Abstract
Lung-protective ventilation and other experimental conditions raise arterial carbon dioxide tension (PaCO2) and alter pH. Short-term benefits are reported in non-infectious settings, whereas infection and/or prolonged exposure are typically harmful. This scoping review systematically maps immune-mediated effects of hypercapnia on innate immunity and alveolar epithelial repair. Scoping review per Levac et al. and PRISMA Extension for Scoping Reviews (Open Science Framework protocol: 10.17605/OSF.IO/WV85T; post hoc). We searched original preclinical studies (in vivo/in vitro) in PubMed, Web of Science, ScienceDirect, Cochrane Reviews, and SciELO (2008–2023). PaCO2 (mmHg) was prioritized; %Fraction of inspired Carbon Dioxide (%FiCO2) was recorded when PaCO2 was unavailable; pH was classified as buffered/unbuffered. Data were organized by context, PaCO2, and exposure duration; synthesis used heat maps (0–120 h) and a narrative description for >120 h. Mechanistic axes extracted the following: NF-κB (canonical/non-canonical), Bcl-2/Bcl-xL–Beclin-1/autophagy, AMPK/PKA/CaMKKβ/ERK1/2 and ENaC/Na,K-ATPase trafficking, Wnt/β-catenin in AT2 cells, and miR-183/IDH2/ATP. Thirty-five studies met the inclusion criteria. In non-infectious models, a “protective window” emerged, with moderate PaCO2 and brief exposure (65–95 mmHg; ≤4–6 h), featuring NF-κB attenuation and preserved epithelial ion transport. In infectious models and/or with prolonged exposure or higher PaCO2, harmful signals predominated: reduced phagocytosis/autophagy (Bcl-2/Bcl-xL–Beclin-1 axis), AMPK/PKA/ERK1/2-mediated internalization of ENaC/Na,K-ATPase, depressed β-catenin signaling in AT2 cells, impaired alveolar fluid clearance, and increased bacterial burden. Chronic exposures (>120 h) reinforced injury. Hypercapnia is a context-, dose-, time-, and pH-dependent double-edged modulator. The safe window is narrow; standardized, parallel reporting of PaCO2 and pH—with explicit comparisons of buffered vs. unbuffered hypercapnia—is essential to guide clinical translation.
1. Introduction
Acute lung injury (ALI), clinically represented by acute respiratory distress syndrome (ARDS), remains associated with high mortality and substantial global health-system burden [1,2,3]. Lung-protective ventilation strategies have reduced ventilator-induced lung injury (VILI) and ARDS mortality [4,5,6]; however, they often lead to alveolar accumulation of carbon dioxide (CO2) and a compensatory fall in pH over prolonged periods [4,5]. This condition—permissive hypercapnia—has been considered useful in selected ARDS scenarios and in obstructive lung diseases [7,8,9]. Yet recent studies suggest that hypercapnia may act not only as a severity marker but also as an independent predictor of mortality in specific clinical contexts [10,11,12,13]. Clinical evidence remains heterogeneous, and mechanistic uncertainties persist regarding how elevated CO2 affects lung tissue and host responses [14,15].
In preclinical models, hypercapnia modulates key immune and epithelial axes: it attenuates or reprograms NF-κB signaling (canonical and non-canonical) [16,17] and activates CaMKKβ/AMPK/PKA/ERK1/2 cascades that promote endocytosis of epithelial ion transporters (ENaC and Na,K-ATPase), with a direct impact on alveolar fluid clearance (AFC) [18,19]. The direction of effect depends on the biological context (infectious vs. non-infectious), dose (arterial carbon dioxide tension [PaCO2]), exposure duration, and acid–base status (buffered vs. unbuffered hypercapnia) [20]. Thus, a “double-edged” profile emerges: a potentially protective window with brief exposures and moderate PaCO2 [20] versus a detrimental profile—functional immunosuppression and epithelial dysfunction—when exposure is prolonged, PaCO2 is high, or active infection is present [21,22].
Beyond ARDS, chronic conditions such as chronic obstructive pulmonary disease (COPD), status asthmaticus, and cystic fibrosis can evolve to acute hypercapnic respiratory failure [23], particularly during infectious exacerbations, and are linked to poor outcomes [21,24]. These observations suggest that elevated CO2 may act as a causal modulator of innate immunity and epithelial homeostasis—rather than a mere epiphenomenon [23,25].
Despite the clinical and biological relevance, no integrative synthesis has specifically centered on the immune-mediated effects of hypercapnia on the pulmonary epithelium while standardizing exposure metrics (PaCO2 versus %FiCO2) and pH. To address this gap, we conducted a PRISMA-ScR–conformant scoping review that maps experimental evidence published between 2008 and 2023, stratified by an infectious/non-infectious context, PaCO2, exposure duration, and pH buffering. Our objective was to identify and characterize how hypercapnia modulates innate immunity and alveolar epithelial repair in experimental models. Specifically, we asked the following: How do these effects vary by context (infectious vs. non-infectious), PaCO2, and exposure time?
2. Results
The electronic search identified 3843 records, plus 116 from other sources (total 3959). After de-duplication (EndNote X8), 2576 unique references remained. Title/abstract screening excluded 2435 records. We assessed 141 full texts and excluded 106 that did not meet the research question. In total, 35 studies met the inclusion criteria (Figure 1). General characteristics (author, year, country, and design) are summarized in Table 1.
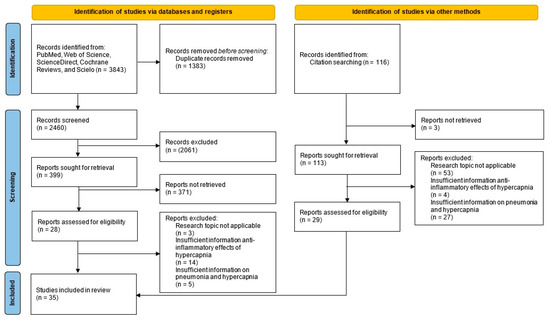
Figure 1.
PRISMA-ScR flow diagram.

Table 1.
General characteristics of included studies (chronological order by year of publication).
2.1. Dose- and Time-Dependent Effects by Context
2.1.1. Non-Infectious Models
In non-infectious models (VILI/sterile mechanical stress), the median PaCO2 was 90.3 mmHg (IQR: 68.4–120), and median exposure was 4 h (IQR: 1–10.5) [16,17,18,19,20,26,27,29,31,32,34,35,36,37,38,39,41,42,43,44,45,47,48,49,50]. Within this setting, a potentially protective window emerged with moderate PaCO2 (~65–95 mmHg) and brief exposures (≤4–6 h), associated with NF-κB attenuation and epithelial preservation [20,32,36,37,41]. Conversely, a harmful window appeared at PaCO2 ≥ 110–120 mmHg, even with minutes-to-hours exposures, consistent with ENaC/Na,K-ATPase endocytosis and reduced AFC [18,26,38,42,47,48]. See Figure 2A.
