuan, M.F.; Guo, Q.; Luo, Z.B.; Wang, J.X.; Kang, J.D.; Yin, X.J. Semen quality and fertilization ability of myostatin-knockout boars. Theriogenology 2019, 135, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zhao, Y.; Li, Y.; Chen, T.; Zhou, W.; Zhang, X.; Deng, S.; Xu, X.; Wu, S.; Liu, Z.; et al. Reproduction and viscera organ characteristics of MSTN and FGF5 dual-gene knockout sheep. Front. Vet. Sci. 2023, 10, 1119312. [Google Scholar] [CrossRef]
- Lin, Z.; Hsu, P.J.; Xing, X.; Fang, J.; Lu, Z.; Zou, Q.; Zhang, K.J.; Zhang, X.; Zhou, Y.; Zhang, T.; et al. Mettl3-/Mettl14-mediated mRNA N6-methyladenosine modulates murine spermatogenesis. Cell Res. 2017, 27, 1216–1230. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Pei, Y.; Zhou, R.; Tang, Z.; Yang, Y. Regulation of RNA N6-methyladenosine modification and its emerging roles in skeletal muscle development. Int. J. Biol. Sci. 2021, 17, 1682–1692. [Google Scholar] [CrossRef] [PubMed]
- Petrosino, J.M.; Hinger, S.A.; Golubeva, V.A.; Barajas, J.M.; Dorn, L.E.; Iyer, C.C.; Sun, H.L.; Arnold, W.D.; He, C.; Accornero, F. The m6A methyltransferase METTL3 regulates muscle maintenance and growth in mice. Nat. Commun. 2022, 13, 168. [Google Scholar] [CrossRef]
- Dang, Y.; Dong, Q.; Wu, B.; Yang, S.; Sun, J.; Cui, G.; Xu, W.; Zhao, M.; Zhang, Y.; Li, P.; et al. Global Landscape of m6A Methylation of Differently Expressed Genes in Muscle Tissue of Liaoyu White Cattle and Simmental Cattle. Front. Cell Dev. Biol. 2022, 10, 840513. [Google Scholar] [CrossRef]
- Yang, X.; Mei, C.; Ma, X.; Du, J.; Wang, J.; Zan, L. m6A Methylases Regulate Myoblast Proliferation, Apoptosis and Differentiation. Animals 2022, 12, 773. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Yan, M.; Cao, Z.; Li, X.; Zhang, Y.; Shi, J.; Feng, G.-h.; Peng, H.; Zhang, X.; Zhang, Y.; et al. Sperm tsRNAs contribute to intergenerational inheritance of an acquired metabolic disorder. Science 2016, 351, 397–400. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, X.; Shi, J.; Tuorto, F.; Li, X.; Liu, Y.; Liebers, R.; Zhang, L.; Qu, Y.; Qian, J.; et al. Dnmt2 mediates intergenerational transmission of paternally acquired metabolic disorders through sperm small non-coding RNAs. Nat. Cell Biol. 2018, 20, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Zheng, C.; Zhuang, Y.; Jin, P.; Wang, F. The m6A reader PRRC2A is essential for meiosis I completion during spermatogenesis. Nat. Commun. 2023, 14, 1636. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.H.; Ma, X.Y.; Yue, T.T.; Wang, Z.C.; Qi, K.L.; Li, J.C.; Lin, F.; Rushdi, H.E.; Gao, Y.Y.; Fu, T.; et al. Transcriptome-Wide m6A Analysis Provides Novel Insights Into Testicular Development and Spermatogenesis in Xia-Nan Cattle. Front. Cell Dev. Biol. 2021, 9, 791221. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Klukovich, R.; Peng, H.; Wang, Z.; Yu, T.; Zhang, Y.; Zheng, H.; Klungland, A.; Yan, W. ALKBH5-dependent m6A demethylation controls splicing and stability of long 3′-UTR mRNAs in male germ cells. Proc. Natl. Acad. Sci. USA 2018, 115, E325–E333. [Google Scholar] [CrossRef] [PubMed]
