
Ectopic Recruitment of the CTCF N-Terminal Domain with Two Proximal Zinc-Finger Domains as a Tool for 3D Genome Engineering

 , , , ,
, , , ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
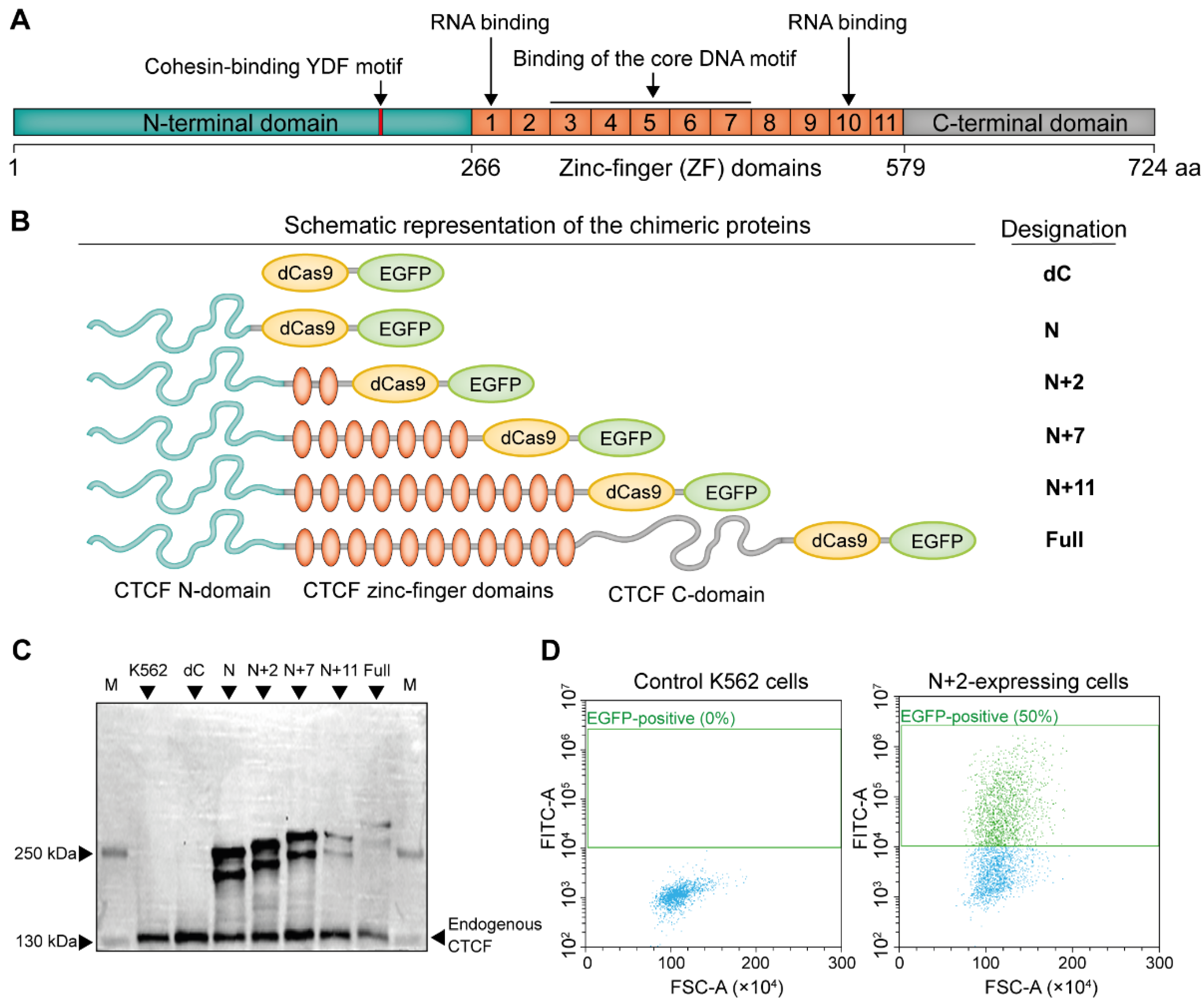
2.1. Construction and Expression of the CTCF-Based Chimeric Proteins
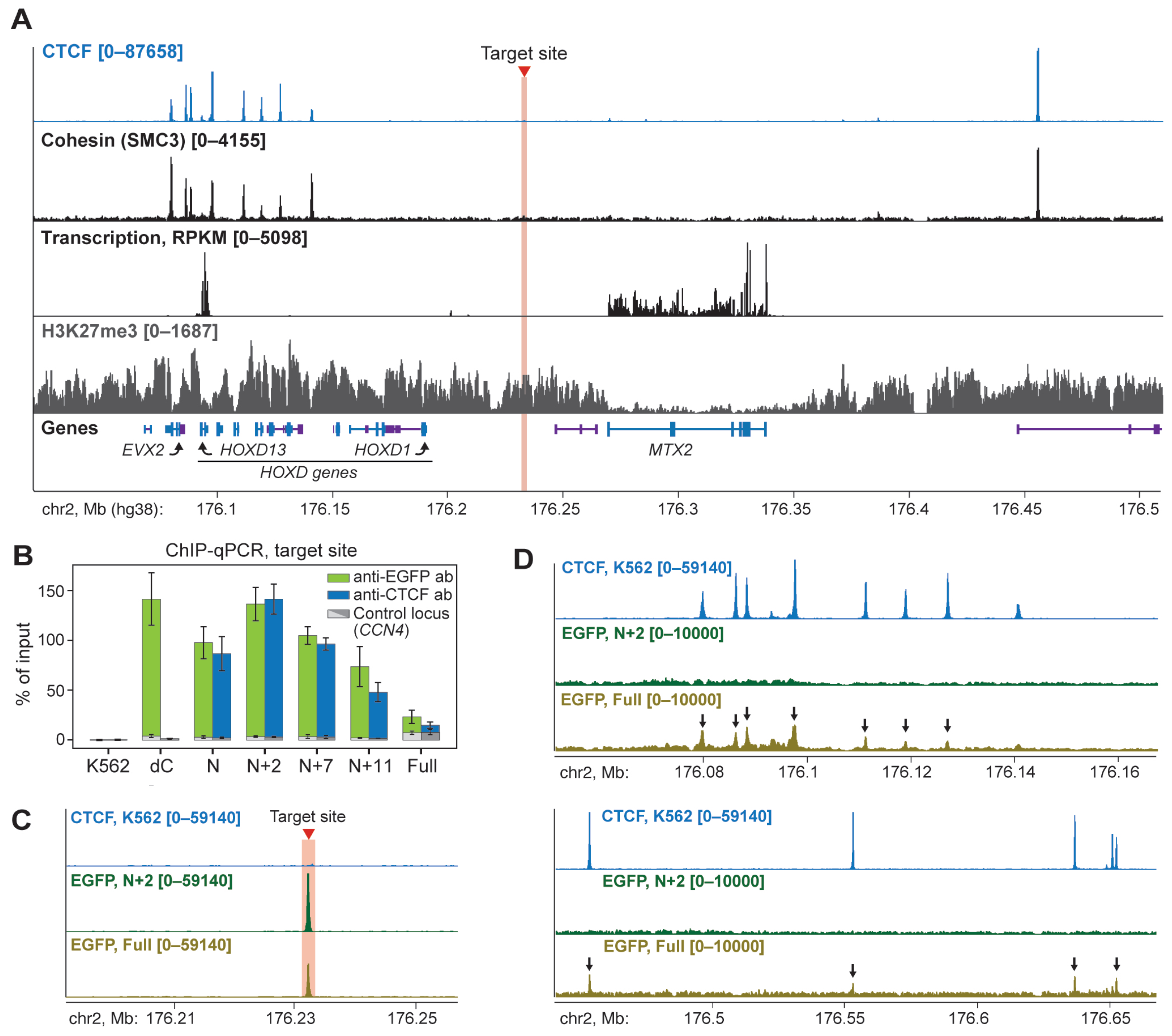
2.2. N+2 Chimeric Protein Effectively and Specifically Binds to the Target Site
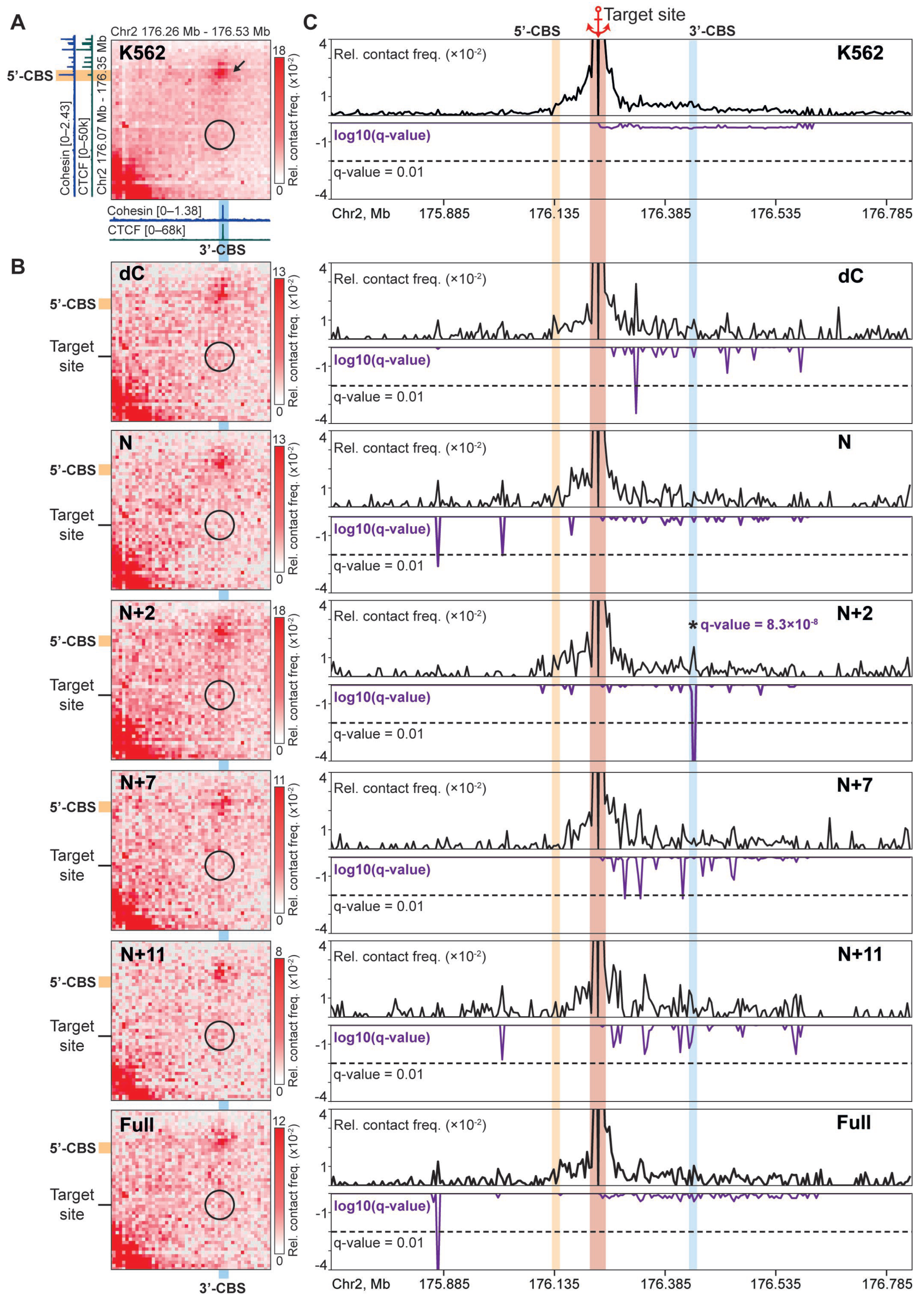
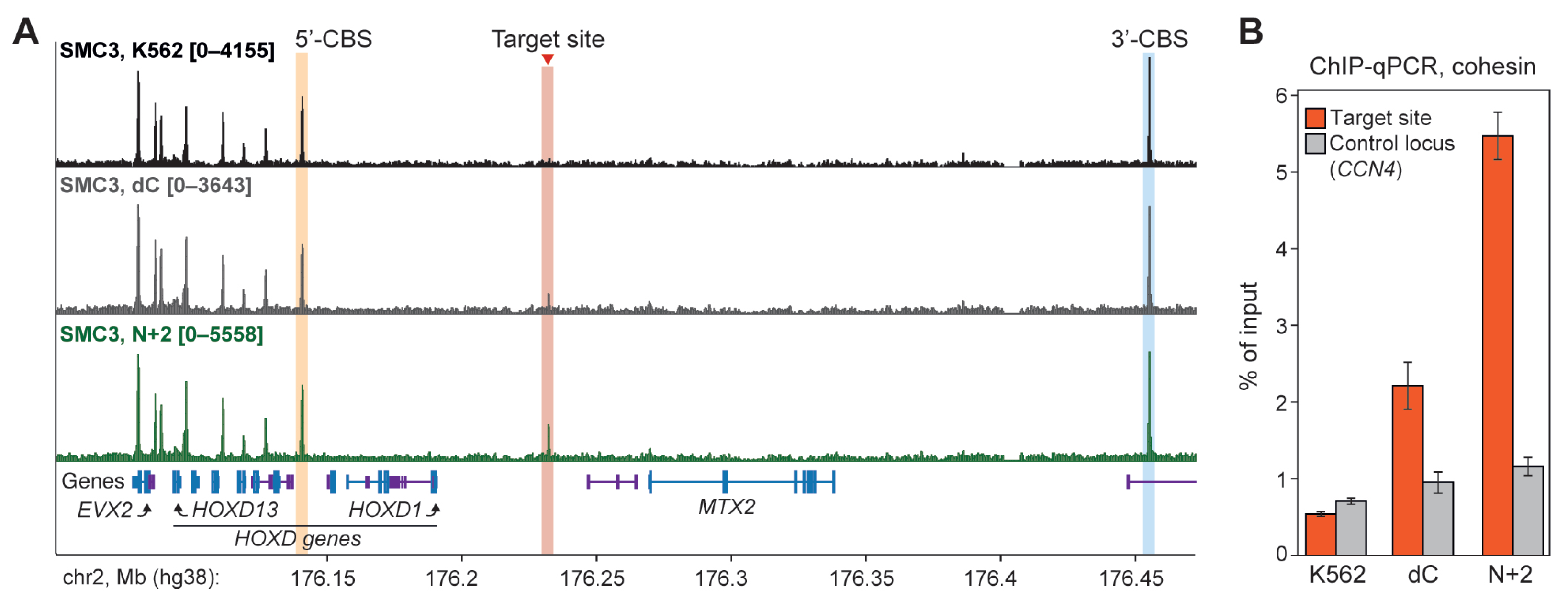
2.3. N+2 Chimeric Protein Mediates Cohesin-Dependent Chromatin Looping
2.4. N+2 Binding Partially Restores Chromatin Loops Lost After Deletion of the Endogenous CBS
3. Discussion
4. Materials and Methods
4.1. Cell Cultures
4.2. Electroporation
4.3. Constructing Plasmids Encoding Parts of the CTCF Protein Fused with dCas9 and EGFP
4.4. Preparation of Cells for the Experiments
4.5. Western Blot
4.6. C-TALE Library Preparation
4.7. C-TALE Data Processing and Analysis
4.8. ChIP-qPCR and ChIP-Seq Library Preparation
4.9. ChIP-Seq Data Processing
4.10. RNA-Seq Library Preparation
4.11. RNA-Seq Data Processing and Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jerkovic, I.; Cavalli, G. Understanding 3D Genome Organization by Multidisciplinary Methods. Nat. Rev. Mol. Cell Biol. 2021, 22, 511–528. [Google Scholar] [CrossRef] [PubMed]
- Finn, E.H.; Pegoraro, G.; Brandão, H.B.; Valton, A.-L.; Oomen, M.E.; Dekker, J.; Mirny, L.; Misteli, T. Extensive Heterogeneity and Intrinsic Variation in Spatial Genome Organization. Cell 2019, 176, 1502–1515.e10. [Google Scholar] [CrossRef] [PubMed]
- Ulianov, S.V.; Razin, S.V. The Two Waves in Single-Cell 3D Genomics. Semin. Cell Dev. Biol. 2022, 121, 143–152. [Google Scholar] [CrossRef]
- Acemel, R.D.; Lupiáñez, D.G. Evolution of 3D Chromatin Organization at Different Scales. Curr. Opin. Genet. Dev. 2023, 78, 102019. [Google Scholar] [CrossRef]
