
A Natural Language Processing Method Identifies an Association Between Bacterial Communities in the Upper Genital Tract and Ovarian Cancer

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
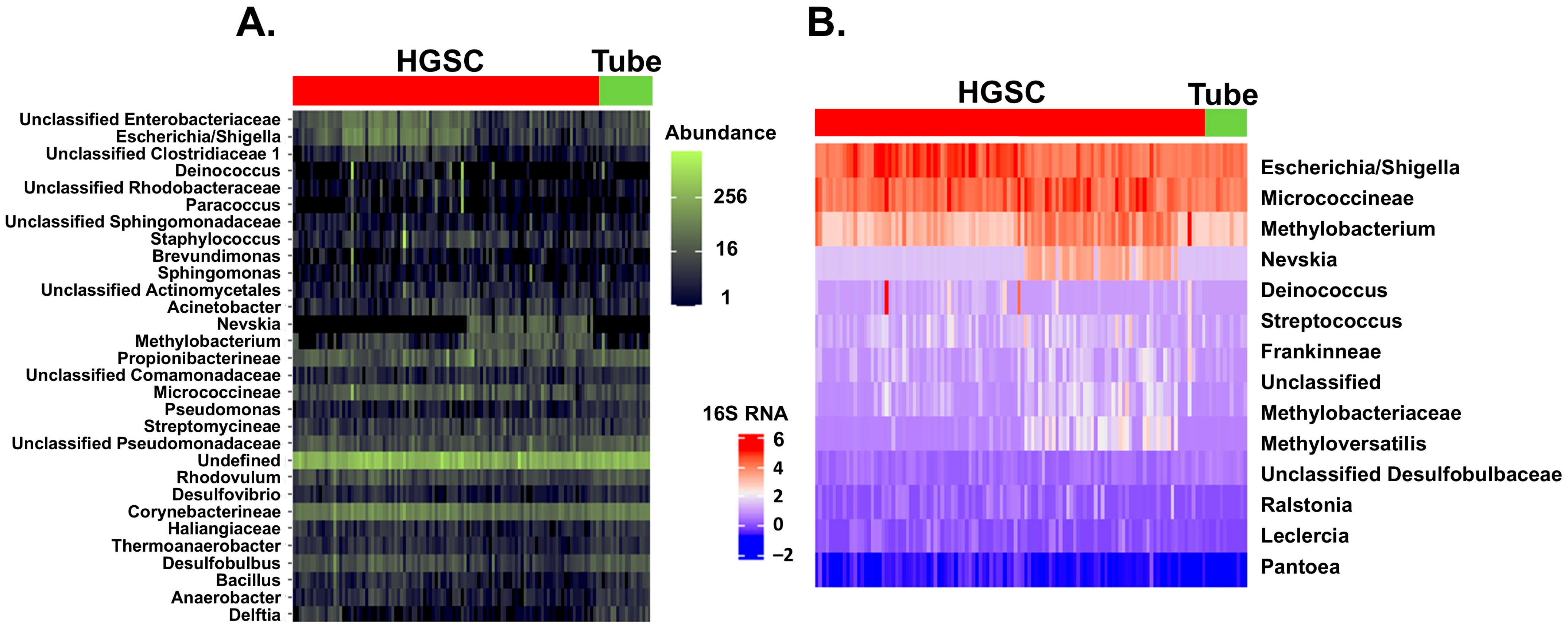
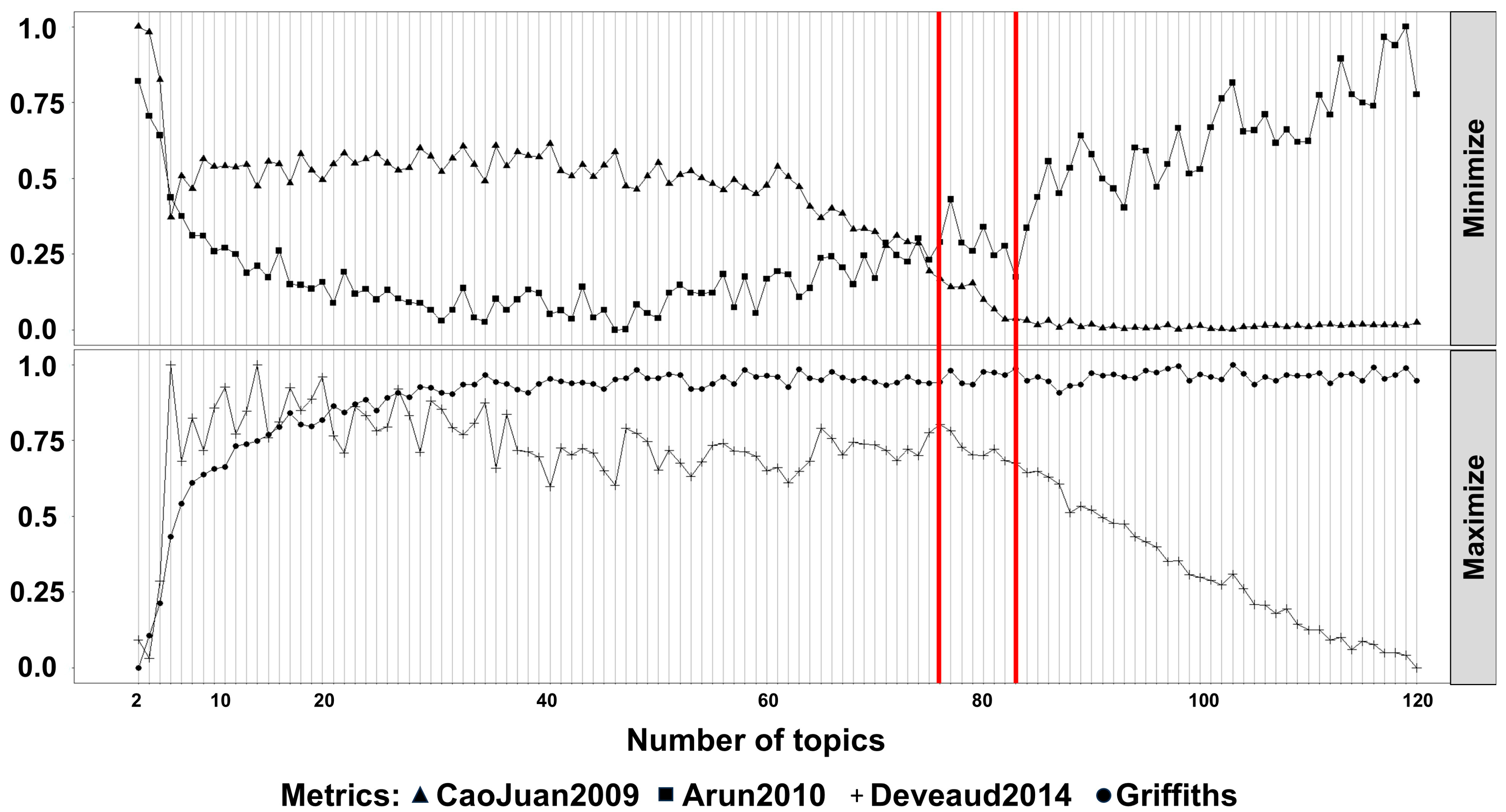
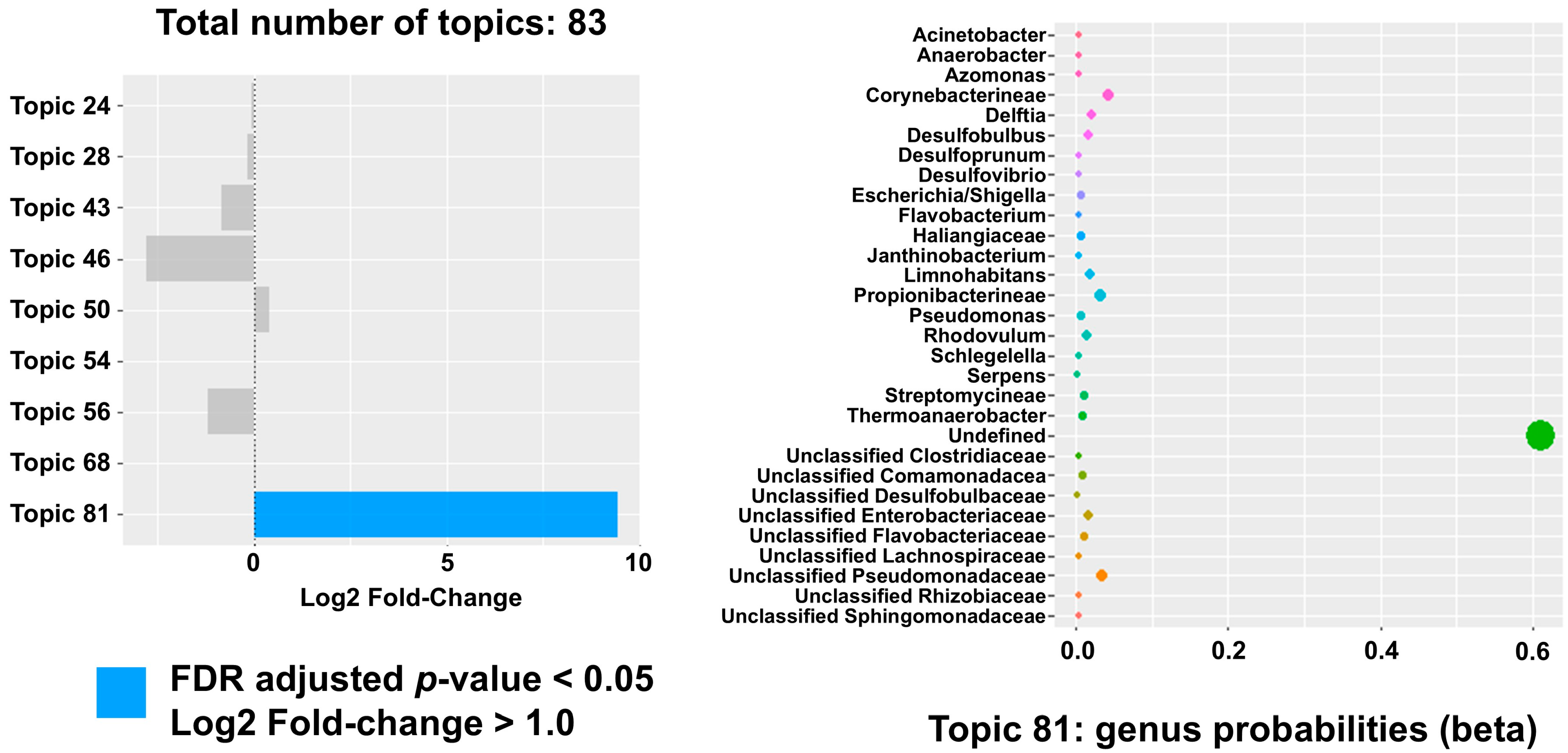
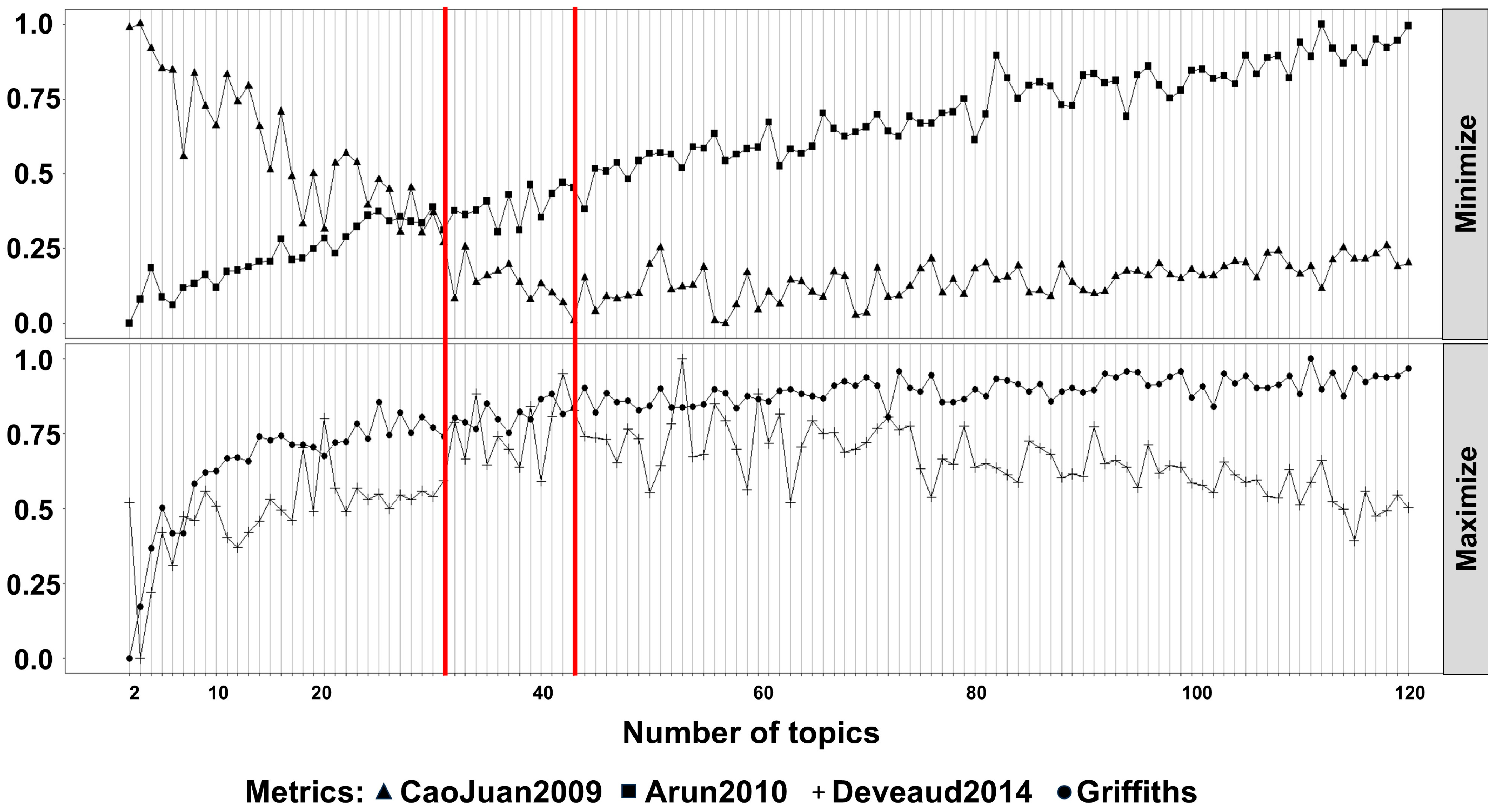
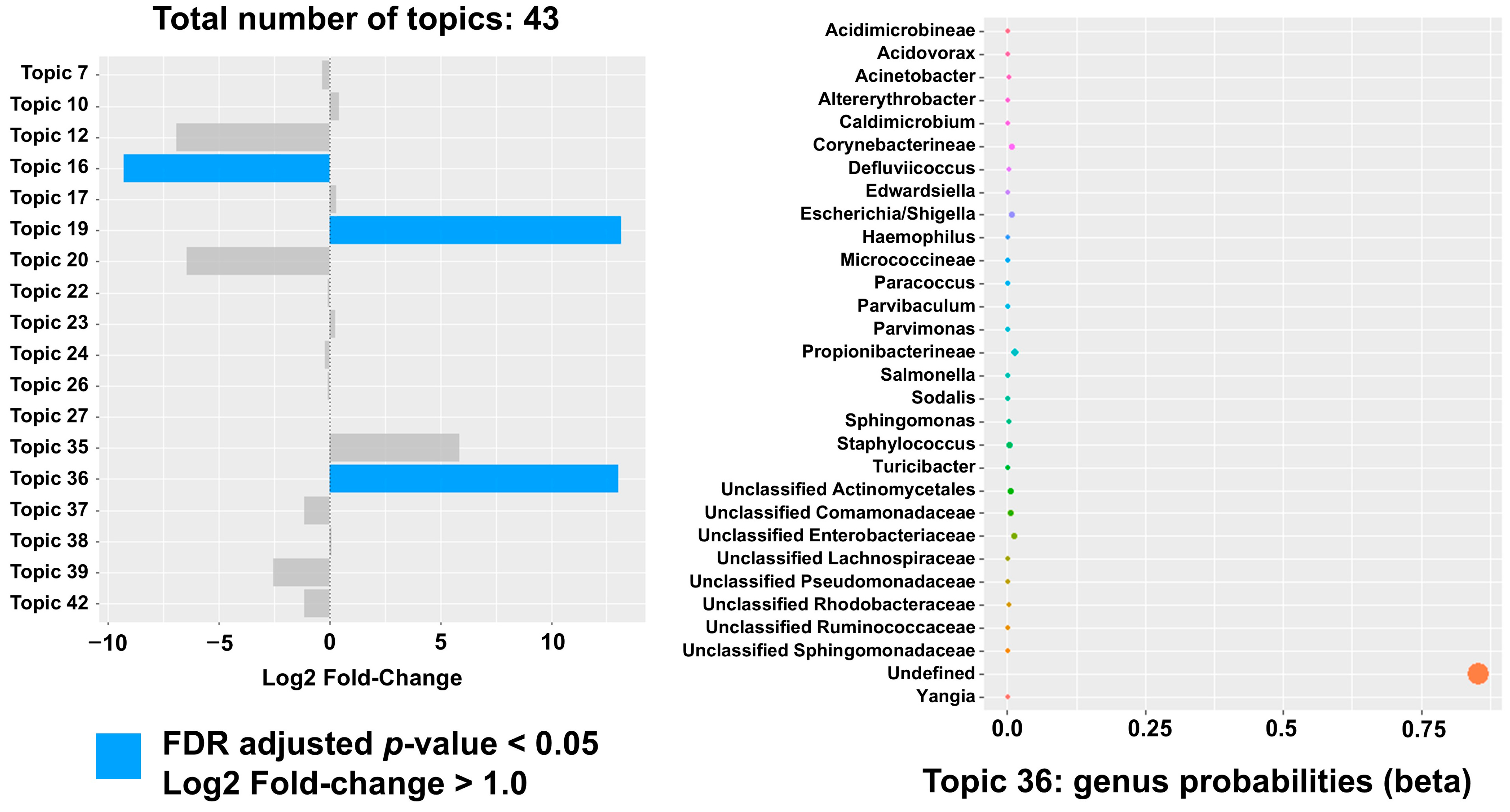
2. Results
3. Discussion
4. Materials and Methods
4.1. Specimen Acquisition
4.2. RNA Sequencing and Metagenomic Analysis
4.3. Natural Language Processing Analysis
4.4. Prediction of Functional Profiles
4.5. Analysis Validation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aggarwal, N.; Kitano, S.; Puah, G.R.Y.; Kittelmann, S.; Hwang, I.Y.; Chang, M.W. Microbiome and Human Health: Current Understanding, Engineering, and Enabling Technologies. Chem. Rev. 2022, 123, 31. [Google Scholar] [CrossRef] [PubMed]
- Madhogaria, B.; Bhowmik, P.; Kundu, A. Correlation between human gut microbiome and diseases. Infect. Med. 2022, 1, 180–191. [Google Scholar] [CrossRef]