- Fry, N.J.; Law, B.A.; Ilkayeva, O.R.; Holley, C.L.; Mansfield, K.D. N6-methyladenosine is required for the hypoxic stabilization of specific mRNAs. RNA 2017, 23, 1444–1455. [Google Scholar] [CrossRef]
- Cooke, P.S.; Nanjappa, M.K.; Ko, C.; Prins, G.S.; Hess, R.A. Estrogens in Male Physiology. Physiol. Rev. 2017, 97, 995–1043. [Google Scholar] [CrossRef]
- Fiedler, S.E.; Bajpai, M.; Carr, D.W. Identification and characterization of RHOA-interacting proteins in bovine spermatozoa. Biol. Reprod. 2008, 78, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Doxtader, K.A.; Nam, Y. Structural Basis for Cooperative Function of Mettl3 and Mettl14 Methyltransferases. Mol. Cell 2016, 63, 306–317. [Google Scholar] [CrossRef]
- Jiang, X.; Liu, B.; Nie, Z.; Duan, L.; Xiong, Q.; Jin, Z.; Yang, C.; Chen, Y. The role of m6A modification in the biological functions and diseases. Signal Transduct. Target. Ther. 2021, 6, 74. [Google Scholar] [CrossRef] [PubMed]
- Saletore, Y.; Meyer, K.; Korlach, J.; Vilfan, I.D.; Jaffrey, S.; Mason, C.E. The birth of the Epitranscriptome: Deciphering the function of RNA modifications. Genome Biol. 2012, 13, 175. [Google Scholar] [CrossRef]
- Ma, S.; Chen, C.; Ji, X.; Liu, J.; Zhou, Q.; Wang, G.; Yuan, W.; Kan, Q.; Sun, Z. The interplay between m6A RNA methylation and noncoding RNA in cancer. J. Hematol. Oncol. 2019, 12, 121. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Zhao, X.; Wu, Y.-S.; Li, M.-M.; Wang, X.-J.; Yang, Y.-G. N6-methyl-adenosine (m6A) in RNA: An Old Modification with A Novel Epigenetic Function. Genom. Proteom. Bioinform. 2013, 11, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Jian, D.; Wang, Y.; Jian, L.; Tang, H.; Rao, L.; Chen, K.; Jia, Z.; Zhang, W.; Liu, Y.; Chen, X.; et al. METTL14 aggravates endothelial inflammation and atherosclerosis by increasing FOXO1 N6-methyladeosine modifications. Theranostics 2020, 10, 8939–8956. [Google Scholar] [CrossRef]
- Kretschmer, J.; Rao, H.; Hackert, P.; Sloan, K.E.; Höbartner, C.; Bohnsack, M.T. The m6A reader protein YTHDC2 interacts with the small ribosomal subunit and the 5′–3′ exoribonuclease XRN1. RNA 2018, 24, 1339–1350. [Google Scholar] [CrossRef] [PubMed]
- Bushkin, G.G.; Pincus, D.; Morgan, J.T.; Richardson, K.; Lewis, C.; Chan, S.H.; Bartel, D.P.; Fink, G.R. m6A modification of a 3′ UTR site reduces RME1 mRNA levels to promote meiosis. Nat. Commun. 2019, 10, 3414. [Google Scholar] [CrossRef]
- Zhou, L.; Tian, S.; Qin, G. RNA methylomes reveal the m6A-mediated regulation of DNA demethylase gene SlDML2 in tomato fruit ripening. Genome Biol. 2019, 20, 156. [Google Scholar] [CrossRef]
- Roundtree, I.A.; Evans, M.E.; Pan, T.; He, C. Dynamic RNA Modifications in Gene Expression Regulation. Cell 2017, 169, 1187–1200. [Google Scholar] [CrossRef]
