
Insights into the Molecular Mechanisms and Signaling Pathways of Epithelial to Mesenchymal Transition (EMT) in the Pathophysiology of Endometriosis


Abstract
1. Introduction
2. Review Methods
3. From Discovery to Classification: Understanding EMT Subtypes
4. Molecular Drivers and Mechanisms of EMT in Endometriosis
4.1. Factors That Trigger EMT in Endometriosis
4.1.1. Growth Factors
4.1.2. Hormonal Regulation
4.1.3. Environmental and Metabolic Factors
4.1.4. Lipid Signaling
4.1.5. Genetic and Epigenetic Modulators
4.2. EMT-Related Transcription Factors in Endometriosis
4.3. Molecular Alteration in EMT in Endometriosis
4.3.1. Cadherin Switch
4.3.2. Loss of Cytokeratin
4.3.3. Upregulation of Vimentin
4.3.4. Suppression of Claudins
5. Fibrosis
6. Lesional Differences
6.1. Lesional Patterns of EMT Expression
6.2. Microenvironmental Influences on EMT Dynamics
6.3. Molecular Feedback Loops and Lesion-Specific Regulation
6.4. Intra-Lesional Heterogeneity
7. Biomarkers
7.1. EMT-Associated Serum microRNAs
7.1.1. miR-17-5p, miR-20a, and miR-22
7.1.2. miR-200 Family
7.1.3. Combined Serum miRNA Panels
7.2. EMT-Related Serum Protein Markers
8. Emerging Interventions That Target EMT
8.1. Isoflavonoids
8.2. Plant Alkaloids
8.3. Terpenes
8.4. Polysaccharides
8.5. Biguanides
8.6. S1P Receptor Modulators
9. Conclusions
10. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| αSMA | Alpha smooth muscle actin |
| Akt | Protein kinase B |
| ARID1A | AT-rich interaction domain-containing protein 1A |
| BMPs | Bone morphogenetic proteins |
| CK | Cytokeratin |
| circRNA | Circular RNA |
| CXCL12 | Chemokine ligand 12 |
| CXCR4 | Chemokine receptor 4 |
| DIE | Deeply infiltrating endometriosis |
| ECM | Extracellular matrix |
| EMT | Epithelial-to-mesenchymal transition |
| EpCAM | Epithelial cell adhesion molecule |
| E2 | 17Î2 estradiol |
| FGFs | Fibroblast growth factors |
| FOXC2 | Forkhead box protein C2 |
| FMT | Fibroblast-to-myofibroblast transition |
| GSK3β | Glycogen synthase kinase 3 beta |
| HGF | Hepatocyte growth factor |
| HIF | Hypoxia inducible factor |
| HMGA2 | High mobility group AT hook 2 |
| IGF1 | Insulin-like growth factor 1 |
| IL | Interleukin |
| lncRNA | Long non-coding RNA |
| MALAT1 | Metastasis-associated lung adenocarcinoma transcript 1 |
| MAPK | Mitogen-activated protein kinase |
| MET | Mesenchymal-to-epithelial transition |
| miRNA | MicroRNA |
| MMT | Mesothelial-to-mesenchymal transition |
| mTOR | Mechanistic target of rapamycin |
| NFκB | Nuclear factor kappa B |
| NICD | Notch intracellular domain |
| OMA | Ovarian endometrioma |
| PDGF | Platelet-derived growth factor |
| PI3K | Phosphoinositide 3 kinase |
| PR | Progesterone receptor |
| PTEN | Phosphatase and tensin homolog |
| ROCK | Rho-associated coiled-coil containing protein kinase |
| S1P | Sphingosine 1 phosphate |
| S1PR | Sphingosine 1 phosphate receptor |
| SCID | Severe combined immunodeficient |
| SMAD | Mothers against decapentaplegic homolog |
| Snail SNAI1 | Zinc-finger protein SNAI1 |
| Slug SNAI2 | Zinc-finger protein SNAI2 |
| SP | Superficial peritoneal endometriosis |
| TGF-β | Transforming growth factor beta |
| TWIST | Twist-related protein |
| VEGF | Vascular endothelial growth factor |
| vWF | von Willebrand factor |
| ZEB1 ZEB2 | Zinc-finger E-box-binding homeobox 1 Zinc-finger E-box-binding homeobox 2 |
References
- Smolarz, B.; Szyłło, K.; Romanowicz, H. Endometriosis: Epidemiology, Classification, Pathogenesis, Treatment and Genetics (Review of Literature). Int. J. Mol. Sci. 2021, 22, 10554. [Google Scholar] [CrossRef] [PubMed]
- Giudice, L.C.; Kao, L.C. Endometriosis. Lancet 2004, 364, 1789–1799. [Google Scholar] [CrossRef]
- Adilbayeva, A.; Kunz, J. Pathogenesis of Endometriosis and Endometriosis-Associated Cancers. Int. J. Mol. Sci. 2024, 25, 7624. [Google Scholar] [CrossRef]
- Bailey, F.; Gaughran, J.; Mitchell, S.; Ovadia, C.; Holland, T.K. Diagnosis of superficial endometriosis on transvaginal ultrasound by visualization of peritoneum of pouch of Douglas. Ultrasound Obstet. Gynecol. 2024, 63, 105–112. [Google Scholar] [CrossRef]
- D’Hooghe, T.M.; Debrock, S.; Hill, J.A.; Meuleman, C. Endometriosis and subfertility: Is the relationship resolved? Semin. Reprod. Med. 2003, 21, 243–254. [Google Scholar] [CrossRef]
- Nisolle, M.; Donnez, J. Peritoneal endometriosis, ovarian endometriosis, and adenomyotic nodules of the rectovaginal septum are three different entities. Fertil. Steril. 1997, 68, 585–596. [Google Scholar] [CrossRef]
- Koninckx, P.R.; Meuleman, C.; Demeyere, S.; Lesaffre, E.; Cornillie, F.J. Suggestive evidence that pelvic endometriosis is a progressive disease, whereas deeply infiltrating endometriosis is associated with pelvic pain. Fertil. Steril. 1991, 55, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Lamceva, J.; Uljanovs, R.; Strumfa, I. The Main Theories on the Pathogenesis of Endometriosis. Int. J. Mol. Sci. 2023, 24, 4254. [Google Scholar] [CrossRef]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Ong, S.L.; Tran, L.M.; Jing, Z.; Liu, B.; Park, S.J.; Huang, Z.L.; Walser, T.C.; Heinrich, E.L.; Lee, G.; et al. Chronic IL-1β-induced inflammation regulates epithelial-to-mesenchymal transition memory phenotypes via epigenetic modifications in non-small cell lung cancer. Sci. Rep. 2020, 10, 377. [Google Scholar] [CrossRef]