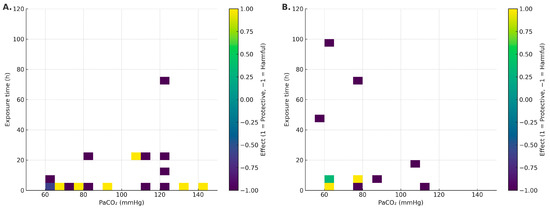
Figure 2.
Heat maps of effect direction (−1 harmful, 0 mixed/neutral, +1 protective) within the acute window (0–120 h), stratified by context: (A) non-infectious; (B) infectious. Axes: PaCO2 (mmHg) and duration (h).
2.1.2. Infectious Models
In infectious models, the median PaCO2 was 69.8 mmHg (IQR: 60–78.3) and median exposure was 6 h (IQR: 5.5–25.5) [10,21,22,23,25,28,30,33,40,46]. A short favorable window (≈4–6 h) was observed in some models [28,30,40]. However, with longer duration (≥24–96 h) and/or higher PaCO2, harmful effects predominated: higher bacterial burden, impaired phagocytosis, and worse physiological outcomes [21,22,25]. Detrimental signals were more consistent under unbuffered conditions and without antibiotics [21,23,25]; with antibiotics and/or buffered pH, the signal often attenuated or became mixed [28,30], with notable exceptions even under buffering [22]. See Figure 2B.
Chronic exposures (>120 h) reinforced the harmful window in a hypercapnia-level-dependent manner [21,47,49].
2.2. Convergent Mechanisms
2.2.1. NF-κB Signaling (Canonical and Non-Canonical), Stress-Kinase Signaling (ASK1/JNK/p38), and Innate Immunity Outputs (Cytokines, Phagocytosis, Autophagy)
Transcriptional alterations in innate immunity were reported in 34.3% (12/35) of studies [16,17,20,27,29,31,33,36,39,40,41,50]. Canonical NF-κB attenuation was the most frequent mechanism (6/12) [20,33,39,40,41,50]. Innate immune impairment (reduced phagocytosis and poorer microbial control) was documented in 51.4% (18/35) [10,17,21,22,23,25,27,28,29,30,31,32,33,36,37,39,43,46], especially in infectious in vivo models [10,21,23]. One cell study showed reduced autophagy and bacterial killing under hypercapnia [25]. While the global signal suggests immunosuppression with infection and/or prolonged exposure [17,23,32,33,36,37,39,43,46], some results were variable, reflecting model heterogeneity [10,28,31,50]. In sterile settings, early, moderate hypercapnia (PaCO2 80–100 mmHg; ≤4 h) inhibits ASK1 and downstream JNK/p38 [39]. See Table 2.
Table 2.
Pathway-level results (canonical/non-canonical NF-κB, AMPK/PKA), innate immunity outputs (cytokines, phagocytosis, autophagy), and epithelial outcomes (ENaC, Na,K-ATPase, fluid clearance), with effect direction and mechanistic notes per study.
2.2.2. cAMP/PKA–AMPK Pathways and Epithelial Transport (ENaC; Na,K-ATPase; PKC-ζ; CaMKKβ)
Alveolar epithelial disruption/resealing with hypercapnia was observed in 37.1% (13/35) [18,19,22,26,27,34,35,42,44,45,47,48,50]. Multiple studies show that elevated CO2 promotes Na,K-ATPase (and ENaC) endocytosis, reduces membrane density, and impairs AFC [18,26,35,38,45,47,48]. Additional findings highlight the effects on epithelial repair, including suppressed Wnt/β-catenin in AT2 and mitochondrial/miR-183 signals [34,49]. In an infectious in vivo model with hypercapnic ventilation, alveolar transudation, septal edema, and greater alveolar damage were reported [27]. See Table 2.
3. Discussion
Hypercapnia is defined as an elevation of PaCO2 beyond the physiological range [51]. It occurs in chronic pulmonary diseases [52] and as a consequence of lung-protective ventilation in ARDS [4,5]. Multiple studies link hypercapnia to worse outcomes in the critically ill [12,13,53,54]. Our synthesis maps, across preclinical models, a context–dose–time–pH-dependent “double-edged” effect: in non-infectious settings, moderate PaCO2 with brief exposures associates with protective signals (attenuated inflammatory pathways and epithelial preservation); whereas in infectious models and/or with prolonged duration or high PaCO2, detrimental signals predominate (oxidative stress, impaired microbial control, epithelial dysfunction, and reduced AFC). See Figure 3.
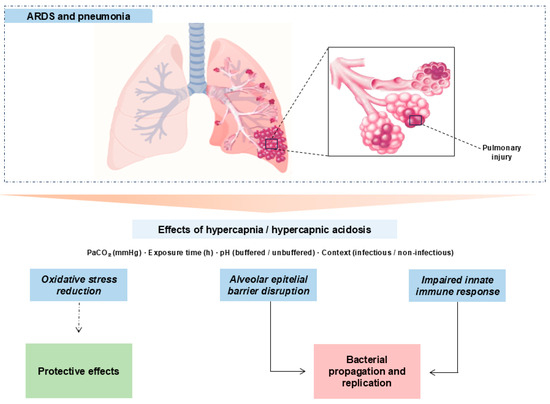
Figure 3.
Mechanistic schema of hypercapnia/hypercapnic acidosis in experimental lung injury. Hypercapnia, modulated by PaCO2, exposure time, pH (buffered vs. unbuffered), and context (infectious vs. non-infectious), can transiently dampen inflammatory tone (potentially protective) but also disrupt the alveolar epithelial barrier and compromise innate immunity. In infection and/or with prolonged exposures or high PaCO2, detrimental pathways dominate with bacterial proliferation and dissemination. Solid arrows: primary relationships; dashed arrows: context-contingent protective links.
3.1. Non-Infectious Models
In the absence of pathogens (VILI/sterile mechanical stress), the picture is nuanced and suggests a potentially protective window around PaCO2 ~65–95 mmHg with brief exposure (≤4–6 h). Within this range, several studies report canonical NF-κB attenuation, decreased IL-6/CXCL2, and epithelial preservation with improved microvascular leak/oxidation markers [32,36,41]. These effects track with ASK1–JNK/p38 inhibition, caspase-3 modulation, and reduced oxidative damage [32,36]. Still, signals are not uniform: PaCO2 55–65 mmHg for 4 h has been associated with increased IL-8, VCAM-1, E-selectin/P-selectin, and macrophage alterations [36]; at 24 h, epithelial chemokines (CXCL1/2/6, CCL28, CXCL14) shift [43]. Nitrotyrosine elevations have also been reported despite anti-inflammatory signals [32,36]. Overall—even without infection—directionality depends on dose–time, species/cell line, sterile stimulus, and pH. In the heat maps (Figure 2A), the protective sector clusters at short exposure + moderate PaCO2, whereas higher thresholds or longer durations tip toward harm. Mechanistic integration in Figure 4A,B.