- Tiukacheva, E.A.; Ulianov, S.V.; Karpukhina, A.; Razin, S.V.; Vassetzky, Y. 3D Genome Alterations and Editing in Pathology. Mol. Ther. J. Am. Soc. Gene Ther. 2023, 31, 924–933. [Google Scholar] [CrossRef]
- Kantidze, O.L.; Gurova, K.V.; Studitsky, V.M.; Razin, S.V. The 3D Genome as a Target for Anticancer Therapy. Trends Mol. Med. 2020, 26, 141–149. [Google Scholar] [CrossRef]
- Hildebrand, E.M.; Dekker, J. Mechanisms and Functions of Chromosome Compartmentalization. Trends Biochem. Sci. 2020, 45, 385–396. [Google Scholar] [CrossRef]
- Davidson, I.F.; Peters, J.-M. Genome Folding through Loop Extrusion by SMC Complexes. Nat. Rev. Mol. Cell Biol. 2021, 22, 445–464. [Google Scholar] [CrossRef]
- Magnitov, M.; de Wit, E. Attraction and Disruption: How Loop Extrusion and Compartmentalisation Shape the Nuclear Genome. Curr. Opin. Genet. Dev. 2024, 86, 102194. [Google Scholar] [CrossRef]
- Dekker, J.; Mirny, L.A. The Chromosome Folding Problem and How Cells Solve It. Cell 2024, 187, 6424–6450. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Shi, Z.; Zhang, H.; Finkelstein, I.J.; Yu, H. Human Cohesin Compacts DNA by Loop Extrusion. Science 2019, 366, 1345–1349. [Google Scholar] [CrossRef]
- Davidson, I.F.; Bauer, B.; Goetz, D.; Tang, W.; Wutz, G.; Peters, J.-M. DNA Loop Extrusion by Human Cohesin. Science 2019, 366, 1338–1345. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Shi, Z.; Banigan, E.J.; Kim, Y.; Yu, H.; Bai, X.; Finkelstein, I.J. CTCF and R-Loops Are Boundaries of Cohesin-Mediated DNA Looping. Mol. Cell 2023, 83, 2856–2871.e8. [Google Scholar] [CrossRef]
- Mach, P.; Kos, P.I.; Zhan, Y.; Cramard, J.; Gaudin, S.; Tünnermann, J.; Marchi, E.; Eglinger, J.; Zuin, J.; Kryzhanovska, M.; et al. Cohesin and CTCF Control the Dynamics of Chromosome Folding. Nat. Genet. 2022, 54, 1907–1918. [Google Scholar] [CrossRef]
- Hansen, A.S. CTCF as a Boundary Factor for Cohesin-Mediated Loop Extrusion: Evidence for a Multi-Step Mechanism. Nucleus 2020, 11, 132–148. [Google Scholar] [CrossRef]