- Li, C.; Feng, Y.; Yang, C.; Wang, D.; Zhang, D.; Luo, X.; Zhang, H.; Huang, H.; Zhang, H.; Jiang, Y.; et al. Association between vaginal microbiota and the progression of ovarian cancer. J. Med. Virol. 2023, 95, e28898. [Google Scholar] [CrossRef]
- Laniewski, P.; Ilhan, Z.E.; Herbst-Kralovetz, M.M. The microbiome and gynaecological cancer development, prevention and therapy. Nat. Rev. Urol. 2020, 17, 232–250. [Google Scholar] [CrossRef]
- Chambers, L.M.; Bussies, P.; Vargas, R.; Esakov, E.; Tewari, S.; Reizes, O.; Michener, C. The Microbiome and Gynecologic Cancer: Current Evidence and Future Opportunities. Curr. Oncol. Rep. 2021, 23, 92. [Google Scholar] [CrossRef] [PubMed]
- Sanschagrin, S.; Yergeau, E. Next-generation sequencing of 16S ribosomal RNA gene amplicons. J. Vis. Exp. 2014, 90, 51709. [Google Scholar]
- Wensel, C.R.; Pluznick, J.L.; Salzberg, S.L.; Sears, C.L. Next-generation sequencing: Insights to advance clinical investigations of the microbiome. J. Clin. Investig. 2022, 132, e154944. [Google Scholar] [CrossRef]
- Shrode, R.L.; Ollberding, N.J.; Mangalam, A.K. Looking at the Full Picture: Utilizing Topic Modeling to Determine Disease-Associated Microbiome Communities. bioRxiv 2023. [Google Scholar] [CrossRef]