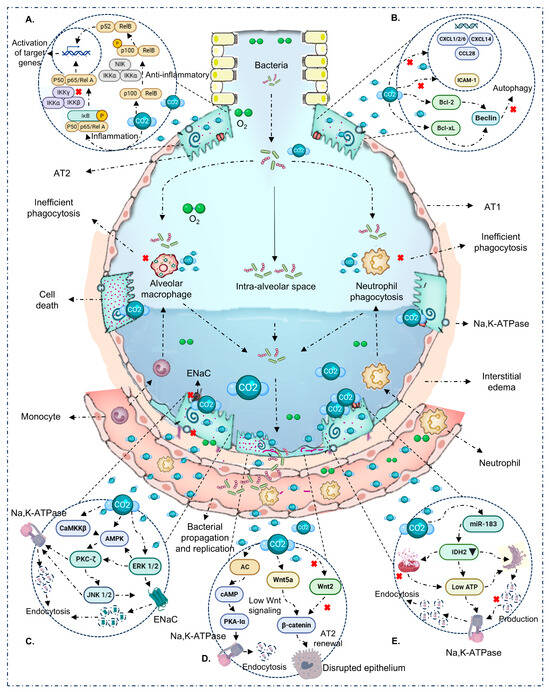
Figure 4.
Immuno-epithelial mechanisms modulated by hypercapnia/hypercapnic acidosis. (A) NF-κB: CO2 dampens the canonical arm (↓ IKK/p65, ↑ IκBα) and can activate the non-canonical arm (p100 → p52/RelB), reprogramming inflammatory output. (B) Autophagy: CO2 promotes Bcl-2/Bcl-xL binding to Beclin-1, inhibiting class III PI3K and autophagosome initiation; this associates with inefficient phagocytosis/bacterial clearance during infection. (C) Ion transport: activation of AMPK/PKA/CaMKKβ/PKC-ζ/ERK1/2 promotes ENaC and Na,K-ATPase endocytosis, reducing AFC. (D) Parallel routes: (i) adenylyl cyclase raises cAMP, phosphorylating PKA-Iα and driving Na,K-ATPase endocytosis/barrier disruption; (ii) depressed Wnt/β-catenin signaling (↓ β-cat, ↓ AT2 proliferation) compromises resealing/repair. (E) Mitochondrial energy/miR-183: ATP/IDH2 alterations and miR-183 favor Na,K-ATPase endocytosis and epithelial dysfunction. With prolonged exposure and/or high PaCO2, these routes converge on barrier disruption and bacterial spread; with brief, moderate exposure without infection, protective effects may prevail.
3.2. Infectious Models
Sepsis (pneumonia/systemic infection) is a major cause of severe ALI [55]. In this context, hypercapnia remodels host–pathogen interactions via intracellular pathways that affect pro-inflammatory cytokines (TNF and IL-6), phagocytosis, and autophagy [23,25,28]. Although a narrow favorable window (~4–6 h) is described in some models, the overall effect in the presence of infection is predominantly detrimental as exposure lengthens (≥24–96 h) or PaCO2 rises. This pattern—consistent with the 0–120 h heat maps (Figure 2) and reinforced by chronic exposures (>120 h)—presents as increased bacterial burden, depressed neutrophil/macrophage phagocytosis, autophagy inhibition (increased Bcl-2/Bcl-xL binding to Beclin-1), and deterioration of lung architecture [21,23,25,28]. Acid–base status is a key modulator: effects are seen with both buffered and unbuffered hypercapnia, but magnitude and even direction can differ [22,28]. We therefore separated these conditions and prioritized PaCO2 over inspired %CO2 whenever possible. In untreated infection, hypercapnia fosters bacterial propagation/replication, impairs host defense, and aggravates ALI—arguing for caution with prolonged/intense CO2 exposure in infectious settings [21,23,25]. Mechanistic integration in Figure 4A,B.
The safety of hypercapnia in pulmonary sepsis is a critical question in ICU patients [28]. Early hypercapnic acidosis within the first 24 h of mechanical ventilation has been associated with higher mortality in ARDS [54], alongside increased lung stiffness and airway pressures [56]. In severe pneumonia, “permissive hypercapnia” did not reduce mortality and was linked to greater morbidity [57]. Rigorous clinical trials are needed to define indications and safety thresholds.
3.3. Immunologic Effects of Hypercapnia
The NF-κB family provides an explanatory scaffold for lung injury, inflammation, and repair [41,58]. Across multiple models, CO2 ≥ 10% for ≥60 min inhibits the canonical pathway by reducing IKK complex phosphorylation, preserving IκBα, and preventing p65 nuclear translocation—dampening pro-inflammatory transcription [12,20,33,39,40,41,50,51,59,60]. In parallel, the non-canonical arm can be engaged via p100 processing to p52 and RelB nuclear localization, producing an immunosuppressive tone that persists for hours [16,17]. After stimulus withdrawal, many changes reverse within minutes, underscoring exposure-time dependence [16,17,33,40,50]. Context-dependent observations (PP2A–p65 axis with pro-inflammatory readout) emphasize modulation by pH, cell type, and microenvironment [16,17,18,19,20,26,27,29,31,32,34,35,36,37,38,39,41,42,43,44,45,47,48,49,50], contrasting PP2A’s classic negative regulation of NF-κB and suggesting key roles in intracellular homeostasis [61]. These axes are summarized in Figure 4A.
3.4. Alveolar Epithelial Repair/Healing
A second critical layer is ion-transporter trafficking and epithelial homeostasis. Hypercapnia activates CaMKKβ→AMPK and, in parallel, increases cAMP via adenylyl cyclase (AC)—especially with buffered pH—activating PKA (PKA-Iα). Together with PKC-ζ and ERK1/2, these routes converge to promote Na,K-ATPase and ENaC endocytosis, reduce their basolateral membrane density, and impair AFC [18,26,35,38,42,45,47,48,62,63,64]. Additional effects include assembly defects of Na,K-ATPase and suppression of Wnt/β-catenin in AT2 cells with reduced proliferation/repair after injury [49]. Notably, CO2-triggered AC rapidly elevates scAMP (≈15–30 min), phosphorylating PKA-Iα and initiating Na,K-ATPase internalization; this axis runs in parallel and complements the CaMKKβ→AMPK route [19,34]. These effects can appear from 30 min at PaCO2 ≥ 60 mmHg, intensify with longer exposures, and have been described with or without acidosis [18,26,42,45]. Figure 4C–E synthesizes these routes: AMPK/PKA and ENaC/Na,K-ATPase endocytosis; β-catenin/AT2; AC/cAMP/PKA-Iα; and miR-183/IDH2/ATP.
3.5. Limitations
This scoping review offers a comprehensive view of hypercapnia’s effects on innate immunity and the alveolar epithelium. Several limitations merit consideration: (i) heterogeneity in models and exposure metrics (PaCO2 vs. %CO2) precluded meta-analysis and motivated a direction-of-effect synthesis; (ii) OSF registration post hoc, dual screening, decision log, and sensitivity analyses mitigate—but do not eliminate—bias; (iii) potential publication bias (time window, databases, language); and (iv) limited clinical translation from in vivo/in vitro models. Accordingly, our patterns should be interpreted as operational hypotheses. Even so, these data provide a basis for studies that define thresholds and mechanisms with clinical relevance.