- Dequeker, B.J.H.; Scherr, M.J.; Brandão, H.B.; Gassler, J.; Powell, S.; Gaspar, I.; Flyamer, I.M.; Lalic, A.; Tang, W.; Stocsits, R.; et al. MCM Complexes Are Barriers That Restrict Cohesin-Mediated Loop Extrusion. Nature 2022, 606, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Selivanovskiy, A.V.; Molodova, M.N.; Khrameeva, E.E.; Ulianov, S.V.; Razin, S.V. Liquid Condensates: A New Barrier to Loop Extrusion? Cell. Mol. Life Sci. 2025, 82, 80. [Google Scholar] [CrossRef]
- Liu, M.; Maurano, M.T.; Wang, H.; Qi, H.; Song, C.-Z.; Navas, P.A.; Emery, D.W.; Stamatoyannopoulos, J.A.; Stamatoyannopoulos, G. Genomic Discovery of Potent Chromatin Insulators for Human Gene Therapy. Nat. Biotechnol. 2015, 33, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.S.P.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A Three-Dimensional Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef]
- Kubo, N.; Ishii, H.; Xiong, X.; Bianco, S.; Meitinger, F.; Hu, R.; Hocker, J.D.; Conte, M.; Gorkin, D.; Yu, M.; et al. Promoter-Proximal CTCF Binding Promotes Distal Enhancer-Dependent Gene Activation. Nat. Struct. Mol. Biol. 2021, 28, 152–161. [Google Scholar] [CrossRef]
- Oh, S.; Shao, J.; Mitra, J.; Xiong, F.; D’Antonio, M.; Wang, R.; Garcia-Bassets, I.; Ma, Q.; Zhu, X.; Lee, J.-H.; et al. Enhancer Release and Retargeting Activates Disease-Susceptibility Genes. Nature 2021, 595, 735–740. [Google Scholar] [CrossRef]
- Sept, C.E.; Tak, Y.E.; Goel, V.; Bhakta, M.S.; Cerda-Smith, C.G.; Hutchinson, H.M.; Blanchette, M.; Eyler, C.E.; Johnstone, S.E.; Joung, J.K.; et al. High-Resolution CTCF Footprinting Reveals Impact of Chromatin State on Cohesin Extrusion. Nat. Commun. 2025, 16, 4506. [Google Scholar] [CrossRef]
- Hashimoto, H.; Wang, D.; Horton, J.R.; Zhang, X.; Corces, V.G.; Cheng, X. Structural Basis for the Versatile and Methylation-Dependent Binding of CTCF to DNA. Mol. Cell 2017, 66, 711–720.e3. [Google Scholar] [CrossRef]