- Asangba, A.E.; Chen, J.; Goergen, K.M.; Larson, M.C.; Oberg, A.L.; Casarin, J.; Multinu, F.; Kaufmann, S.H.; Mariani, A.; Chia, N.; et al. Diagnostic and prognostic potential of the microbiome in ovarian cancer treatment response. Sci. Rep. 2023, 13, 730. [Google Scholar] [CrossRef]
- Miao, R.; Badger, T.C.; Groesch, K.; Diaz-Sylvester, P.L.; Wilson, T.; Ghareeb, A.; Martin, J.A.; Cregger, M.; Welge, M.; Bushell, C.; et al. Assessment of peritoneal microbial features and tumor marker levels as potential diagnostic tools for ovarian cancer. PLoS ONE 2020, 15, e0227707. [Google Scholar] [CrossRef]
- Wang, Q.; Zhao, L.; Han, L.; Fu, G.; Tuo, X.; Ma, S.; Li, Q.; Wang, Y.; Liang, D.; Tang, M.; et al. The differential distribution of bacteria between cancerous and noncancerous ovarian tissues in situ. J. Ovarian Res. 2020, 13, 8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Mo, J.; Huang, W.; Bao, Y.; Luo, X.; Yuan, L. The ovarian cancer-associated microbiome contributes to the tumor’s inflammatory microenvironment. Front. Cell Infect Microbiol. 2024, 21, 1440742. [Google Scholar] [CrossRef]
- Sipos, A.; Ujlaki, G.; Mikó, E.; Maka, E.; Szabó, J.; Uray, K.; Krasznai, Z.; Bai, Z. The role of the microbiome in ovarian cancer: Mechanistic insights into oncobiosis and to bacterial metabolite signaling. Mol. Med. 2021, 27, 33. [Google Scholar] [CrossRef]