3.6. Clinical Implications and Future Directions
Taken together with the visual synthesis (heat maps, Figure 2; mechanistic schema, Figure 4), hypercapnia should be understood as a double-edged modulator whose effect depends on context (infectious vs. non-infectious), dose (PaCO2), exposure time, and pH. In non-infectious settings, we identify a potential protective window at moderate PaCO2 (~65–95 mmHg) and brief exposure (≤4–6 h), where canonical NF-κB inactivation, cytokine reduction, and preservation of epithelial ion transport predominate [10,28,30,39]. In contrast, with active infection and/or prolonged exposure (≥24–96 h) or high PaCO2 (≥110–120 mmHg), the signal shifts toward harm: autophagy inhibition (Bcl-2/Bcl-xL–Beclin-1 axis), depressed phagocytosis, AMPK/PKA/ERK1/2-mediated ENaC/Na,K-ATPase endocytosis, and reduced β-catenin in AT2 cells, with worse AFC and higher bacterial burden [19,25,34,49,50]. pH modulation (buffered vs. unbuffered) contributes to heterogeneity, reinforcing the need to co-report PaCO2 and pH [27,37].
Clinically, these observations favor a prudent, titrated approach. When hypercapnia arises from lung-protective ventilation, tolerance should be time-limited and constrained to a “protective window” of PaCO2, with close monitoring of pH, airway pressures, oxygenation, and infectious context. In uncontrolled infection or when long exposures/high PaCO2 are anticipated, risk–benefit appears unfavorable: Figure 4’s axes predict functional immunosuppression (impaired autophagy/phagocytosis) and epithelial dysfunction (altered ion trafficking and depressed AT2 repair) that may facilitate bacterial proliferation and dissemination. Permissive hypercapnia should therefore be avoided or minimized in untreated infection and, when unavoidable, restricted to brief exposure with appropriate antimicrobials and explicit PaCO2/pH targets.
From a translational standpoint, we propose a PaCO2–time framework, modulated by pH and context, to standardize experimental design and clinical hypotheses: (i) systematically report PaCO2 (mmHg), pH, and exposure time; (ii) explicitly compare buffered vs. unbuffered hypercapnia and their interaction with AMPK/PKA and Bcl-2/Bcl-xL–Beclin-1 axes; (iii) define reproducible dose–time curves to bound protective windows and damage thresholds; (iv) incorporate biomarkers of epithelial dysfunction (membrane density of ENaC/Na,K-ATPase; β-catenin/AT2 markers) and innate immunity (phagocytosis and autophagy flux); and (v) evaluate therapeutic synchrony with antibiotics and pH-buffering strategies. Together, Figure 2 and Figure 4 offer an operational, biologically plausible framework to guide prudent clinical decisions and to design studies that confirm—or refute—the existence of a narrow safety window for hypercapnia.
3.7. The “Double-Edged Sword” of Hypercapnia: A Context-Specific Therapeutic Framework
Before context-specific recommendations, we set two operational principles: (i) CO2 effects depend on dose (PaCO2), exposure time, and pH, shifting the NF-κB–Ca2+/CaMKKβ–AMPK/PKA–β-catenin network between transient anti-inflammatory dampening and immuno-epithelial failure; (ii) clinical decisions are not binary but a titrated, reversible tolerance, guided by biomarkers (AFC, membrane ENaC/Na,K-ATPase density, β-catenin/AT2, phagocytosis/autophagy flux). With these principles, we distinguish the following.
3.7.1. Non-Infectious Context (VILI/Sterile Stress)
Consider time-limited tolerance to moderate PaCO2 (≈65–95 mmHg) for ≤4–6 h, with pH closely monitored/preferably buffered, aiming to dampen canonical NF-κB without triggering epithelial failure loops.
3.7.2. Infectious Context (Pneumonia/Sepsis)
Avoid or minimize hypercapnia—especially prolonged or high (≥110–120 mmHg)—due to functional immunosuppression (autophagy/phagocytosis) and barrier failure (ENaC/Na,K-ATPase). If unavoidable, limit duration, set explicit PaCO2/pH targets, and synchronize with antibiotics and source control.
4. Materials and Methods
This scoping review followed the PRISMA-ScR guidance (Preferred Reporting Items for Systematic Reviews and Meta-Analyses—extension for scoping reviews) and methodological recommendations by Levac et al. [65]. The protocol was publicly registered on the Open Science Framework (OSF) on 30 August 2024 (https://doi.org/10.17605/OSF.IO/WV85T). Because registration occurred after defining the study period, it is considered post hoc; any subsequent adjustments were documented in a decision log (Table S1). The PRISMA-ScR checklist is provided in the Supplementary Table S2.
4.1. Eligibility Criteria
We included original preclinical studies (in vivo/in vitro/ex vivo) evaluating CO2 exposure/intervention and pulmonary immune and/or epithelial outcomes. Given the limited human clinical evidence, we considered experimental animal models and relevant cell lines/cultures (with or without acute lung injury). The preferred exposure metric was PaCO2 (mmHg); when only % inspired CO2 was reported, it was recorded descriptively. We distinguished between buffered and unbuffered hypercapnia. Outcomes covered the following: (i) innate immunity (phagocytosis, cytokines, canonical/non-canonical NF-κB); (ii) stress-kinase pathways (ASK1/JNK/p38); and (iii) CaMKKβ/AMPK/PKA/ERK1/2 axes linked to epithelial transport (ENaC, Na,K-ATPase, fluid clearance/integrity). We included studies published from Jan-2008 to Dec-2023 in English or Spanish. We excluded reviews, communications without primary data, clinical studies without an experimental component, models without documented epithelial impact, or lacking minimal CO2/pH reporting.
4.2. Information Sources and Search Strategy
We implemented a two-pronged search strategy. Electronic searches were run in PubMed, Web of Science, ScienceDirect, Cochrane Reviews, and SciELO. We combined MeSH terms and keywords, including “acute respiratory distress syndrome,” “pneumonia,” “ventilator-associated pneumonia,” “hypercapnia,” and “anti-inflammatory.” The full strategy is reported in Table A1. To broaden coverage, we performed manual reference screening of the included articles. Duplicates were managed in EndNote X8.
4.3. Study Selection
Title/abstract screening and full-text assessment were performed independently and in duplicate by two reviewers; discrepancies were resolved by consensus or a third reviewer. We did not compute κ coefficients, as the aim was to map the evidence (PRISMA-ScR scoping review); we prioritized a systematic, documented resolution of disagreements. We did not conduct a formal risk-of-bias assessment (SYRCLE) given the descriptive nature of the synthesis; relevant design domains were extracted and reported in a structured manner.
4.4. Data Extraction
Data were extracted using standardized templates (double entry with cross-checks). Variables included the following: model type; species/strain or cell line; context (infectious/non-infectious); acid–base status (buffered/unbuffered); exposure metric (PaCO2 in mmHg; %CO2 if only metric available); exposure duration (h); immune outcomes (NF-κB, cytokines, phagocytosis, autophagy); epithelial outcomes (ENaC, Na,K-ATPase, AFC/integrity); mechanistic axes (canonical/non-canonical NF-κB; ASK1/JNK/p38; CaMKKβ/AMPK/PKA/ERK1/2; AC/cAMP/PKA-Iα; miR-183/IDH2; Wnt/β-catenin in AT2 cells); and direction of effect (protective/mixed-neutral/harmful). Effect classification followed a pre-specified operational definition based on primary physiological/biological outcomes.