- Fontana, R.; Mestre-Farrera, A.; Yang, J. Update on Epithelial-Mesenchymal Plasticity in Cancer Progression. Annu. Rev. Pathol. 2024, 19, 133–156. [Google Scholar] [CrossRef]
- Yao, W.; Wang, Z.; Ma, H.; Lin, Y.; Liu, X.; Li, P.; He, X. Epithelial-mesenchymal plasticity (EMP) in wound healing: Exploring EMT mechanisms, regulatory network, and therapeutic opportunities. Heliyon 2024, 10, e34269. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Trelstad, R.L.; Hay, E.D.; Revel, J.D. Cell contact during early morphogenesis in the chick embryo. Dev. Biol. 1967, 16, 78–106. [Google Scholar] [CrossRef] [PubMed]
- Greenburg, G.; Hay, E.D. Epithelia suspended in collagen gels can lose polarity and express characteristics of migrating mesenchymal cells. J. Cell Biol. 1982, 95, 333–339. [Google Scholar] [CrossRef]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef]
- Gaetje, R.; Kotzian, S.; Herrmann, G.; Baumann, R.; Starzinski-Powitz, A. Nonmalignant epithelial cells, potentially invasive in human endometriosis, lack the tumor suppressor molecule E-cadherin. Am. J. Pathol. 1997, 150, 461–467. [Google Scholar] [PubMed]
- Zeitvogel, A.; Baumann, R.; Starzinski-Powitz, A. Identification of an invasive, N-cadherin-expressing epithelial cell type in endometriosis using a new cell culture model. Am. J. Pathol. 2001, 159, 1839–1852. [Google Scholar] [CrossRef]
- Matsuzaki, S.; Darcha, C. Epithelial to mesenchymal transition-like and mesenchymal to epithelial transition-like processes might be involved in the pathogenesis of pelvic endometriosis. Hum. Reprod. 2012, 27, 712–721. [Google Scholar] [CrossRef] [PubMed]
- Han, S.J.; Hawkins, S.M.; Begum, K.; Jung, S.Y.; Kovanci, E.; Qin, J.; Lydon, J.P.; DeMayo, F.J.; O’Malley, B.W. A new isoform of steroid receptor coactivator-1 is crucial for pathogenic progression of endometriosis. Nat. Med. 2012, 18, 1102–1111. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Massague, J. TGF-beta in developmental and fibrogenic EMTs. Semin. Cancer Biol. 2022, 86, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Martin, B.L. Mesoderm induction and patterning: Insights from neuromesodermal progenitors. Semin. Cell Dev. Biol. 2022, 127, 37–45. [Google Scholar] [CrossRef]
- Yang, Y.M.; Yang, W.X. Epithelial-to-mesenchymal transition in the development of endometriosis. Oncotarget 2017, 8, 41679–41689. [Google Scholar] [CrossRef]
- Zubrzycka, A.; Migdalska-Sęk, M.; Jędrzejczyk, S.; Brzeziańska-Lasota, E. The Expression of TGF-β1, SMAD3, ILK and miRNA-21 in the Ectopic and Eutopic Endometrium of Women with Endometriosis. Int. J. Mol. Sci. 2023, 24, 2453. [Google Scholar] [CrossRef]
- Jin, P.; Cai, J.; Chen, N.; Liu, Y.; Zhao, H.; Wang, Y.; Chen, J.; Li, M.; Xiao, T.; Shan, C.; et al. TGF-β/snail-mediated epithelial-to-mesenchymal transition disrupts estradiol metabolism through suppressing the HSD17B2 expression in endometriotic epithelial cells. Biochem. Biophys. Res. Commun. 2025, 771, 151964. [Google Scholar] [CrossRef]
- Anchan, M.M.; Kalthur, G.; Datta, R.; Majumdar, K.; P, K.; Dutta, R. Unveiling the fibrotic puzzle of endometriosis: An overlooked concern calling for prompt action. F1000Research 2024, 13, 721. [Google Scholar] [CrossRef]
- Yan, D.; Liu, X.; Xu, H.; Guo, S.W. Mesothelial Cells Participate in Endometriosis Fibrogenesis Through Platelet-Induced Mesothelial-Mesenchymal Transition. J. Clin. Endocrinol. Metab. 2020, 105, e4124–e4147. [Google Scholar] [CrossRef]
- Guo, S.W.; Du, Y.; Liu, X. Platelet-derived TGF-β1 mediates the down-modulation of NKG2D expression and may be responsible for impaired natural killer (NK) cytotoxicity in women with endometriosis. Hum. Reprod. 2016, 31, 1462–1474. [Google Scholar] [CrossRef]
- Labelle, M.; Begum, S.; Hynes, R.O. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell 2011, 20, 576–590. [Google Scholar] [CrossRef] [PubMed]
- Arablou, T.; Aryaeian, N.; Khodaverdi, S.; Kolahdouz-Mohammadi, R.; Moradi, Z.; Rashidi, N.; Delbandi, A.A. The effects of resveratrol on the expression of VEGF, TGF-β, and MMP-9 in endometrial stromal cells of women with endometriosis. Sci. Rep. 2021, 11, 6054. [Google Scholar] [CrossRef]
- Zhuang, J.; Lu, Q.; Shen, B.; Huang, X.; Shen, L.; Zheng, X.; Huang, R.; Yan, J.; Guo, H. TGFβ1 secreted by cancer-associated fibroblasts induces epithelial-mesenchymal transition of bladder cancer cells through lncRNA-ZEB2NAT. Sci. Rep. 2015, 5, 11924. [Google Scholar] [CrossRef]
- Song, J. EMT or apoptosis: A decision for TGF-beta. Cell Res. 2007, 17, 289–290. [Google Scholar] [CrossRef]
- Sadeghi-Ardebili, M.; Hasannia, S.; Dabirmanesh, B.; Khavari-Nejad, R.A. Functional characterization of the dimeric form of PDGF-derived fusion peptide fabricated based on theoretical arguments. Sci. Rep. 2024, 14, 1003. [Google Scholar] [CrossRef] [PubMed]
- Surrey, E.S.; Halme, J. Effect of platelet-derived growth factor on endometrial stromal cell proliferation in vitro: A model for endometriosis? Fertil. Steril. 1991, 56, 672–679. [Google Scholar] [CrossRef]
- Mohagheghian Yaghoubi, H.; Samadi, M.; Tajik, N.; Babaheidarian, P.; Movahedinia, S.; Rashidi, N.; Delbandi, A.A. Immunomodulatory effects of vitamin D3 on gene expression of MDGF, EGF and PDGFB in endometriosis. Reprod. Biomed. Online 2020, 41, 782–789. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Hou, X.; Xia, J.; Qian, X.; Miele, L.; Sarkar, F.H.; Wang, Z. Emerging roles of PDGF-D in EMT progression during tumorigenesis. Cancer Treat. Rev. 2013, 39, 640–646. [Google Scholar] [CrossRef]