- Sadrekarimi, H.; Gardanova, Z.R.; Bakhshesh, M.; Ebrahimzadeh, F.; Yaseri, A.F.; Thangavelu, L.; Hasanpoor, Z.; Zadeh, F.A.; Kahrizi, M.S. Emerging role of human microbiome in cancer development and response to therapy: Special focus on intestinal microflora. J. Transl. Med. 2022, 20, 301. [Google Scholar] [CrossRef]
- Ervin, S.M.; Li, H.; Lim, L.; Roberts, L.R.; Liang, X.; Mani, S.; Redinbo, M.R. Gut microbial β-glucuronidases reactivate estrogens as components of the estrobolome that reactivate estrogens. J. Biol. Chem. 2019, 294, 18586–18599. [Google Scholar] [CrossRef] [PubMed]
- Parida, S.; Sharma, D. The Microbiome–Estrogen Connection and Breast Cancer Risk. Cells 2019, 8, 1642. [Google Scholar] [CrossRef]
- He, S.; Li, H.; Yu, Z.; Zhang, F.; Liang, S.; Liu, H.; Chen, H.; Lü, M. The Gut Microbiome and Sex Hormone-Related Diseases. Front Microbiol. 2021, 12, 711137. [Google Scholar] [CrossRef]
- Reyes, H.D.; Devor, E.J.; Warrier, A.; Newtson, A.M.; Mattson, J.; Wagner, V.; Duncan, G.N.; Leslie, K.K.; Gonzalez-Bosquet, J. Differential DNA methylation in high-grade serous ovarian cancer (HGSOC) is associated with tumor behavior. Sci. Rep. 2019, 9, 17996. [Google Scholar] [CrossRef]
