
Cancer Stem Cells in Melanoma: Drivers of Tumor Plasticity and Emerging Therapeutic Strategies

 , , , and
, , , and 
Abstract
1. Introduction
2. Identification and Characterization of Melanoma Tumor Stem Cells
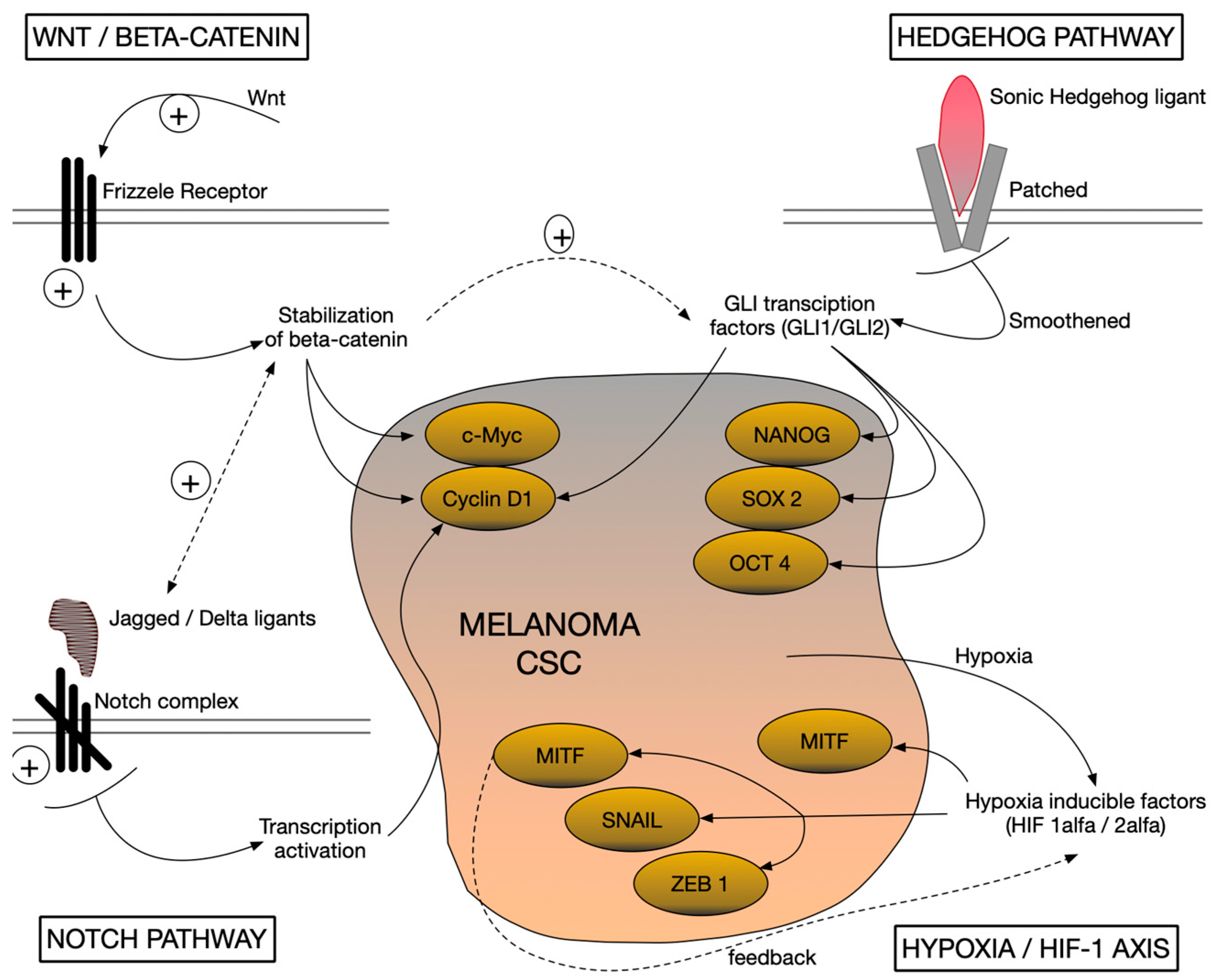
3. Signaling Pathways Involved in Maintaining Stemness in Melanoma
4. Tumor Plasticity and Cellular Origin in Melanoma
5. Interaction with the Tumor Microenvironment (TME)
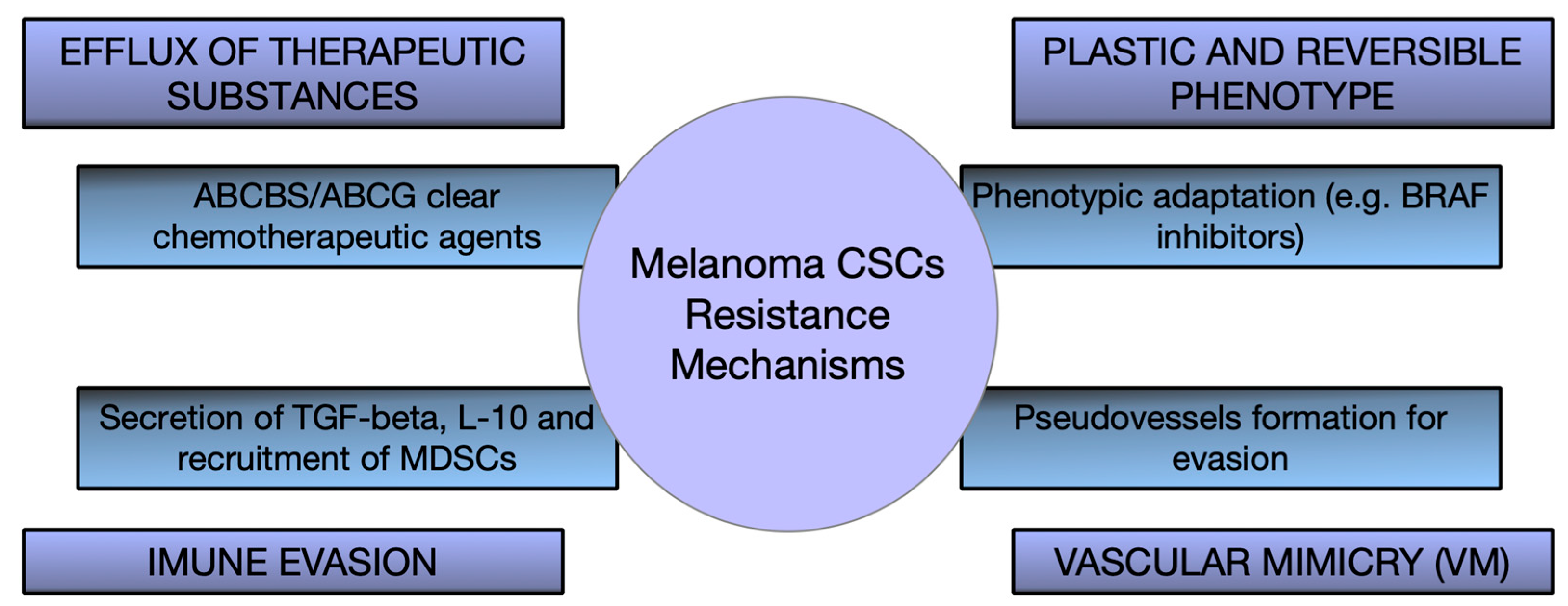
6. Therapeutic Implications and Targeting Strategies of CSCs
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lopes, J.; Rodrigues, C.M.P.; Gaspar, M.M.; Reis, C.P. Melanoma Management: From Epidemiology to Treatment and Latest Advances. Cancers 2022, 14, 4652. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Garbe, C.; Amaral, T.; Peris, K.; Hauschild, A.; Arenberger, P.; Basset-Seguin, N.; Bastholt, L.; Bataille, V.; Del Marmol, V.; Dréno, B.; et al. European consensus-based interdisciplinary guideline for melanoma. Part 1: Diagnostics: Update 2022. Eur. J. Cancer 2022, 170, 236–255. [Google Scholar] [CrossRef]