4.5. Evidence Synthesis and Mapping
Extracted data were tabulated for a comprehensive descriptive synthesis aligned with the review’s objective and research question. For the acute window (0–120 h), we built heat maps stratified by context (infectious/non-infectious), with PaCO2 and time discretization and effect coding (+1 = protective; 0 = mixed/neutral; −1 = harmful). When multiple data points were mapped to one cell, we averaged the score; missing cells were left blank. Chronic exposures (>120 h) were described separately and not integrated into the acute maps. Additionally, we created mechanistic panels for canonical/non-canonical NF-κB, ASK1/JNK/p38, and CaMKKβ/AMPK/PKA/ERK1/2 with epithelial transport (ENaC/Na,K-ATPase/AFC). Descriptive statistics (medians [p25–p75]) were produced in Jamovi 2.2.5. Operational details (PaCO2/time discretization, coding rules, sensitivity analyses) are provided in the Supplementary Material.
4.6. Transparency and Data Availability
5. Conclusions
This scoping review, organized within a PaCO2–time–pH framework and stratified by context (infectious vs. non-infectious), synthesizes the effects of hypercapnia on innate immunity and alveolar epithelial integrity/repair. Taken together, the evidence supports a narrow, context-dependent window in which moderate PaCO2 and brief exposure are associated with inflammatory attenuation and preservation of epithelial ion transport. Outside this window—particularly with active infection, prolonged exposure, or high PaCO2—a detrimental phenotype predominates, characterized by functional immunosuppression (autophagy blockade) and epithelial dysfunction (endocytosis of ENaC and Na,K-ATPase with depressed AT2-mediated repair), with potential adverse prognostic implications. These findings support hypercapnia as a dose-, time-, and pH-dependent double-edged modulator and underscore the need to co-report PaCO2 (mmHg), pH, and exposure time and to stratify by infection. The integrative framework presented here provides operational criteria to guide prudent clinical decision-making and to design translational studies that validate—or refute—the existence of a safe window for hypercapnia and delineate its damage thresholds with precision.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/ijms26199622/s1.
Author Contributions
Conceptualization, E.O.-R. and J.C.-G.; data curation, E.O.-R., D.R.-B., M.B.-L., M.A.-G. and M.G.-O.; formal analysis, E.O.-R.; methodology, E.O.-R., J.C.-G., C.R.-M. and C.D.-C.; project administration, E.O.-R.; visualization, E.O.-R., C.R.-M. and C.D.-C.; writing—original draft preparation, E.O.-R., J.C.-G., D.R.-B., M.B.-L., C.R.-M., J.P.-P., J.V.-T., M.A.-G., M.G.-O., D.G.-B., Y.N.-B., G.S.-S. and C.D.-C.; writing—review and editing, E.O.-R., J.C.-G., D.R.-B., M.B.-L., C.R.-M., J.P.-P., J.V.-T., M.A.-G., M.G.-O., D.G.-B., Y.N.-B., G.S.-S. and C.D.-C. The corresponding author attests that all listed authors meet authorship criteria and that no others meeting the criteria have been omitted. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data, extraction templates, decision log, PRISMA-ScR checklist, and full search strategies are openly available at OSF (https://doi.org/10.17605/OSF.IO/WV85T).
Conflicts of Interest
The authors declare no conflicts of interest.
Appendix A
Table A1.
Search Strategy.
Table A1.
Search Strategy.
| Search Number | Search Terms |
|---|---|
| 1 | Pneumonia |
| 2 | Hypercapnia |
| 3 | Acute respiratory distress syndrome (ARDS) |
| 4 | Ventilator-Associated Pneumonia (VAP) |
| 5 | Anti-Inflammatories |
| 6 | 1 AND 2 AND 3 |
| 7 | 4 AND 2 |
| 8 | 5 AND 2 AND 1 |
References
- Bellani, G.; Laffey, J.G.; Pham, T.; Fan, E.; Brochard, L.; Esteban, A.; Gattinoni, L.; van Haren, F.; Larsson, A.; McAuley, D.F.; et al. Epidemiology, Patterns of Care, and Mortality for Patients With Acute Respiratory Distress Syndrome in Intensive Care Units in 50 Countries. JAMA 2016, 315, 788. [Google Scholar] [CrossRef]
- Máca, J.; Jor, O.; Holub, M.; Sklienka, P.; Burša, F.; Burda, M.; Janout, V.; Ševčík, P. Past and Present ARDS Mortality Rates: A Systematic Review. Respir. Care 2017, 62, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Hanrahan, J.; Bornstein, J.; Chen, S.-Y. Healthcare Costs Utilization and Costs of Patients Hospitalized with Acute Respiratory Distress Syndrome (ARDS) in US Commercially-Insured Individuals and Medicare Beneficiaries. In Proceedings of the 2.1 Acute Critical Care; European Respiratory Society: Lausanne, Switzerland, 2015; p. PA2139. [Google Scholar]
- Amato, M.B.P.; Barbas, C.S.V.; Medeiros, D.M.; Magaldi, R.B.; Schettino, G.P.; Lorenzi-Filho, G.; Kairalla, R.A.; Deheinzelin, D.; Munoz, C.; Oliveira, R.; et al. Effect of a Protective-Ventilation Strategy on Mortality in the Acute Respiratory Distress Syndrome. N. Engl. J. Med. 1998, 338, 347–354. [Google Scholar] [CrossRef]
- The Acute Respiratory Distress Syndrome Network Ventilation with Lower Tidal Volumes as Compared with Traditional Tidal Volumes for Acute Lung Injury and the Acute Respiratory Distress Syndrome. N. Engl. J. Med. 2000, 342, 1301–1308. [CrossRef]
- Petrucci, N.; De Feo, C. Lung Protective Ventilation Strategy for the Acute Respiratory Distress Syndrome. Cochrane Database Syst. Rev. 2013, 2013, CD003844. [Google Scholar] [CrossRef]