- Lih Yuan, T.; Sulaiman, N.; Nur Azurah, A.G.; Maarof, M.; Rabiatul Adawiyah, R.; Yazid, M.D. Oestrogen-induced epithelial-mesenchymal transition (EMT) in endometriosis: Aetiology of vaginal agenesis in Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome. Front. Physiol. 2022, 13, 937988. [Google Scholar] [CrossRef]
- Wu, R.F.; Chen, Z.X.; Zhou, W.D.; Li, Y.Z.; Huang, Z.X.; Lin, D.C.; Ren, L.L.; Chen, Q.X.; Chen, Q.H. High expression of ZEB1 in endometriosis and its role in 17β-estradiol-induced epithelial-mesenchymal transition. Int. J. Clin. Exp. Pathol. 2018, 11, 4744–4758. [Google Scholar]
- Du, Y.; Zhang, Z.; Xiong, W.; Li, N.; Liu, H.; He, H.; Li, Q.; Liu, Y.; Zhang, L. Estradiol promotes EMT in endometriosis via MALAT1/miR200s sponge function. Reproduction 2019, 157, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Oviedo, P.J.; Sobrino, A.; Laguna-Fernandez, A.; Novella, S.; Tarín, J.J.; García-Pérez, M.-A.; Sanchís, J.; Cano, A.; Hermenegildo, C. Estradiol induces endothelial cell migration and proliferation through estrogen receptor-enhanced RhoA/ROCK pathway. Mol. Cell. Endocrinol. 2011, 335, 96–103. [Google Scholar] [CrossRef]
- Ono, Y.J.; Hayashi, M.; Tanabe, A.; Hayashi, A.; Kanemura, M.; Terai, Y.; Ohmichi, M. Estradiol-mediated hepatocyte growth factor is involved in the implantation of endometriotic cells via the mesothelial-to-mesenchymal transition in the peritoneum. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E950–E959. [Google Scholar] [CrossRef]
- Lu, J.; Ling, X.; Liu, L.; Jiang, A.; Ren, C.; Lu, C.; Yu, Z. Emerging hallmarks of endometriosis metabolism: A promising target for the treatment of endometriosis. Biochim. Biophys. Acta BBA-Mol. Cell Res. 2023, 1870, 119381. [Google Scholar] [CrossRef]
- Ma, L.; Andrieu, T.; McKinnon, B.; Duempelmann, L.; Peng, R.W.; Wotzkow, C.; Müller, C.; Mueller, M.D. Epithelial-to-mesenchymal transition contributes to the downregulation of progesterone receptor expression in endometriosis lesions. J. Steroid Biochem. Mol. Biol. 2021, 212, 105943. [Google Scholar] [CrossRef]
- Zhang, P.; Wang, G. Progesterone Resistance in Endometriosis: Current Evidence and Putative Mechanisms. Int. J. Mol. Sci. 2023, 24, 6992. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, K.Y.; Lin, S.C.; Wu, M.H.; Tsai, S.J. Pathological functions of hypoxia in endometriosis. Front. Biosci. Elite Ed. 2015, 7, 309–321. [Google Scholar] [CrossRef]
- Filippi, I.; Carrarelli, P.; Luisi, S.; Batteux, F.; Chapron, C.; Naldini, A.; Petraglia, F. Different Expression of Hypoxic and Angiogenic Factors in Human Endometriotic Lesions. Reprod. Sci. 2016, 23, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Liu, Y.; Xiong, W.; Zhang, L.; Liu, H.; Du, Y.; Li, N. Hypoxia-inducible factor 1α-induced epithelial-mesenchymal transition of endometrial epithelial cells may contribute to the development of endometriosis. Hum. Reprod. 2016, 31, 1327–1338. [Google Scholar] [CrossRef]
- Zhang, F.; Liu, X.L.; Wang, W.; Dong, H.L.; Xia, Y.F.; Ruan, L.P.; Liu, L.P. Expression of MMIF, HIF-1α and VEGF in Serum and Endometrial Tissues of Patients with Endometriosis. Curr. Med. Sci. 2018, 38, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Tirpe, A.A.; Gulei, D.; Ciortea, S.M.; Crivii, C.; Berindan-Neagoe, I. Hypoxia: Overview on Hypoxia-Mediated Mechanisms with a Focus on the Role of HIF Genes. Int. J. Mol. Sci. 2019, 20, 6140. [Google Scholar] [CrossRef]
- Zhu, H.; Wang, D.; Zhang, L.; Xie, X.; Wu, Y.; Liu, Y.; Shao, G.; Su, Z. Upregulation of autophagy by hypoxia-inducible factor-1α promotes EMT and metastatic ability of CD133+ pancreatic cancer stem-like cells during intermittent hypoxia. Oncol. Rep. 2014, 32, 935–942. [Google Scholar] [CrossRef] [PubMed]
- Kusama, K.; Fukushima, Y.; Yoshida, K.; Sakakibara, H.; Tsubata, N.; Yoshie, M.; Kojima, J.; Nishi, H.; Tamura, K. Endometrial epithelial-mesenchymal transition (EMT) by menstruation-related inflammatory factors during hypoxia. Mol. Hum. Reprod. 2021, 27, gaab036. [Google Scholar] [CrossRef]
- Bowers, L.W.; Rossi, E.L.; McDonell, S.B.; Doerstling, S.S.; Khatib, S.A.; Lineberger, C.G.; Albright, J.E.; Tang, X.; deGraffenried, L.A.; Hursting, S.D. Leptin Signaling Mediates Obesity-Associated CSC Enrichment and EMT in Preclinical TNBC Models. Mol. Cancer Res. 2018, 16, 869–879. [Google Scholar] [CrossRef]
- Villanueva-Duque, A.; Zuniga-Eulogio, M.D.; Dena-Beltran, J.; Castaneda-Saucedo, E.; Calixto-Galvez, M.; Mendoza-Catalán, M.A.; Ortuno-Pineda, C.; Navarro-Tito, N. Leptin induces partial epithelial-mesenchymal transition in a FAK-ERK dependent pathway in MCF10A mammary non-tumorigenic cells. Int. J. Clin. Exp. Pathol. 2017, 10, 10334–10342. [Google Scholar]
- Akrida, I.; Papadaki, H. Adipokines and epithelial-mesenchymal transition (EMT) in cancer. Mol. Cell. Biochem. 2023, 478, 2419–2433. [Google Scholar] [CrossRef]
- De Angulo, A.; Travis, P.; Galvan, G.C.; Jolly, C.; deGraffenried, L. Obesity-Modified CD4+ T-Cells Promote an Epithelial-Mesenchymal Transition Phenotype in Prostate Cancer Cells. Nutr. Cancer 2022, 74, 650–659. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Zhang, C. Role of the gut microbiota in the pathogenesis of endometriosis: A review. Front. Microbiol. 2024, 15, 1363455. [Google Scholar] [CrossRef] [PubMed]