- Rowland, I.; Gibson, G.; Heinken, A.; Scott, K.; Swann, J.; Thiele, I.; Tuohy, K. microbiota functions: Metabolism of nutrients and other food components. Eur. J. Nutr. 2018, 57, 1–24. [Google Scholar] [CrossRef]
- Kim, A.; Sevanto, S.; Moore, E.R.; Lubbers, N. Latent Dirichlet Allocation modeling of environmental microbiomes. PLoS Comput. Biol. 2023, 19, e1011075. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Xu, R.; Chen, J.; Wang, S.; Hu, P.; Wu, Y.; Que, Y.; Du, W.; Cai, X.; Chen, H.; et al. The epithelial Na+ channel (ENaC) in ovarian granulosa cells modulates Ca2+ mobilization and gonadotrophin signaling for estrogen homeostasis and female fertility. Cell Commun. Signal. 2024, 22, 398. [Google Scholar] [CrossRef] [PubMed]
- Ramos Meyers, G.; Samouda, H.; Bohn, T. Short Chain Fatty Acid Metabolism in Relation to Gut Microbiota and Genetic Variability. Nutrients 2022, 14, 5361. [Google Scholar] [CrossRef] [PubMed]
- Erickson, B.K.; Conner, M.G.; Landen, C.N., Jr. The role of the fallopian tube in the origin of ovarian cancer. Am. J. Obstet. Gynecol. 2013, 209, 409–414. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Le, S.; Li, Y.; Hu, F. SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS ONE 2016, 11, e0163962. [Google Scholar] [CrossRef]
- Yilmaz, P.; Parfrey, L.W.; Yarza, P.; Gerken, J.; Pruesse, E.; Quast, C.; Schweer, T.; Peplies, J.; Ludwig, W.; Glöckner, F.O. The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res. 2014, 42, D643–D648. [Google Scholar] [CrossRef]
- Mori, H.; Maruyama, T.; Yano, M.; Yamada, T.; Kurokawa, K. VITCOMIC2: Visualization tool for the phylogenetic composition of microbial communities based on 16S rRNA gene amplicons and metagenomic shotgun sequencing. BMC Syst. Biol. 2018, 12 (Suppl. 2), 30. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef] [PubMed Central]
- Yang, C.; Mai, J.; Cao, X.; Burberry, A.; Cominelli, F.; Zhang, L. ggpicrust2: An R package for PICRUSt2 predicted functional profile analysis and visualization. Bioinformatics 2023, 39, btad470. [Google Scholar] [CrossRef] [PubMed Central]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Polio, A.; Wagner, V.; Bender, D.P.; Goodheart, M.J.; Gonzalez Bosquet, J. A Natural Language