- Tuxen, D.V.; Williams, T.J.; Scheinkestel, C.D.; Czarny, D.; Bowes, G. Use of a Measurement of Pulmonary Hyperinflation to Control the Level of Mechanical Ventilation in Patients with Acute Severe Asthma. Am. Rev. Respir. Dis. 1992, 146, 1136–1142. [Google Scholar] [CrossRef]
- Hickling, K.G.; Henderson, S.J.; Jackson, R. Low Mortality Associated with Low Volume Pressure Limited Ventilation with Permissive Hypercapnia in Severe Adult Respiratory Distress Syndrome. Intensive Care Med. 1990, 16, 372–377. [Google Scholar] [CrossRef]
- Hickling, K.G.; Walsh, J.; Henderson, S.; Jackson, R. Low Mortality Rate in Adult Respiratory Distress Syndrome Using Low-Volume, Pressure-Limited Ventilation with Permissive Hypercapnia: A Prospective Study. Crit. Care Med. 1994, 22, 1568–1578. [Google Scholar] [CrossRef]
- Chonghaile, M.N.; Higgins, B.D.; Costello, J.; Laffey, J.G. Hypercapnic Acidosis Attenuates Lung Injury Induced by Established Bacterial Pneumonia. Anesthesiology 2008, 109, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Madotto, F.; Rezoagli, E.; McNicholas, B.A.; Pham, T.; Slutsky, A.S.; Bellani, G.; Laffey, J.G. Patterns and Impact of Arterial CO2 Management in Patients With Acute Respiratory Distress Syndrome. Chest 2020, 158, 1967–1982. [Google Scholar] [CrossRef]
- Shigemura, M.; Lecuona, E.; Sznajder, J.I. Effects of Hypercapnia on the Lung. J. Physiol. 2017, 595, 2431–2437. [Google Scholar] [CrossRef] [PubMed]
- Nin, N.; Muriel, A.; Peñuelas, O.; Brochard, L.; Lorente, J.A.; Ferguson, N.D.; Raymondos, K.; Ríos, F.; Violi, D.A.; Thille, A.W.; et al. Severe Hypercapnia and Outcome of Mechanically Ventilated Patients with Moderate or Severe Acute Respiratory Distress Syndrome. Intensive Care Med. 2017, 43, 200–208. [Google Scholar] [CrossRef]
- Gendreau, S.; Geri, G.; Pham, T.; Vieillard-Baron, A.; Mekontso Dessap, A. The Role of Acute Hypercapnia on Mortality and Short-Term Physiology in Patients Mechanically Ventilated for ARDS: A Systematic Review and Meta-Analysis. Intensive Care Med. 2022, 48, 517–534. [Google Scholar] [CrossRef]
- Masterson, C.; Horie, S.; McCarthy, S.D.; Gonzalez, H.; Byrnes, D.; Brady, J.; Fandiño, J.; Laffey, J.G.; O’Toole, D. Hypercapnia in the Critically Ill: Insights from the Bench to the Bedside. Interface Focus 2021, 11, 20200032. [Google Scholar] [CrossRef]
- Keogh, C.E.; Scholz, C.C.; Rodriguez, J.; Selfridge, A.C.; von Kriegsheim, A.; Cummins, E.P. Carbon Dioxide-Dependent Regulation of NF-κB Family Members RelB and p100 Gives Molecular Insight into CO2-Dependent Immune Regulation. J. Biol. Chem. 2017, 292, 11561–11571. [Google Scholar] [CrossRef] [PubMed]
- Oliver, K.M.; Lenihan, C.R.; Bruning, U.; Cheong, A.; Laffey, J.G.; McLoughlin, P.; Taylor, C.T.; Cummins, E.P. Hypercapnia Induces Cleavage and Nuclear Localization of RelB Protein, Giving Insight into CO2 Sensing and Signaling. J. Biol. Chem. 2012, 287, 14004–14011. [Google Scholar] [CrossRef]
- Welch, L.C.; Lecuona, E.; Briva, A.; Trejo, H.E.; Dada, L.A.; Sznajder, J.I. Extracellular Signal-Regulated Kinase (ERK) Participates in the Hypercapnia-Induced Na,K-ATPase Downregulation. FEBS Lett. 2010, 584, 3985–3989. [Google Scholar] [CrossRef]
- Lecuona, E.; Sun, H.; Chen, J.; Trejo, H.E.; Baker, M.A.; Sznajder, J.I. Protein Kinase A-Iα Regulates Na,K-ATPase Endocytosis in Alveolar Epithelial Cells Exposed to High CO2 Concentrations. Am. J. Respir. Cell Mol. Biol. 2013, 48, 626–634. [Google Scholar] [CrossRef]
- Contreras, M.; Ansari, B.; Curley, G.; Higgins, B.D.; Hassett, P.; O’Toole, D.; Laffey, J.G. Hypercapnic Acidosis Attenuates Ventilation-Induced Lung Injury by a Nuclear Factor-κB–dependent Mechanism. Crit. Care Med. 2012, 40, 2622–2630. [Google Scholar] [CrossRef]
- O’Croinin, D.F.; Nichol, A.D.; Hopkins, N.; Boylan, J.; O’Brien, S.; O’Connor, C.; Laffey, J.G.; McLoughlin, P. Sustained Hypercapnic Acidosis during Pulmonary Infection Increases Bacterial Load and Worsens Lung Injury. Crit. Care Med. 2008, 36, 2128–2135. [Google Scholar] [CrossRef] [PubMed]
- Nichol, A.D.; O’Cronin, D.F.; Howell, K.; Naughton, F.; O’Brien, S.; Boylan, J.; O’Connor, C.; O’Toole, D.; Laffey, J.G.; McLoughlin, P. Infection-Induced Lung Injury Is Worsened after Renal Buffering of Hypercapnic Acidosis. Crit. Care Med. 2009, 37, 2953–2961. [Google Scholar] [CrossRef]
- Gates, K.L.; Howell, H.A.; Nair, A.; Vohwinkel, C.U.; Welch, L.C.; Beitel, G.J.; Hauser, A.R.; Sznajder, J.I.; Sporn, P.H.S. Hypercapnia Impairs Lung Neutrophil Function and Increases Mortality in Murine Pseudomonas Pneumonia. Am. J. Respir. Cell Mol. Biol. 2013, 49, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, H.S.; Tiangco, N.D.; Harrell, C.; Vender, R.L. Severe Hypercapnia in Critically Ill Adult Cystic Fibrosis Patients. J. Clin. Med. Res. 2011, 3, 209–212. [Google Scholar] [CrossRef][Green Version]
- Casalino-Matsuda, S.M.; Nair, A.; Beitel, G.J.; Gates, K.L.; Sporn, P.H.S. Hypercapnia Inhibits Autophagy and Bacterial Killing in Human Macrophages by Increasing Expression of Bcl-2 and Bcl-xL. J. Immunol. 2015, 194, 5388–5396. [Google Scholar] [CrossRef] [PubMed]