- Gupta, I.; Pedersen, S.; Vranic, S.; Al Moustafa, A.E. Implications of Gut Microbiota in Epithelial-Mesenchymal Transition and Cancer Progression: A Concise Review. Cancers 2022, 14, 2964. [Google Scholar] [CrossRef]
- Zhang, S.; Li, C.; Liu, J.; Geng, F.; Shi, X.; Li, Q.; Lu, Z.; Pan, Y. Fusobacterium nucleatum promotes epithelial-mesenchymal transiton through regulation of the lncRNA MIR4435-2HG/miR-296-5p/Akt2/SNAI1 signaling pathway. FEBS J. 2020, 287, 4032–4047. [Google Scholar] [CrossRef]
- Miller, A.V.; Alvarez, S.E.; Spiegel, S.; Lebman, D.A. Sphingosine kinases and sphingosine-1-phosphate are critical for transforming growth factor beta-induced extracellular signal-regulated kinase 1 and 2 activation and promotion of migration and invasion of esophageal cancer cells. Mol. Cell. Biol. 2008, 28, 4142–4151. [Google Scholar] [CrossRef]
- Bernacchioni, C.; Capezzuoli, T.; Vannuzzi, V.; Malentacchi, F.; Castiglione, F.; Cencetti, F.; Ceccaroni, M.; Donati, C.; Bruni, P.; Petraglia, F. Sphingosine 1-phosphate receptors are dysregulated in endometriosis: Possible implication in transforming growth factor beta-induced fibrosis. Fertil. Steril. 2021, 115, 501–511. [Google Scholar] [CrossRef]
- Ono, Y.; Kawakita, T.; Yoshino, O.; Sato, E.; Kano, K.; Ohba, M.; Okuno, T.; Ito, M.; Koga, K.; Honda, M.; et al. Sphingosine 1-Phosphate (S1P) in the Peritoneal Fluid Skews M2 Macrophage and Contributes to the Development of Endometriosis. Biomedicines 2021, 9, 1519. [Google Scholar] [CrossRef]
- Bernacchioni, C.; Rossi, M.; Vannuzzi, V.; Prisinzano, M.; Seidita, I.; Raeispour, M.; Muccilli, A.; Castiglione, F.; Bruni, P.; Petraglia, F.; et al. Sphingosine-1-phosphate receptor 3 is a non-hormonal target to counteract endometriosis-associated fibrosis. Fertil. Steril. 2024, 121, 631–641. [Google Scholar] [CrossRef]
- Santulli, P.; Marcellin, L.; Noel, J.C.; Borghese, B.; Fayt, I.; Vaiman, D.; Chapron, C.; Mehats, C. Sphingosine pathway deregulation in endometriotic tissues. Fertil. Steril. 2012, 97, 904–911. [Google Scholar] [CrossRef]
- Fedorova, O.; Parfenyev, S.; Daks, A.; Shuvalov, O.; Barlev, N.A. The Role of PTEN in Epithelial-Mesenchymal Transition. Cancers 2022, 14, 3786. [Google Scholar] [CrossRef]
- Ma, X.; Hui, Y.; Lin, L.; Wu, Y.; Zhang, X.; Qin, X. Possible relevance of tumor-related genes mutation to malignant transformation of endometriosis. Eur. J. Gynaecol. Oncol. 2016, 37, 89–94. [Google Scholar] [PubMed]
- Kitai, H.; Ebi, H. Key roles of EMT for adaptive resistance to MEK inhibitor in KRAS mutant lung cancer. Small GTPases 2017, 8, 172–176. [Google Scholar] [CrossRef]
- Anglesio, M.S.; Papadopoulos, N.; Ayhan, A.; Nazeran, T.M.; Noe, M.; Horlings, H.M.; Lum, A.; Jones, S.; Senz, J.; Seckin, T.; et al. Cancer-Associated Mutations in Endometriosis without Cancer. N. Engl. J. Med. 2017, 376, 1835–1848. [Google Scholar] [CrossRef]
- Wilson, M.R.; Reske, J.J.; Holladay, J.; Wilber, G.E.; Rhodes, M.; Koeman, J.; Adams, M.; Johnson, B.; Su, R.W.; Joshi, N.R.; et al. ARID1A and PI3-kinase pathway mutations in the endometrium drive epithelial transdifferentiation and collective invasion. Nat. Commun. 2019, 10, 3554. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Wang, D.; Chen, S.; Yang, Q. The role of miR-34c-5p/Notch in epithelial-mesenchymal transition (EMT) in endometriosis. Cell. Signal. 2020, 72, 109666. [Google Scholar] [CrossRef] [PubMed]
- Cavallari, I.; Ciccarese, F.; Sharova, E.; Urso, L.; Raimondi, V.; Silic-Benussi, M.; D’Agostino, D.M.; Ciminale, V. The miR-200 Family of microRNAs: Fine Tuners of Epithelial-Mesenchymal Transition and Circulating Cancer Biomarkers. Cancers 2021, 13, 5874. [Google Scholar] [CrossRef]
- Rekker, K.; Saare, M.; Roost, A.M.; Kaart, T.; Sõritsa, D.; Karro, H.; Sõritsa, A.; Simón, C.; Salumets, A.; Peters, M. Circulating miR-200-family micro-RNAs have altered plasma levels in patients with endometriosis and vary with blood collection time. Fertil. Steril. 2015, 104, 938–946.e2. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Chen, Y.; Zhao, Y.; Xu, C.; Zhang, A.; Zhang, Q.; Wang, D.; He, J.; Hua, W.; Duan, P. miR-200c suppresses endometriosis by targeting MALAT1 in vitro and in vivo. Stem Cell Res. Ther. 2017, 8, 251. [Google Scholar] [CrossRef]
- Liu, Y.; Lu, C.; Fan, L.; Wang, J.; Li, T.; Liu, Z.; Sheng, J.; Qian, R.; Duan, A.; Lu, D. MiR-199a-5p Targets ZEB1 to Inhibit the Epithelial-Mesenchymal Transition of Ovarian Ectopic Endometrial Stromal Cells via PI3K/Akt/mTOR Signal Pathway In Vitro and In Vivo. Reprod. Sci. 2020, 27, 110–118. [Google Scholar] [CrossRef]
- Meng, X.; Liu, J.; Wang, H.; Chen, P.; Wang, D. MicroRNA-126-5p downregulates BCAR3 expression to promote cell migration and invasion in endometriosis. Mol. Cell. Endocrinol. 2019, 494, 110486. [Google Scholar] [CrossRef]
- He, X.; Liu, N.; Mu, T.; Lu, D.; Jia, C.; Wang, S.; Yin, C.; Liu, L.; Zhou, L.; Huang, X.; et al. Oestrogen induces epithelial-mesenchymal transition in endometriosis via circ_0004712/miR-148a-3p sponge function. J. Cell. Mol. Med. 2020, 24, 9658–9666. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Liu, M. CircATRNL1 increases acid-sensing ion channel 1 to advance epithelial-mesenchymal transition in endometriosis by binding to microRNA-103a-3p. Reprod. Biol. 2022, 22, 100643. [Google Scholar] [CrossRef] [PubMed]