- Yin, M.; Wang, J.; Wang, M.; Li, X.; Zhang, M.; Wu, Q.; Wang, Y. Molecular Mechanism of Directional CTCF Recognition of a Diverse Range of Genomic Sites. Cell Res. 2017, 27, 1365–1377. [Google Scholar] [CrossRef] [PubMed]
- Pugacheva, E.M.; Kubo, N.; Loukinov, D.; Tajmul, M.; Kang, S.; Kovalchuk, A.L.; Strunnikov, A.V.; Zentner, G.E.; Ren, B.; Lobanenkov, V.V. CTCF Mediates Chromatin Looping via N-Terminal Domain-Dependent Cohesin Retention. Proc. Natl. Acad. Sci. USA 2020, 117, 2020–2031. [Google Scholar] [CrossRef]
- Li, Y.; Haarhuis, J.H.I.; Cacciatore, Á.S.; Oldenkamp, R.; van Ruiten, M.S.; Willems, L.; Teunissen, H.; Muir, K.W.; de Wit, E.; Rowland, B.D.; et al. The Structural Basis for Cohesin-CTCF Anchored Loops. Nature 2020, 578, 472–476. [Google Scholar] [CrossRef]
- Saldaña-Meyer, R.; Rodriguez-Hernaez, J.; Escobar, T.; Nishana, M.; Jácome-López, K.; Nora, E.P.; Bruneau, B.G.; Tsirigos, A.; Furlan-Magaril, M.; Skok, J.; et al. RNA Interactions Are Essential for CTCF-Mediated Genome Organization. Mol. Cell 2019, 76, 412–422.e5. [Google Scholar] [CrossRef]
- Farrar, D.; Rai, S.; Chernukhin, I.; Jagodic, M.; Ito, Y.; Yammine, S.; Ohlsson, R.; Murrell, A.; Klenova, E. Mutational Analysis of the Poly(ADP-Ribosyl)Ation Sites of the Transcription Factor CTCF Provides an Insight into the Mechanism of Its Regulation by Poly(ADP-Ribosyl)Ation. Mol. Cell. Biol. 2010, 30, 1199–1216. [Google Scholar] [CrossRef] [PubMed]
- Pavlaki, I.; Docquier, F.; Chernukhin, I.; Kita, G.; Gretton, S.; Clarkson, C.T.; Teif, V.B.; Klenova, E. Poly(ADP-Ribosyl)Ation Associated Changes in CTCF-Chromatin Binding and Gene Expression in Breast Cells. Biochim. Biophys. Acta 2018, 1861, 718–730. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Ginjala, V.; Pant, V.; Chernukhin, I.; Whitehead, J.; Docquier, F.; Farrar, D.; Tavoosidana, G.; Mukhopadhyay, R.; Kanduri, C.; et al. Poly(ADP-Ribosyl)Ation Regulates CTCF-Dependent Chromatin Insulation. Nat. Genet. 2004, 36, 1105–1110. [Google Scholar] [CrossRef]