- Vadász, I.; Dada, L.A.; Briva, A.; Trejo, H.E.; Welch, L.C.; Chen, J.; Tóth, P.T.; Lecuona, E.; Witters, L.A.; Schumacker, P.T.; et al. AMP-Activated Protein Kinase Regulates CO2-Induced Alveolar Epithelial Dysfunction in Rats and Human Cells by Promoting Na,K-ATPase Endocytosis. J. Clin. Investig. 2008. [Google Scholar] [CrossRef]
- Liu, Y.; Chacko, B.K.; Ricksecker, A.; Shingarev, R.; Andrews, E.; Patel, R.P.; Lang, J.D. Modulatory Effects of Hypercapnia on in Vitro and in Vivo Pulmonary Endothelial-Neutrophil Adhesive Responses during Inflammation. Cytokine 2008, 44, 108–117. [Google Scholar] [CrossRef]
- Ni Chonghaile, M.; Higgins, B.D.; Costello, J.F.; Laffey, J.G. Hypercapnic Acidosis Attenuates Severe Acute Bacterial Pneumonia-Induced Lung Injury by a Neutrophil-Independent Mechanism. Crit. Care Med. 2008, 36, 3135–3144. [Google Scholar] [CrossRef]
- Abolhassani, M.; Guais, A.; Chaumet-Riffaud, P.; Sasco, A.J.; Schwartz, L. Carbon Dioxide Inhalation Causes Pulmonary Inflammation. Am. J. Physiol. Cell. Mol. Physiol. 2009, 296, L657–L665. [Google Scholar] [CrossRef] [PubMed]
- Higgins, B.D.; Costello, J.; Contreras, M.; Hassett, P.; O’ Toole, D.; Laffey, J.G. Differential Effects of Buffered Hypercapnia versus Hypercapnic Acidosis on Shock and Lung Injury Induced by Systemic Sepsis. Anesthesiology 2009, 111, 1317–1326. [Google Scholar] [CrossRef]
- Wang, N.; Gates, K.L.; Trejo, H.; Favoreto, S.; Schleimer, R.P.; Sznajder, J.I.; Beitel, G.J.; Sporn, P.H.S. Elevated CO2 Selectively Inhibits interleukin-6 and Tumor Necrosis Factor Expression and Decreases Phagocytosis in the Macrophage. FASEB J. 2010, 24, 2178–2190. [Google Scholar] [CrossRef]
- Peltekova, V.; Engelberts, D.; Otulakowski, G.; Uematsu, S.; Post, M.; Kavanagh, B.P. Hypercapnic Acidosis in Ventilator-Induced Lung Injury. Intensive Care Med. 2010, 36, 869–878. [Google Scholar] [CrossRef]
- Cummins, E.P.; Oliver, K.M.; Lenihan, C.R.; Fitzpatrick, S.F.; Bruning, U.; Scholz, C.C.; Slattery, C.; Leonard, M.O.; McLoughlin, P.; Taylor, C.T. NF-κB Links CO2 Sensing to Innate Immunity and Inflammation in Mammalian Cells. J. Immunol. 2010, 185, 4439–4445. [Google Scholar] [CrossRef]
- Vohwinkel, C.U.; Lecuona, E.; Sun, H.; Sommer, N.; Vadász, I.; Chandel, N.S.; Sznajder, J.I. Elevated CO2 Levels Cause Mitochondrial Dysfunction and Impair Cell Proliferation. J. Biol. Chem. 2011, 286, 37067–37076. [Google Scholar] [CrossRef]
- Vadász, I.; Dada, L.A.; Briva, A.; Helenius, I.T.; Sharabi, K.; Welch, L.C.; Kelly, A.M.; Grzesik, B.A.; Budinger, G.R.S.; Liu, J.; et al. Evolutionary Conserved Role of c-Jun-N-Terminal Kinase in CO2-Induced Epithelial Dysfunction. PLoS ONE 2012, 7, e46696. [Google Scholar] [CrossRef]
- Yang, W.-C.; Song, C.-Y.; Wang, N.; Zhang, L.-L.; Yue, Z.-Y.; Cui, X.-G.; Zhou, H.-C. Hypercapnic Acidosis Confers Antioxidant and Anti-Apoptosis Effects against Ventilator-Induced Lung Injury. Lab. Investig. 2013, 93, 1339–1349. [Google Scholar] [CrossRef]
- Nardelli, L.M.; Rzezinski, A.; Silva, J.D.; Maron-Gutierrez, T.; Ornellas, D.S.; Henriques, I.; Capelozzi, V.L.; Teodoro, W.; Morales, M.M.; Silva, P.L.; et al. Effects of Acute Hypercapnia with and without Acidosis on Lung Inflammation and Apoptosis in Experimental Acute Lung Injury. Respir. Physiol. Neurobiol. 2015, 205, 1–6. [Google Scholar] [CrossRef]
- Dada, L.A.; Trejo Bittar, H.E.; Welch, L.C.; Vagin, O.; Deiss-Yehiely, N.; Kelly, A.M.; Baker, M.R.; Capri, J.; Cohn, W.; Whitelegge, J.P.; et al. High CO2 Leads to Na,K-ATPase Endocytosis via c-Jun Amino-Terminal Kinase-Induced LMO7b Phosphorylation. Mol. Cell. Biol. 2015, 35, 3962–3973. [Google Scholar] [CrossRef]
- Yang, W.; Yue, Z.; Cui, X.; Guo, Y.; Zhang, L.; Zhou, H.; Li, W. Comparison of the Effects of Moderate and Severe Hypercapnic Acidosis on Ventilation-Induced Lung Injury. BMC Anesthesiol. 2015, 15, 67. [Google Scholar] [CrossRef] [PubMed]
- Masterson, C.; O’Toole, D.; Leo, A.; McHale, P.; Horie, S.; Devaney, J.; Laffey, J.G. Effects and Mechanisms by Which Hypercapnic Acidosis Inhibits Sepsis-Induced Canonical Nuclear Factor-κB Signaling in the Lung. Crit. Care Med. 2016, 44, e207–e217. [Google Scholar] [CrossRef] [PubMed]
- Horie, S.; Ansari, B.; Masterson, C.; Devaney, J.; Scully, M.; O’Toole, D.; Laffey, J.G. Hypercapnic Acidosis Attenuates Pulmonary Epithelial Stretch-Induced Injury via Inhibition of the Canonical NF-κB Pathway. Intensive Care Med. Exp. 2016, 4, 8. [Google Scholar] [CrossRef]
- Gwoździńska, P.; Buchbinder, B.A.; Mayer, K.; Herold, S.; Morty, R.E.; Seeger, W.; Vadász, I. Hypercapnia Impairs ENaC Cell Surface Stability by Promoting Phosphorylation, Polyubiquitination and Endocytosis of β-ENaC in a Human Alveolar Epithelial Cell Line. Front. Immunol. 2017, 8, 591. [Google Scholar] [CrossRef]
- Casalino-Matsuda, S.M.; Wang, N.; Ruhoff, P.T.; Matsuda, H.; Nlend, M.C.; Nair, A.; Szleifer, I.; Beitel, G.J.; Sznajder, J.I.; Sporn, P.H.S. Hypercapnia Alters Expression of Immune Response, Nucleosome Assembly and Lipid Metabolism Genes in Differentiated Human Bronchial Epithelial Cells. Sci. Rep. 2018, 8, 13508. [Google Scholar] [CrossRef]
- Cortes-Puentes, G.A.; Westerly, B.; Schiavo, D.; Wang, S.; Stroetz, R.; Walters, B.; Hubmayr, R.D.; Oeckler, R.A. Hypercapnia Alters Alveolar Epithelial Repair by a pH-Dependent and Adenylate Cyclase-Mediated Mechanism. Sci. Rep. 2019, 9, 349. [Google Scholar] [CrossRef]