- Kazmi, I.; Alharbi, K.S.; Al-Abbasi, F.A.; Almalki, W.H.; G, S.K.; Yasmeen, A.; Khan, A.; Gupta, G. Role of Epithelial-to-Mesenchymal Transition Markers in Different Stages of Endometriosis: Expression of the Snail, Slug, ZEB1, and Twist Genes. Crit. Rev. Eukaryot. Gene Expr. 2021, 31, 89–95. [Google Scholar] [CrossRef]
- Lin, L.L.; Makwana, S.; Chen, M.; Wang, C.M.; Gillette, L.H.; Huang, T.H.; Burney, R.O.; Nicholson, B.J.; Kirma, N.B. Cellular junction and mesenchymal factors delineate an endometriosis-specific response of endometrial stromal cells to the mesothelium. Mol. Cell. Endocrinol. 2022, 539, 111481. [Google Scholar] [CrossRef]
- Serrano-Gomez, S.J.; Maziveyi, M.; Alahari, S.K. Regulation of epithelial-mesenchymal transition through epigenetic and post-translational modifications. Mol. Cancer 2016, 15, 18. [Google Scholar] [CrossRef]
- Bartley, J.; Jülicher, A.; Hotz, B.; Mechsner, S.; Hotz, H. Epithelial to mesenchymal transition (EMT) seems to be regulated differently in endometriosis and the endometrium. Arch. Gynecol. Obstet. 2014, 289, 871–881. [Google Scholar] [CrossRef]
- Furuya, M.; Masuda, H.; Hara, K.; Uchida, H.; Sato, K.; Sato, S.; Asada, H.; Maruyama, T.; Yoshimura, Y.; Katabuchi, H.; et al. ZEB1 expression is a potential indicator of invasive endometriosis. Acta Obstet. Gynecol. Scand. 2017, 96, 1128–1135. [Google Scholar] [CrossRef]
- Hugo, H.J.; Kokkinos, M.I.; Blick, T.; Ackland, M.L.; Thompson, E.W.; Newgreen, D.F. Defining the E-cadherin repressor interactome in epithelial-mesenchymal transition: The PMC42 model as a case study. Cells Tissues Organs 2011, 193, 23–40. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Tang, M.; Wu, L. Expression of combined interference of slug and FoxC2 in endometrial carcinoma and its clinicopathological relationship. Transl. Cancer Res. 2020, 9, 5268–5280. [Google Scholar] [CrossRef]
- Hao, M.; Liu, X.; Guo, S.W. Activation of α7 nicotinic acetylcholine receptor retards the development of endometriosis. Reprod. Biol. Endocrinol. 2022, 20, 85. [Google Scholar] [CrossRef] [PubMed]
- Konrad, L.; Dietze, R.; Riaz, M.A.; Scheiner-Bobis, G.; Behnke, J.; Horné, F.; Hoerscher, A.; Reising, C.; Meinhold-Heerlein, I. Epithelial-Mesenchymal Transition in Endometriosis-When Does It Happen? J. Clin. Med. 2020, 9, 1915. [Google Scholar] [CrossRef] [PubMed]
- Loh, C.Y.; Chai, J.Y.; Tang, T.F.; Wong, W.F.; Sethi, G.; Shanmugam, M.K.; Chong, P.P.; Looi, C.Y. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells 2019, 8, 1118. [Google Scholar] [CrossRef]
- Cai, X.; Shen, M.; Liu, X.; Guo, S.W. Reduced Expression of Eukaryotic Translation Initiation Factor 3 Subunit e and Its Possible Involvement in the Epithelial-Mesenchymal Transition in Endometriosis. Reprod. Sci. 2018, 25, 102–109. [Google Scholar] [CrossRef]
- Liu, D.; Yang, N.; Liang, Y.; Chen, M.; Yang, F.; Liu, L.; Yao, S. Increased expression of epithelial cell adhesion molecule and its possible role in epithelial-mesenchymal transition in endometriosis. J. Obstet. Gynaecol. Res. 2020, 46, 2066–2075. [Google Scholar] [CrossRef]
- Alieva, I.B.; Shakhov, A.S.; Dayal, A.A.; Churkina, A.S.; Parfenteva, O.I.; Minin, A.A. Unique Role of Vimentin in the Intermediate Filament Proteins Family. Biochemistry 2024, 89, 726–736. [Google Scholar]
- Usman, S.; Waseem, N.H.; Nguyen, T.K.N.; Mohsin, S.; Jamal, A.; Teh, M.T.; Waseem, A. Vimentin Is at the Heart of Epithelial Mesenchymal Transition (EMT) Mediated Metastasis. Cancers 2021, 13, 4985. [Google Scholar] [CrossRef]
- Chang, L.C.; Chiang, Y.F.; Chen, H.Y.; Huang, Y.J.; Liu, A.C.; Hsia, S.M. The Potential Effect of Fucoidan on Inhibiting Epithelial-to-Mesenchymal Transition, Proliferation, and Increase in Apoptosis for Endometriosis Treatment: In Vivo and In Vitro Study. Biomedicines 2020, 8, 528. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Lin, X.; Shi, T.; Tian, Y. miRNA-223 expression in patient-derived eutopic and ectopic endometrial stromal cells and its effect on epithelial-to-mesenchymal transition in endometriosis. Clinics 2022, 77, 100112. [Google Scholar] [CrossRef] [PubMed]
- Horné, F.; Dietze, R.; Berkes, E.; Oehmke, F.; Tinneberg, H.R.; Meinhold-Heerlein, I.; Konrad, L. Impaired Localization of Claudin-11 in Endometriotic Epithelial Cells Compared to Endometrial Cells. Reprod. Sci. 2019, 26, 1181–1192. [Google Scholar] [CrossRef]
- Gaetje, R.; Holtrich, U.; Engels, K.; Kissler, S.; Rody, A.; Karn, T.; Kaufmann, M. Differential expression of claudins in human endometrium and endometriosis. Gynecol. Endocrinol. 2008, 24, 442–449. [Google Scholar] [CrossRef]
- Pan, X.Y.; Li, X.; Weng, Z.P.; Wang, B. Altered expression of claudin-3 and claudin-4 in ectopic endometrium of women with endometriosis. Fertil. Steril. 2009, 91, 1692–1699. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Zhang, W.; You, S.; Cui, X.; Tu, H.; Yi, Q.; Wu, J.; Liu, O. The role of epithelial cells in fibrosis: Mechanisms and treatment. Pharmacol. Res. 2024, 202, 107144. [Google Scholar] [CrossRef]
- Vincent-Mistiaen, Z.I. Epithelial-mesenchymal transition links inflammation and fibrosis in the pathogenesis of endometriosis: A narrative review. F S Rev. 2025, 6, 100089. [Google Scholar] [CrossRef]
- Luo, X.; Wen, S.; Zeng, J.; Liu, J.; Ye, W.; Wu, J.; Huang, S.; Xie, W.; Wen, H.; Sun, Y.; et al. AOPPs induces EMT and fibrosis by activating oxidative stress through ERK/p38 MAPK signaling pathway in endometriosis. Reprod. Biol. 2024, 24, 100950. [Google Scholar] [CrossRef]