- MacPherson, M.J.; Beatty, L.G.; Zhou, W.; Du, M.; Sadowski, P.D. The CTCF Insulator Protein Is Posttranslationally Modified by SUMO. Mol. Cell. Biol. 2009, 29, 714–725. [Google Scholar] [CrossRef]
- Luo, H.; Yu, Q.; Liu, Y.; Tang, M.; Liang, M.; Zhang, D.; Xiao, T.S.; Wu, L.; Tan, M.; Ruan, Y.; et al. LATS Kinase–Mediated CTCF Phosphorylation and Selective Loss of Genomic Binding. Sci. Adv. 2020, 6, eaaw4651. [Google Scholar] [CrossRef]
- Del Rosario, B.C.; Kriz, A.J.; Del Rosario, A.M.; Anselmo, A.; Fry, C.J.; White, F.M.; Sadreyev, R.I.; Lee, J.T. Exploration of CTCF Post-Translation Modifications Uncovers Serine-224 Phosphorylation by PLK1 at Pericentric Regions during the G2/M Transition. eLife 2019, 8, e42341. [Google Scholar] [CrossRef]
- El-Kady, A.; Klenova, E. Regulation of the Transcription Factor, CTCF, by Phosphorylation with Protein Kinase CK2. FEBS Lett. 2005, 579, 1424–1434. [Google Scholar] [CrossRef] [PubMed]
- Gong, S.; Hu, G.; Guo, R.; Zhang, J.; Yang, Y.; Ji, B.; Li, G.; Yao, H. CTCF Acetylation at Lysine 20 Is Required for the Early Cardiac Mesoderm Differentiation of Embryonic Stem Cells. Cell Regen. Lond. Engl. 2022, 11, 34. [Google Scholar] [CrossRef] [PubMed]
- Willemin, A.; Lopez-Delisle, L.; Bolt, C.C.; Gadolini, M.-L.; Duboule, D.; Rodriguez-Carballo, E. Induction of a Chromatin Boundary in Vivo upon Insertion of a TAD Border. PLoS Genet. 2021, 17, e1009691. [Google Scholar] [CrossRef]
- Jia, Z.; Li, J.; Ge, X.; Wu, Y.; Guo, Y.; Wu, Q. Tandem CTCF Sites Function as Insulators to Balance Spatial Chromatin Contacts and Topological Enhancer-Promoter Selection. Genome Biol. 2020, 21, 75. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.S.; Wu, H.; Ji, X.; Stelzer, Y.; Wu, X.; Czauderna, S.; Shu, J.; Dadon, D.; Young, R.A.; Jaenisch, R. Editing DNA Methylation in the Mammalian Genome. Cell 2016, 167, 233–247.e17. [Google Scholar] [CrossRef]