- Kryvenko, V.; Wessendorf, M.; Morty, R.E.; Herold, S.; Seeger, W.; Vagin, O.; Dada, L.A.; Sznajder, J.I.; Vadász, I. Hypercapnia Impairs Na,K-ATPase Function by Inducing Endoplasmic Reticulum Retention of the β-Subunit of the Enzyme in Alveolar Epithelial Cells. Int. J. Mol. Sci. 2020, 21, 1467. [Google Scholar] [CrossRef]
- Casalino-Matsuda, S.M.; Berdnikovs, S.; Wang, N.; Nair, A.; Gates, K.L.; Beitel, G.J.; Sporn, P.H.S. Hypercapnia Selectively Modulates LPS-Induced Changes in Innate Immune and DNA Replication-Related Gene Transcription in the Macrophage. Interface Focus 2021, 11, 20200039. [Google Scholar] [CrossRef] [PubMed]
- Gabrielli, N.M.; Mazzocchi, L.C.; Kryvenko, V.; Tello, K.; Herold, S.; Morty, R.E.; Grimminger, F.; Dada, L.A.; Seeger, W.; Sznajder, J.I.; et al. TRAF2 Is a Novel Ubiquitin E3 Ligase for the Na,K-ATPase β-Subunit That Drives Alveolar Epithelial Dysfunction in Hypercapnia. Front. Cell Dev. Biol. 2021, 9, 689983. [Google Scholar] [CrossRef]
- Kryvenko, V.; Wessendorf, M.; Tello, K.; Herold, S.; Morty, R.E.; Seeger, W.; Vadász, I. Hypercapnia Induces Inositol-Requiring Enzyme 1α–Driven Endoplasmic Reticulum–associated Degradation of the Na,K-ATPase β-Subunit. Am. J. Respir. Cell Mol. Biol. 2021, 65, 615–629. [Google Scholar] [CrossRef] [PubMed]
- Dada, L.A.; Welch, L.C.; Magnani, N.D.; Ren, Z.; Han, H.; Brazee, P.L.; Celli, D.; Flozak, A.S.; Weng, A.; Herrerias, M.M.; et al. Hypercapnia Alters Stroma-Derived Wnt Production to Limit β-Catenin Signaling and Proliferation in AT2 Cells. JCI Insight 2023, 8, e159331. [Google Scholar] [CrossRef]
- O’Toole, D.; Hassett, P.; Contreras, M.; Higgins, B.D.; McKeown, S.T.W.; McAuley, D.F.; O’Brien, T.; Laffey, J.G. Hypercapnic Acidosis Attenuates Pulmonary Epithelial Wound Repair by an NF- B Dependent Mechanism. Thorax 2009, 64, 976–982. [Google Scholar] [CrossRef]
- Curley, G.; Laffey, J.G.; Kavanagh, B.P. Bench-to-Bedside Review: Carbon Dioxide. Crit. Care 2010, 14, 220. [Google Scholar] [CrossRef] [PubMed]
- Crummy, F.; Buchan, C.; Miller, B.; Toghill, J.; Naughton, M.T. The Use of Noninvasive Mechanical Ventilation in COPD with Severe Hypercapnic Acidosis. Respir. Med. 2007, 101, 53–61. [Google Scholar] [CrossRef]
- Groenewegen, K.H.; Schols, A.M.W.J.; Wouters, E.F.M. Mortality and Mortality-Related Factors After Hospitalization for Acute Exacerbation of COPD. Chest 2003, 124, 459–467. [Google Scholar] [CrossRef]
- Tiruvoipati, R.; Pilcher, D.; Buscher, H.; Botha, J.; Bailey, M. Effects of Hypercapnia and Hypercapnic Acidosis on Hospital Mortality in Mechanically Ventilated Patients. Crit. Care Med. 2017, 45, e649–e656. [Google Scholar] [CrossRef]
- Hsieh, P.-C.; Wu, Y.-K.; Yang, M.-C.; Su, W.-L.; Kuo, C.-Y.; Lan, C.-C. Deciphering the Role of Damage-Associated Molecular Patterns and Inflammatory Responses in Acute Lung Injury. Life Sci. 2022, 305, 120782. [Google Scholar] [CrossRef] [PubMed]
- Morales-Quinteros, L.; Camprubí-Rimblas, M.; Bringué, J.; Bos, L.D.; Schultz, M.J.; Artigas, A. The Role of Hypercapnia in Acute Respiratory Failure. Intensive Care Med. Exp. 2019, 7, 39. [Google Scholar] [CrossRef]
- Stewart, T.E.; Meade, M.O.; Cook, D.J.; Granton, J.T.; Hodder, R.V.; Lapinsky, S.E.; Mazer, C.D.; McLean, R.F.; Rogovein, T.S.; Schouten, B.D.; et al. Evaluation of a Ventilation Strategy to Prevent Barotrauma in Patients at High Risk for Acute Respiratory Distress Syndrome. Pressure- and Volume-Limited Ventilation Strategy Group. N. Engl. J. Med. 1998, 338, 355–361. [Google Scholar] [CrossRef]
- Ghosh, S.; Hayden, M.S. New Regulators of NF-κB in Inflammation. Nat. Rev. Immunol. 2008, 8, 837–848. [Google Scholar] [CrossRef]
- Morales Quinteros, L.; Bringué Roque, J.; Kaufman, D.; Artigas Raventós, A. Importance of Carbon Dioxide in the Critical Patient: Implications at the Cellular and Clinical Levels. Med. Intensiv. 2019, 43, 234–242. [Google Scholar] [CrossRef]
- Vadász, I.; Hubmayr, R.D.; Nin, N.; Sporn, P.H.S.; Sznajder, J.I. Hypercapnia: A Nonpermissive Environment for the Lung. Am. J. Respir. Cell Mol. Biol. 2012, 46, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, Y.; Osaki, K.; Kanamoto, M.; Nakao, Y.; Takahashi, E.; Higuchi, T.; Kamata, H. Distinct B Subunits of PP2A Regulate the NF-κB Signalling Pathway through Dephosphorylation of IKKβ, IκBα and RelA. FEBS Lett. 2017, 591, 4083–4094. [Google Scholar] [CrossRef]
- Hamacher, J.; Hadizamani, Y.; Borgmann, M.; Mohaupt, M.; Männel, D.N.; Moehrlen, U.; Lucas, R.; Stammberger, U. Cytokine–Ion Channel Interactions in Pulmonary Inflammation. Front. Immunol. 2018, 8, 1644. [Google Scholar] [CrossRef] [PubMed]
- Baloğlu, E.; Mairbäurl, H. In Search of a Sensor: How Does CO2 Regulate Alveolar Ion Transport? Am. J. Respir. Cell Mol. Biol. 2021, 65, 571–572. [Google Scholar] [CrossRef]
- Vadász, I.; Sznajder, J.I. Gas Exchange Disturbances Regulate Alveolar Fluid Clearance during Acute Lung Injury. Front. Immunol. 2017, 8, 757. [Google Scholar] [CrossRef] [PubMed]
- Tricco, A.C.; Lillie, E.; Zarin, W.; O’Brien, K.K.; Colquhoun, H.; Levac, D.; Moher, D.; Peters, M.D.J.; Horsley, T.; Weeks, L.; et al. PRISMA Extension for Scoping Reviews (PRISMA-ScR): Checklist and Explanation. Ann. Intern. Med. 2018, 169, 467–473. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).