- Itoga, T.; Matsumoto, T.; Takeuchi, H.; Yamasaki, S.; Sasahara, N.; Hoshi, T.; Kinoshita, K. Fibrosis and smooth muscle metaplasia in rectovaginal endometriosis. Pathol. Int. 2003, 53, 371–375. [Google Scholar] [CrossRef]
- Vissers, G.; Giacomozzi, M.; Verdurmen, W.; Peek, R.; Nap, A. The role of fibrosis in endometriosis: A systematic review. Hum. Reprod. Update 2024, 30, 706–750. [Google Scholar] [CrossRef]
- Vallée, A.; Lecarpentier, Y. TGF-β in fibrosis by acting as a conductor for contractile properties of myofibroblasts. Cell Biosci. 2019, 9, 98. [Google Scholar] [CrossRef]
- Duan, J.; Liu, X.; Wang, H.; Guo, S.W. The M2a macrophage subset may be critically involved in the fibrogenesis of endometriosis in mice. Reprod. Biomed. Online 2018, 37, 254–268. [Google Scholar] [CrossRef]
- Matsuzaki, S.; Darcha, C. Involvement of the Wnt/β-catenin signaling pathway in the cellular and molecular mechanisms of fibrosis in endometriosis. PLoS ONE 2013, 8, e76808. [Google Scholar] [CrossRef]
- Matsuzaki, S.; Darcha, C.; Pouly, J.L.; Canis, M. Effects of matrix stiffness on epithelial to mesenchymal transition-like processes of endometrial epithelial cells: Implications for the pathogenesis of endometriosis. Sci. Rep. 2017, 7, 44616. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, Q.; Guo, S.W. Histological and Immunohistochemical Characterization of the Similarity and Difference Between Ovarian Endometriomas and Deep Infiltrating Endometriosis. Reprod. Sci. 2018, 25, 329–340. [Google Scholar] [CrossRef]
- Ntzeros, K.; Mavrogianni, D.; Blontzos, N.; Soyhan, N.; Kathopoulis, N.; Papamentzelopoulou, M.S.; Chatzipapas, I.; Protopapas, A. Expression of ZEB1 in different forms of endometriosis: A pilot study. Eur. J. Obstet. Gynecol. Reprod. Biol. 2023, 286, 121–125. [Google Scholar] [CrossRef]
- Donnez, O.; Orellana, R.; Van Kerk, O.; Dehoux, J.P.; Donnez, J.; Dolmans, M.M. Invasion process of induced deep nodular endometriosis in an experimental baboon model: Similarities with collective cell migration? Fertil. Steril. 2015, 104, 491–497.e492. [Google Scholar] [CrossRef] [PubMed]
- Gratton, S.M.; Choudhry, A.J.; Vilos, G.A.; Vilos, A.; Baier, K.; Holubeshen, S.; Medor, M.C.; Mercier, S.; Nguyen, V.; Chen, I. Diagnosis of Endometriosis at Laparoscopy: A Validation Study Comparing Surgeon Visualization with Histologic Findings. J. Obstet. Gynaecol. Can. 2022, 44, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Jia, S.Z.; Yang, Y.; Lang, J.; Sun, P.; Leng, J. Plasma miR-17-5p, miR-20a and miR-22 are down-regulated in women with endometriosis. Hum. Reprod. 2013, 28, 322–330. [Google Scholar] [CrossRef]
- Chen, G.; Guo, J.; Li, W.; Zheng, R.; Shang, H.; Wang, Y. Diagnostic value of the combination of circulating serum miRNAs and CA125 in endometriosis. Medicine 2023, 102, e36339. [Google Scholar] [CrossRef]
- Wang, W.T.; Zhao, Y.N.; Han, B.W.; Hong, S.J.; Chen, Y.Q. Circulating microRNAs identified in a genome-wide serum microRNA expression analysis as noninvasive biomarkers for endometriosis. J. Clin. Endocrinol. Metab. 2013, 98, 281–289. [Google Scholar] [CrossRef]
- Maged, A.M.; Deeb, W.S.; El Amir, A.; Zaki, S.S.; El Sawah, H.; Al Mohamady, M.; Metwally, A.A.; Katta, M.A. Diagnostic accuracy of serum miR-122 and miR-199a in women with endometriosis. Int. J. Gynaecol. Obstet. 2018, 141, 14–19. [Google Scholar] [CrossRef]
- Kim, T.W.; Lee, Y.S.; Yun, N.H.; Shin, C.H.; Hong, H.K.; Kim, H.H.; Cho, Y.B. MicroRNA-17-5p regulates EMT by targeting vimentin in colorectal cancer. Br. J. Cancer 2020, 123, 1123–1130. [Google Scholar] [CrossRef]
- De, S.; Das, S.; Mukherjee, S.; Das, S.; Sengupta Bandyopadhyay, S. Establishment of twist-1 and TGFBR2 as direct targets of microRNA-20a in mesenchymal to epithelial transition of breast cancer cell-line MDA-MB-231. Exp. Cell Res. 2017, 361, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Li, J.; Wang, X.; Meng, S.; Shen, J.; Wang, S.; Xu, X.; Xie, B.; Liu, B.; Xie, L. MiR-22 suppresses epithelial-mesenchymal transition in bladder cancer by inhibiting Snail and MAPK1/Slug/vimentin feedback loop. Cell Death Dis. 2018, 9, 209. [Google Scholar] [CrossRef]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Cela, V.; Malacarne, E.; Obino, M.E.R.; Marzi, I.; Papini, F.; Vergine, F.; Pisacreta, E.; Zappelli, E.; Pietrobono, D.; Scarfò, G.; et al. Exploring Epithelial-Mesenchymal Transition Signals in Endometriosis Diagnosis and In Vitro Fertilization Outcomes. Biomedicines 2021, 9, 1681. [Google Scholar] [CrossRef]
- Kuokkanen, S.; Chen, B.; Ojalvo, L.; Benard, L.; Santoro, N.; Pollard, J.W. Genomic profiling of microRNAs and messenger RNAs reveals hormonal regulation in microRNA expression in human endometrium. Biol. Reprod. 2010, 82, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Kalaitzopoulos, D.R.; Samartzis, N.; Kolovos, G.N.; Mareti, E.; Samartzis, E.P.; Eberhard, M.; Dinas, K.; Daniilidis, A. Treatment of endometriosis: A review with comparison of 8 guidelines. BMC Womens Health 2021, 21, 397. [Google Scholar] [CrossRef]