- Lupiáñez, D.G.; Kraft, K.; Heinrich, V.; Krawitz, P.; Brancati, F.; Klopocki, E.; Horn, D.; Kayserili, H.; Opitz, J.M.; Laxova, R.; et al. Disruptions of Topological Chromatin Domains Cause Pathogenic Rewiring of Gene-Enhancer Interactions. Cell 2015, 161, 1012–1025. [Google Scholar] [CrossRef]
- Wei, Z.; Wang, S.; Xu, Y.; Wang, W.; Soares, F.; Ahmed, M.; Su, P.; Wang, T.; Orouji, E.; Xu, X.; et al. MYC Reshapes CTCF-Mediated Chromatin Architecture in Prostate Cancer. Nat. Commun. 2023, 14, 1787. [Google Scholar] [CrossRef]
- Guo, Y.; Perez, A.A.; Hazelett, D.J.; Coetzee, G.A.; Rhie, S.K.; Farnham, P.J. CRISPR-Mediated Deletion of Prostate Cancer Risk-Associated CTCF Loop Anchors Identifies Repressive Chromatin Loops. Genome Biol. 2018, 19, 160. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Xu, Q.; Canzio, D.; Shou, J.; Li, J.; Gorkin, D.U.; Jung, I.; Wu, H.; Zhai, Y.; Tang, Y.; et al. CRISPR Inversion of CTCF Sites Alters Genome Topology and Enhancer/Promoter Function. Cell 2015, 162, 900–910. [Google Scholar] [CrossRef] [PubMed]
- Korkmaz, G.; Manber, Z.; Lopes, R.; Prekovic, S.; Schuurman, K.; Kim, Y.; Teunissen, H.; Flach, K.; de Wit, E.; Galli, G.G.; et al. A CRISPR-Cas9 Screen Identifies Essential CTCF Anchor Sites for Estrogen Receptor-Driven Breast Cancer Cell Proliferation. Nucleic Acids Res. 2019, 47, 9557–9572. [Google Scholar] [CrossRef]
- Samuelson, C.; Radtke, S.; Zhu, H.; Llewellyn, M.; Fields, E.; Cook, S.; Huang, M.-L.W.; Jerome, K.R.; Kiem, H.-P.; Humbert, O. Multiplex CRISPR/Cas9 Genome Editing in Hematopoietic Stem Cells for Fetal Hemoglobin Reinduction Generates Chromosomal Translocations. Mol. Ther. Methods Clin. Dev. 2021, 23, 507–523. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, Q. CCCTC-Binding Factor N-Terminal Domain Regulates Clustered Protocadherin Gene Expression by Enhancing Cohesin Processivity. J. Biol. Chem. 2025, 301, 108337. [Google Scholar] [CrossRef] [PubMed]
- Two CTCF Motifs Impede Cohesin-Mediated DNA Loop Extrusion|bioRxiv. Available online: https://www.biorxiv.org/content/10.1101/2025.01.26.634934v1 (accessed on 27 May 2025).