- Moura, F.H.; Macias-Franco, A.; Pena-Bello, C.A.; Archilia, E.C.; Batalha, I.M.; Silva, A.E.M.; Moreira, G.M.; Norris, A.B.; Schütz, L.F.; Fonseca, M.A. Sperm DNA 5-methyl cytosine and RNA N6-methyladenosine methylation are differently affected during periods of body weight losses and body weight gain of young and mature breeding bulls. J. Anim. Sci. 2022, 100, skab362. [Google Scholar] [CrossRef]
- Smith, L.B.; Walker, W.H. The regulation of spermatogenesis by androgens. Semin. Cell Dev. Biol. 2014, 30, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Harper, A.P.; Finger, B.J.; Green, M.P. Chronic Atrazine Exposure Beginning Prenatally Impacts Liver Function and Sperm Concentration With Multi-Generational Consequences in Mice. Front. Endocrinol. 2020, 11, 580124. [Google Scholar] [CrossRef]
- Kiezun, J.; Kaminska, B.; Jankowski, J.; Dusza, L. Concentrations of the adrenocorticotropic hormone, corticosterone and sex steroid hormones and the expression of the androgen receptor in the pituitary and adrenal glands of male turkeys (Meleagris gallopavo) during growth and development. Gen. Comp. Endocrinol. 2015, 217–218, 62–70. [Google Scholar] [CrossRef]
- Frost, C.R.; Goss, G.G. Absence of some cytochrome P450 (CYP) and hydroxysteroid dehydrogenase (HSD) enzymes in hagfishes. Gen. Comp. Endocrinol. 2022, 323–324, 114045. [Google Scholar] [CrossRef]
- Guengerich, F.P.; Yoshimoto, F.K. Formation and Cleavage of C-C Bonds by Enzymatic Oxidation-Reduction Reactions. Chem. Rev. 2018, 118, 6573–6655. [Google Scholar] [CrossRef]
- Prasad, M.; Pawlak, K.J.; Burak, W.E.; Perry, E.E.; Marshall, B.; Whittal, R.M.; Bose, H.S. Mitochondrial metabolic regulation by GRP78. Sci. Adv. 2017, 3, e1602038. [Google Scholar] [CrossRef]
- Wang, Z.; Fang, K.; Wan, Y.; Yin, Y.; Li, M.; Xu, K.; Li, T.; Cao, Y.; Lv, Y.; Lu, G.; et al. TTC6-Mediated Stabilization of the Flagellum Annulus Ensures the Rapid and Directed Motion of Sperm. Cells 2023, 12, 2091. [Google Scholar] [CrossRef]
- Reyes-Miguel, T.; Roa-Espitia, A.L.; Baltiérrez-Hoyos, R.; Hernández-González, E.O. CDC42 drives RHOA activity and actin polymerization during capacitation. Reproduction 2020, 160, 393–404. [Google Scholar] [CrossRef]
- Mehaisen, G.M.K.; Elomda, A.M.; Hamad, S.K.; Ghaly, M.M.; Sun, Y.; Li, Y.; Zong, Y.; Chen, J.; Partyka, A.; Nazmi, A.; et al. Effect of Dimethylacetamide Concentration on Motility, Quality, Antioxidant Biomarkers, Anti-Freeze Gene Expression, and Fertilizing Ability of Frozen/Thawed Rooster Sperm. Animals 2022, 12, 2739. [Google Scholar] [CrossRef]
- Siudzińska, A.; Łukaszewicz, E. Effect of Semen Extenders and Storage Time on Sperm Morphology of Four Chicken Breeds. J. Appl. Poult. Res. 2008, 17, 101–108. [Google Scholar] [CrossRef]
- Zhang, J.; Xiong, Y.-W.; Tan, L.-L.; Zheng, X.-M.; Zhang, Y.-F.; Ling, Q.; Zhang, C.; Zhu, H.-L.; Chang, W.; Wang, H. Sperm Rhoa m6A modification mediates intergenerational transmission of paternally acquired hippocampal neuronal senescence and cognitive deficits after combined exposure to environmental cadmium and high-fat diet in mice. J. Hazard. Mater. 2023, 458, 131891. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Cao, J.; Zhang, H.; Wu, J.; Yin, J. Profiling of Transcriptome-Wide N6-Methyladenosine (m6A) Modifications and Identifying m6A Associated Regulation in Sperm Tail Formation in Anopheles sinensis. Int. J. Mol. Sci. 2022, 23, 4630. [Google Scholar] [CrossRef] [PubMed]