Processing Method Identifies an Association Between Bacterial Communities in the Upper Genital Tract and Ovarian Cancer. Int. J. Mol. Sci. 2025, 26, 7432. https://doi.org/10.3390/ijms26157432
Polio A, Wagner V, Bender DP, Goodheart MJ, Gonzalez Bosquet J. A Natural Language Processing Method Identifies an Association Between Bacterial Communities in the Upper Genital Tract and Ovarian Cancer. International Journal of Molecular Sciences. 2025; 26(15):7432. https://doi.org/10.3390/ijms26157432
Chicago/Turabian StylePolio, Andrew, Vincent Wagner, David P. Bender, Michael J. Goodheart, and Jesus Gonzalez Bosquet. 2025. "A Natural Language Processing Method Identifies an Association Between Bacterial Communities in the Upper Genital Tract and Ovarian Cancer" International Journal of Molecular Sciences 26, no. 15: 7432. https://doi.org/10.3390/ijms26157432
APA StylePolio, A., Wagner, V., Bender, D. P., Goodheart, M. J., & Gonzalez Bosquet, J. (2025). A Natural Language Processing Method Identifies an Association Between Bacterial Communities in the Upper Genital Tract and Ovarian Cancer. International Journal of Molecular Sciences, 26(15), 7432. https://doi.org/10.3390/ijms26157432