- Huber, J.; Tanasie, N.-L.; Zernia, S.; Stigler, J. Single-Molecule Imaging Reveals a Direct Role of CTCF’s Zinc Fingers in SA Interaction and Cluster-Dependent RNA Recruitment. Nucleic Acids Res. 2024, 52, 6490–6506. [Google Scholar] [CrossRef]
- Rasko, J.E.; Klenova, E.M.; Leon, J.; Filippova, G.N.; Loukinov, D.I.; Vatolin, S.; Robinson, A.F.; Hu, Y.J.; Ulmer, J.; Ward, M.D.; et al. Cell Growth Inhibition by the Multifunctional Multivalent Zinc-Finger Factor CTCF. Cancer Res. 2001, 61, 6002–6007. [Google Scholar]
- Xiao, T.; Wallace, J.; Felsenfeld, G. Specific Sites in the C Terminus of CTCF Interact with the SA2 Subunit of the Cohesin Complex and Are Required for Cohesin-Dependent Insulation Activity. Mol. Cell. Biol. 2011, 31, 2174–2183. [Google Scholar] [CrossRef]
- Torrano, V.; Navascués, J.; Docquier, F.; Zhang, R.; Burke, L.J.; Chernukhin, I.; Farrar, D.; León, J.; Berciano, M.T.; Renkawitz, R.; et al. Targeting of CTCF to the Nucleolus Inhibits Nucleolar Transcription through a Poly(ADP-Ribosyl)Ation-Dependent Mechanism. J. Cell Sci. 2006, 119, 1746–1759. [Google Scholar] [CrossRef]
- Golov, A.K.; Ulianov, S.V.; Luzhin, A.V.; Kalabusheva, E.P.; Kantidze, O.L.; Flyamer, I.M.; Razin, S.V.; Gavrilov, A.A. C-TALE, a New Cost-Effective Method for Targeted Enrichment of Hi-C/3C-Seq Libraries. Methods 2020, 170, 48–60. [Google Scholar] [CrossRef]
- Fan, J.; Lee, H.-O.; Lee, S.; Ryu, D.-E.; Lee, S.; Xue, C.; Kim, S.J.; Kim, K.; Barkas, N.; Park, P.J.; et al. Linking Transcriptional and Genetic Tumor Heterogeneity through Allele Analysis of Single-Cell RNA-Seq Data. Genome Res. 2018, 28, 1217–1227. [Google Scholar] [CrossRef]
- Kasai, F.; Mizukoshi, K.; Nakamura, Y. Variable Characteristics Overlooked in Human K-562 Leukemia Cell Lines with a Common Signature. Sci. Rep. 2024, 14, 9619. [Google Scholar] [CrossRef]
- Perez, G.; Barber, G.P.; Benet-Pages, A.; Casper, J.; Clawson, H.; Diekhans, M.; Fischer, C.; Gonzalez, J.N.; Hinrichs, A.S.; Lee, C.M.; et al. The UCSC Genome Browser Database: 2025 Update. Nucleic Acids Res. 2025, 53, D1243–D1249. [Google Scholar] [CrossRef]
- Bonev, B.; Mendelson Cohen, N.; Szabo, Q.; Fritsch, L.; Papadopoulos, G.L.; Lubling, Y.; Xu, X.; Lv, X.; Hugnot, J.-P.; Tanay, A.; et al. Multiscale 3D Genome Rewiring during Mouse Neural Development. Cell 2017, 171, 557–572.e24. [Google Scholar] [CrossRef] [PubMed]
- Strohkendl, I.; Saifuddin, F.A.; Gibson, B.A.; Rosen, M.K.; Russell, R.; Finkelstein, I.J. Inhibition of CRISPR-Cas12a DNA Targeting by Nucleosomes and Chromatin. Sci. Adv. 2021, 7, eabd6030. [Google Scholar] [CrossRef] [PubMed]
- Amândio, A.R.; Beccari, L.; Lopez-Delisle, L.; Mascrez, B.; Zakany, J.; Gitto, S.; Duboule, D. Sequential in Cis Mutagenesis in Vivo Reveals Various Functions for CTCF Sites at the Mouse HoxD Cluster. Genes Dev. 2021, 35, 1490–1509. [Google Scholar] [CrossRef]
- Kim, J.H.; Rege, M.; Valeri, J.; Dunagin, M.C.; Metzger, A.; Titus, K.R.; Gilgenast, T.G.; Gong, W.; Beagan, J.A.; Raj, A.; et al. LADL: Light-Activated Dynamic Looping for Endogenous Gene Expression Control. Nat. Methods 2019, 16, 633–639. [Google Scholar] [CrossRef]
- Morgan, S.L.; Mariano, N.C.; Bermudez, A.; Arruda, N.L.; Wu, F.; Luo, Y.; Shankar, G.; Jia, L.; Chen, H.; Hu, J.-F.; et al. Manipulation of Nuclear Architecture through CRISPR-Mediated Chromosomal Looping. Nat. Commun. 2017, 8, 15993. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Rupon, J.W.; Krivega, I.; Breda, L.; Motta, I.; Jahn, K.S.; Reik, A.; Gregory, P.D.; Rivella, S.; Dean, A.; et al. Reactivation of Developmentally Silenced Globin Genes by Forced Chromatin Looping. Cell 2014, 158, 849–860. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Liang, Z.; Zhang, J.; Hu, G.; Wang, J.; Li, Y.; Guo, R.; Dong, X.; Babarinde, I.A.; Ping, W.; et al. CTCF Functions as an Insulator for Somatic Genes and a Chromatin Remodeler for Pluripotency Genes during Reprogramming. Cell Rep. 2022, 39, 110626. [Google Scholar] [CrossRef]