- Su, G.; Wu, S.; Wu, M.; Wang, L.; Yang, L.; Du, M.; Zhao, X.; Su, X.; Liu, X.; Bai, C.; et al. Melatonin improves the quality of frozen bull semen and influences gene expression related to embryo genome activation. Theriogenology 2021, 176, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Meng, J.; Lu, Z.; Liu, H.; Zhang, L.; Zhang, S.; Chen, Y.; Rao, M.K.; Huang, Y. A protocol for RNA methylation differential analysis with MeRIP-Seq data and exomePeak R/Bioconductor package. Methods 2014, 69, 274–281. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | m6A Diff_Log2 | Gene Diff_Log2 | Region |
|---|---|---|---|
| RHOA | 2.69 | −1.52 | 3′ UTR |
| DAAM1 | −3.49 | −1.53 | 3′ UTR |
| EXOC4 | 1.7 | −1.31 | exonic |
| STXBP5L | 1.94 | −1.39 | exonic |
| GNA12 | −1.55 | 1.52 | 3′ UTR |
| PRICKLE1 | 1.11 | −1.37 | 5′ UTR |
| SCN1A | 2.91 | −1.22 | exonic |
| Semen (Sperm) Parameters | MT (Mean ± SD) | WT (Mean ± SD) | p Value |
|---|---|---|---|
| Semen volume (mL) | 5.6 ± 1.0 | 4.8 ± 1.2 | 0.346 |
| Sperm count (×109/mL) | 1591.67 ± 180.10 | 1319 ± 198.67 | 0.153 |
| MOT (%) | 80.87 ± 5.2 | 78.77 ± 6.1 | 0.675 |
| VCL (μm/s) | 86.05 ± 2.35 | 84.59 ± 3.05 | 0.542 |
| VSL (μm/s) | 52.27 ± 2.40 | 50.97 ± 3.54 | 0.625 |
| VAP (μm/s) | 56.83 ± 1.08 | 54.52 ± 1.32 | 0.212 |
| ALH (μm) | 3.19 ± 0.05 | 3.19 ± 0.03 | 0.837 |
| LIN (%) | 60.74 ± 1.20 | 60.25 ± 1.10 | 0.609 |
| WOB (%) | 66.04 ± 1.28 | 64.45 ± 1.21 | 0.183 |
| SRT (%) | 91.98 ± 2.05 | 93.49 ± 3.34 | 0.540 |
| BCF (Hz) | 5.2 ± 1.3 | 5.0 ± 1.6 | 0.979 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hai, C.; Wang, L.; Wang, S.; Di, A.; Song, L.; Liu, X.; Bai, C.; Su, G.; Yang, L.; Li, G. Loss of Myostatin Affects m6A Modification but Not Semen Characteristics in Bull Spermatozoa. Int. J. Mol. Sci. 2025, 26, 591. https://doi.org/10.3390/ijms26020591
Hai C, Wang L, Wang S, Di A, Song L, Liu X, Bai C, Su G, Yang L, Li G. Loss of Myostatin Affects m6A Modification but Not Semen Characteristics in Bull Spermatozoa. International Journal of Molecular Sciences. 2025; 26(2):591. https://doi.org/10.3390/ijms26020591
Chicago/Turabian StyleHai, Chao, Linfeng Wang, Song Wang, Anqi Di, Lishuang Song, Xuefei Liu, Chunling Bai, Guanghua Su, Lei Yang, and Guangpeng Li. 2025. "Loss of Myostatin Affects m6A Modification but Not Semen Characteristics in Bull Spermatozoa" International Journal of Molecular Sciences 26, no. 2: 591. https://doi.org/10.3390/ijms26020591
APA StyleHai, C., Wang, L., Wang, S., Di, A., Song, L., Liu, X., Bai, C., Su, G., Yang, L., & Li, G. (2025). Loss of Myostatin Affects m6A Modification but Not Semen Characteristics in Bull Spermatozoa. International Journal of Molecular Sciences, 26(2), 591. https://doi.org/10.3390/ijms26020591