- Hsu, Y.W.; Chen, H.Y.; Chiang, Y.F.; Chang, L.C.; Lin, P.H.; Hsia, S.M. The effects of isoliquiritigenin on endometriosis in vivo and in vitro study. Phytomedicine 2020, 77, 153214. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.M.; Zhou, Q.M. 3,6-dihydroxyflavone suppresses the epithelial-mesenchymal transition, migration and invasion in endometrial stromal cells by inhibiting the Notch signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 4009–4017. [Google Scholar] [PubMed]
- Huang, S.; Xiao, F.; Guo, S.W.; Zhang, T. Tetramethylpyrazine Retards the Progression and Fibrogenesis of Endometriosis. Reprod. Sci. 2022, 29, 1170–1187. [Google Scholar] [CrossRef]
- Kabil, S.L.; Rashed, H.E.; Mohamed, N.M.; Elwany, N.E. Parthenolide repressed endometriosis induced surgically in rats: Role of PTEN/PI3Kinase/AKT/GSK-3beta/beta-catenin signaling in inhibition of epithelial mesenchymal transition. Life Sci. 2023, 331, 122037. [Google Scholar] [CrossRef]
- Xie, Y.; Kong, W.; Zhao, X.; Luo, D.; Chen, S.; Zhang, H.E.; Ran, Y. Metformin Inhibits the Estrogen-mediated Epithelial-Mesenchymal Transition of Ectopic Endometrial Stromal Cells in Endometriosis. Vivo 2023, 37, 2490–2497. [Google Scholar] [CrossRef]
- Zhang, F.; Peng, M.; Zheng, X.; Wang, X.; Liu, X.; Chen, C.; Lu, Y. Blocking sphingosine 1-phosphate receptor 1 with modulators reduces immune cells infiltration and alleviates endometriosis in mice. Reprod. Biomed. Online 2023, 47, 103304. [Google Scholar] [CrossRef]
- Chen, H.Y.; Huang, T.C.; Shieh, T.M.; Wu, C.H.; Lin, L.C.; Hsia, S.M. Isoliquiritigenin Induces Autophagy and Inhibits Ovarian Cancer Cell Growth. Int. J. Mol. Sci. 2017, 18, 2025. [Google Scholar] [CrossRef]
- Tian, T.; Sun, J.; Wang, J.; Liu, Y.; Liu, H. Isoliquiritigenin inhibits cell proliferation and migration through the PI3K/AKT signaling pathway in A549 lung cancer cells. Oncol. Lett. 2018, 16, 6133–6139. [Google Scholar] [CrossRef]
- Wu, C.H.; Chen, H.Y.; Wang, C.W.; Shieh, T.M.; Huang, T.C.; Lin, L.C.; Wang, K.L.; Hsia, S.M. Isoliquiritigenin induces apoptosis and autophagy and inhibits endometrial cancer growth in mice. Oncotarget 2016, 7, 73432–73447. [Google Scholar] [CrossRef]
- Chen, J.; Chang, H.; Peng, X.; Gu, Y.; Yi, L.; Zhang, Q.; Zhu, J.; Mi, M. 3,6-dihydroxyflavone suppresses the epithelial-mesenchymal transition in breast cancer cells by inhibiting the Notch signaling pathway. Sci. Rep. 2016, 6, 28858. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Chang, H.; Gu, Y.; Chen, J.; Yi, L.; Xie, Q.; Zhu, J.; Zhang, Q.; Mi, M. 3,6-Dihydroxyflavone Suppresses Breast Carcinogenesis by Epigenetically Regulating miR-34a and miR-21. Cancer Prev. Res. 2015, 8, 509–517. [Google Scholar] [CrossRef]
- Liu, J.W.; Cai, M.X.; Xin, Y.; Wu, Q.S.; Ma, J.; Yang, P.; Xie, H.Y.; Huang, D.S. Parthenolide induces proliferation inhibition and apoptosis of pancreatic cancer cells in vitro. J. Exp. Clin. Cancer Res. 2010, 29, 108. [Google Scholar] [CrossRef]
- Zhu, S.M.; Park, Y.R.; Seo, S.Y.; Kim, I.H.; Lee, S.T.; Kim, S.W. Parthenolide inhibits transforming growth factor beta1-induced epithelial-mesenchymal transition in colorectal cancer cells. Intest. Res. 2019, 17, 527–536. [Google Scholar] [CrossRef]
- Rui, X.; Pan, H.F.; Shao, S.L.; Xu, X.M. Anti-tumor and anti-angiogenic effects of Fucoidan on prostate cancer: Possible JAK-STAT3 pathway. BMC Complement. Altern. Med. 2017, 17, 378. [Google Scholar] [CrossRef]
- van Weelden, G.; Bobinski, M.; Okla, K.; van Weelden, W.J.; Romano, A.; Pijnenborg, J.M.A. Fucoidan Structure and Activity in Relation to Anti-Cancer Mechanisms. Mar. Drugs 2019, 17, 32. [Google Scholar] [CrossRef] [PubMed]
- Saisho, Y. Metformin and Inflammation: Its Potential Beyond Glucose-lowering Effect. Endocr. Metab. Immune Disord. Drug Targets 2015, 15, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Zhang, L.; Liu, H.; Li, N.; Du, Y.; He, H.; Zhang, Z.; Liu, Y. E2-mediated EMT by activation of beta-catenin/Snail signalling during the development of ovarian endometriosis. J. Cell. Mol. Med. 2019, 23, 8035–8045. [Google Scholar] [CrossRef] [PubMed]
- Camm, J.; Hla, T.; Bakshi, R.; Brinkmann, V. Cardiac and vascular effects of fingolimod: Mechanistic basis and clinical implications. Am. Heart J. 2014, 168, 632–644. [Google Scholar] [CrossRef]
- Yoshino, O.; Yamada-Nomoto, K.; Kano, K.; Ono, Y.; Kobayashi, M.; Ito, M.; Yoneda, S.; Nakashima, A.; Shima, T.; Onda, T.; et al. Sphingosine 1 Phosphate (S1P) Increased IL-6 Expression and Cell Growth in Endometriotic Cells. Reprod. Sci. 2019, 26, 1460–1467. [Google Scholar] [CrossRef]
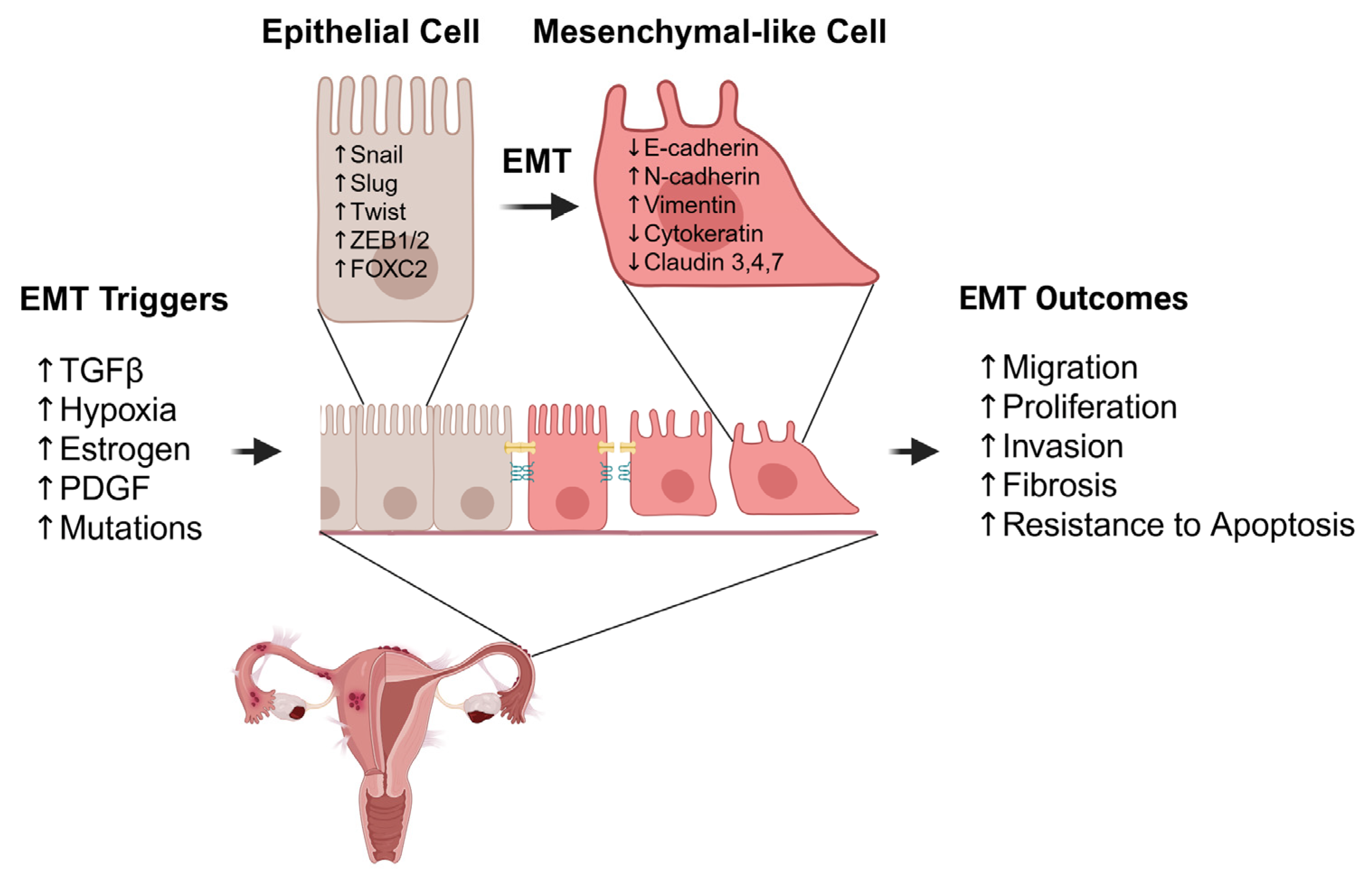

{kind=link}
| Category | Molecule/Factor | Change in Endometriosis | Function in EMT | Reference |
|---|---|---|---|---|
| Key Triggers | TGF-β | ↑ Active in lesions | Master EMT inducer by upregulated EMT-TFs (Snail, ZEB1); involved in fibrosis. | [27,28,29,30] |
| PDGF | ↑ At lesion sites | Platelet-derived signaling promotes invasion under hypoxia. | [36,37] | |
| Estradiol | ↑ Estrogen-rich environment | Upregulates ZEB1, activates RhoA/ROCK and HGF pathways, and promotes EMTEMT gene regulator under stress by activating VEGF, Wnt/β-catenin, and TGF-β1 pathways. | [40,41,42] | |
| Hypoxia (HIF-1α/2α) | ↑ Elevated in ectopic tissue | EMT gene regulator under stress by activating VEGF, Wnt/β-catenin, and TGF-β1 pathways. | [46,47,50] | |
| S1P | ↑ At lesion sites | Cross-talk with factors such as TGF-β. | [60,62] | |
| Genetic mutations in PTEN, KRAS, ARID1A | Frequently mutated in DIE | Promote EMT via loss of suppression or gain of oncogenic signals. | [65,66,67,69] | |