- Chernukhin, I.; Shamsuddin, S.; Kang, S.Y.; Bergström, R.; Kwon, Y.-W.; Yu, W.; Whitehead, J.; Mukhopadhyay, R.; Docquier, F.; Farrar, D.; et al. CTCF Interacts with and Recruits the Largest Subunit of RNA Polymerase II to CTCF Target Sites Genome-Wide. Mol. Cell. Biol. 2007, 27, 1631–1648. [Google Scholar] [CrossRef]
- Klenova, E.M.; Chernukhin, I.V.; El-Kady, A.; Lee, R.E.; Pugacheva, E.M.; Loukinov, D.I.; Goodwin, G.H.; Delgado, D.; Filippova, G.N.; León, J.; et al. Functional Phosphorylation Sites in the C-Terminal Region of the Multivalent Multifunctional Transcriptional Factor CTCF. Mol. Cell. Biol. 2001, 21, 2221–2234. [Google Scholar] [CrossRef]
- Concordet, J.-P.; Haeussler, M. CRISPOR: Intuitive Guide Selection for CRISPR/Cas9 Genome Editing Experiments and Screens. Nucleic Acids Res. 2018, 46, W242–W245. [Google Scholar] [CrossRef]
- Germini, D.; Saada, Y.B.; Tsfasman, T.; Osina, K.; Robin, C.; Lomov, N.; Rubtsov, M.; Sjakste, N.; Lipinski, M.; Vassetzky, Y. A One-Step PCR-Based Assay to Evaluate the Efficiency and Precision of Genomic DNA-Editing Tools. Mol. Ther. Methods Clin. Dev. 2017, 5, 43–50. [Google Scholar] [CrossRef]
- Imakaev, M.; Fudenberg, G.; McCord, R.P.; Naumova, N.; Goloborodko, A.; Lajoie, B.R.; Dekker, J.; Mirny, L.A. Iterative Correction of Hi-C Data Reveals Hallmarks of Chromosome Organization. Nat. Methods 2012, 9, 999–1003. [Google Scholar] [CrossRef]
- Luzhin, A.V.; Golov, A.K.; Gavrilov, A.A.; Velichko, A.K.; Ulianov, S.V.; Razin, S.V.; Kantidze, O.L. LASCA: Loop and Significant Contact Annotation Pipeline. Sci. Rep. 2021, 11, 6361. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinform. Oxf. Engl. 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve Years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, F.; Ryan, D.P.; Grüning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dündar, F.; Manke, T. deepTools2: A next Generation Web Server for Deep-Sequencing Data Analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinform. Oxf. Engl. 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinform. Oxf. Engl. 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tiukacheva, E.A.; Luzhin, A.V.; Kruglova, N.; Shtompel, A.S.; Antonov, G.; Tvorogova, A.; Vassetzky, Y.; Ulianov, S.V.; Razin, S.V. Ectopic Recruitment of the CTCF N-Terminal Domain with Two Proximal Zinc-Finger Domains as a Tool for 3D Genome Engineering. Int. J. Mol. Sci. 2025, 26, 7446. https://doi.org/10.3390/ijms26157446
Tiukacheva EA, Luzhin AV, Kruglova N, Shtompel AS, Antonov G, Tvorogova A, Vassetzky Y, Ulianov SV, Razin SV. Ectopic Recruitment of the CTCF N-Terminal Domain with Two Proximal Zinc-Finger Domains as a Tool for 3D Genome Engineering. International Journal of Molecular Sciences. 2025; 26(15):7446. https://doi.org/10.3390/ijms26157446
Chicago/Turabian StyleTiukacheva, Eugenia A., Artem V. Luzhin, Natalia Kruglova, Anastasia S. Shtompel, Grigorii Antonov, Anna Tvorogova, Yegor Vassetzky, Sergey V. Ulianov, and Sergey V. Razin. 2025. "Ectopic Recruitment of the CTCF N-Terminal Domain with Two Proximal Zinc-Finger Domains as a Tool for 3D Genome Engineering" International Journal of Molecular Sciences 26, no. 15: 7446. https://doi.org/10.3390/ijms26157446
APA StyleTiukacheva, E. A., Luzhin, A. V., Kruglova, N., Shtompel, A. S., Antonov, G., Tvorogova, A., Vassetzky, Y., Ulianov, S. V., & Razin, S. V. (2025). Ectopic Recruitment of the CTCF N-Terminal Domain with Two Proximal Zinc-Finger Domains as a Tool for 3D Genome Engineering. International Journal of Molecular Sciences, 26(15), 7446. https://doi.org/10.3390/ijms26157446