| Transcription Factors | Snail (SNAI1), Slug (SNAI2) | ↑ Upregulated | Induced by TGF-β, hypoxia, estrogen, and repressed by E-cadherin. | [81] |
| ZEB1/ZEB2 | ↑ Upregulated | Targeted by miR-200 family and promotes EMT. | [24] | |
| Twist | ↑ Upregulated | Associated with initiation and invasion of EMT. | [78] | |
| FOXC2 | Likely ↑ | Enhance cell motility. | [83] | |
| Epithelial Markers | E-cadherin | ↓ Downregulated | Repressed by Snail, Slug, ZEB1/2 and weakening of cell–cell adhesion. | [85,86] |
| Cytokeratin | ↓ Downregulated | Reduced expression in ectopic epithelial cells leads to a loss of epithelial structure. | [81,87] | |
| Claudins (e.g., -3, -4, -7) | ↓ Downregulated | Disruption of tight junction proteins in peritoneal lesions contributes to compromised epithelial integrity. | [20] | |
| Mesenchymal Markers | N-cadherin | ↑ Upregulated | Cadherin switches from E- to N-cadherin and promotes cell motility. | [81,87] |
| Vimentin | ↑ Upregulated | Associated with enhanced invasion and fibrosis. | [88,92] |
| Biomarker | Relative Change | AUC (95%CI) | Sensitivity | Specificity | Reference |
|---|---|---|---|---|---|
| miR-17-5p | Decreased | 0.74 (0.58–0.90) | NR | NR | [110] |
| miR-20a | Decreased | 0.79 (0.65–0.93) | NR | NR | [110] |
| miR-22 | Decreased | 0.85 (0.71–0.98) | NR | NR | [110] |
| miR-200a | Decreased | 0.75 (0.62–0.86) | 90.6% | 62.5% | [71,72] |
| miR-200b | Decreased | 0.67 (0.53–0.79) | 90.6% | 45.8% | [71,72] |
| miR-141 | Decreased | 0.71 (0.57–0.82) | 71.9% | 70.8% | [71,72] |
| Decreased | 0.59 (0.51–0.67) | 81.1% | 37.5% | [111] | |
| miR-199a | Increased | 0.83 (0.73–0.92) | 78.3% | 76.0% | [112] |
| Increased | 0.62 (0.55–0.71) | 54.6% | 98.6% | [111] | |
| Increased | 1.00 (no range) | 91.4% | 100% | [113] | |
| miR-122 | Increased | 0.84 (0.75–0.92) | 80.0% | 76.0% | [112] |
| Increased | 0.72 (0.61–0.82) | 45.8% | 97.4% | [111] | |
| Increased | 0.96 (no range) | 95.6% | 100% | [106] | |
| miR-145 | Decreased | 0.88 (0.81–0.95) | 70.0% | 96.0% | [112] |
| Decreased | 0.76 (0.68–0.85) | 85.3% | 38.1% | [111] | |
| miR-141 | Decreased | 0.85 (0.77–0.93) | 71.7% | 96.0% | [112] |
| miR-542-3p | Decreased | 0.85 (0.77–0.94) | 79.7% | 92.0% | [112] |
| E-cadherin | Decreased | 0.63 (0.33–0.81) | NR | NR | [107] |
| N-Cadherin | Increased | 0.71 (0.45–0.86) | NR | NR | [107] |
| HIF-1α | Increased | 0.74 (0.48–0.88) | NR | NR | [107] |
| Biomarker Panel | Relative Change | AUC (95%CI) | Sensitivity | Specificity | Reference |
|---|---|---|---|---|---|
| miR-200a miR-200b miR-141 | Decreased Decreased Decreased | 0.76 (0.63–0.87) | 84.4% | 66.7% | [72] |
| miR-199a miR-122 miR-145 miR-542-3p | Increased Increased Decreased Decreased | 0.99 (0.98-1.00) | 93.2% | 96.0% | [112] |
| miR-141 miR-199a miR-122 miR-145 CA-125 | Decreased Increased Increased Decreased Increased | 0.94 (0.90–0.98) | 81.8% | 92.6% | [111] |
| Class | Molecule | Model | Findings | Reference |
|---|---|---|---|---|
| Isoflavonoids | Isoliquiritigenin 3,6-ihydroxyflavone | End1/E6E7 cells; mouse endometriosis model OMA stromal cells; SCID/rat models | ↑ E-cadherin; ↓ N-cadherin, Snail, Slug; ↓ lesion size; ↑ BAX, caspase-3; ↑ E-cadherin, ↓ Twist, Slug; Notch pathway inhibition. | [121,122] |
| Plant Alkaloids | Tetramethylpyrazine | 11Z cells, OMA stromal cells; mouse model | ↑ E-cadherin; inhibited FMT; ↓ TGF-β1, α-SMA; ↓ p-Smad2/3; ↓ weight/fibrosis. | [123] |
| Terpenes | Parthenolide | Wistar albino rat model | ↓ Lesion area; ↑ E-cadherin, ↓ vimentin; inhibited PI3K/AKT and β-catenin. | [124] |
| Polysaccharides | Fucoidan | End1/E6E7, Vk2/E6E7 cells; mouse model | ↑ Proliferation/migration; ↑ E-cadherin; ↓ N-cadherin, Slug, Snail. | [92] |
| Biguanides | Metformin | OMA stromal cells; Wistar albino rat model | ↓ β-catenin translocation; ↓ proliferation/invasion; ↑ apoptosis. | [125] |
| S1P Receptor Modulators | SEW2871, FTY720, JTE013 | Mouse endometriosis model; Wistar rat model; Primary OMA stromal cells | ↓ Lesion growth and fibrosis; ↓ collagen type I; ↑ E-cadherin; ↓ S1P1 signaling. | [126] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hosseinirad, H.; Jeong, J.-W.; Barrier, B.F. Insights into the Molecular Mechanisms and Signaling Pathways of Epithelial to Mesenchymal Transition (EMT) in the Pathophysiology of Endometriosis. Int. J. Mol. Sci. 2025, 26, 7460. https://doi.org/10.3390/ijms26157460
Hosseinirad H, Jeong J-W, Barrier BF. Insights into the Molecular Mechanisms and Signaling Pathways of Epithelial to Mesenchymal Transition (EMT) in the Pathophysiology of Endometriosis. International Journal of Molecular Sciences. 2025; 26(15):7460. https://doi.org/10.3390/ijms26157460
Chicago/Turabian StyleHosseinirad, Hossein, Jae-Wook Jeong, and Breton F. Barrier. 2025. "Insights into the Molecular Mechanisms and Signaling Pathways of Epithelial to Mesenchymal Transition (EMT) in the Pathophysiology of Endometriosis" International Journal of Molecular Sciences 26, no. 15: 7460. https://doi.org/10.3390/ijms26157460
APA StyleHosseinirad, H., Jeong, J.-W., & Barrier, B. F. (2025). Insights into the Molecular Mechanisms and Signaling Pathways of Epithelial to Mesenchymal Transition (EMT) in the Pathophysiology of Endometriosis. International Journal of Molecular Sciences, 26(15), 7460. https://doi.org/10.3390/ijms26157460