- Merlino, G.; Herlyn, M.; Fisher, D.E.; Bastian, B.C.; Flaherty, K.T.; Davies, M.A.; Wargo, J.A.; Curiel-Lewandrowski, C.; Weber, M.J.; Leachman, S.A.; et al. The state of melanoma: Challenges and opportunities. Pigment. Cell Melanoma Res. 2016, 29, 404–416. [Google Scholar] [CrossRef]
- Michielin, O.; Atkins, M.B.; Koon, H.B.; Dummer, R.; Ascierto, P.A. Evolving impact of long-term survival results on metastatic melanoma treatment. J. Immunother. Cancer 2020, 8, e000948. [Google Scholar] [CrossRef] [PubMed]
- Abbaszadegan, M.R.; Bagheri, V.; Razavi, M.S.; Momtazi, A.A.; Sahebkar, A.; Gholamin, M. Isolation, identification, and characterization of cancer stem cells: A review. J. Cell. Physiol. 2017, 232, 2008–2018. [Google Scholar] [CrossRef]
- Lodestijn, S.C.; Lenos, K.J.; Miedema, D.M.; Bijlsma, M.F.; Vermeulen, L. Cancer stem cells: Here, there, and everywhere. Mol. Cell. Oncol. 2018, 6, 1540235. [Google Scholar] [CrossRef]
- Almanaa, T.N.; Geusz, M.E.; Jamasbi, R.J. A new method for identifying stem-like cells in esophageal cancer cell lines. J. Cancer 2013, 4, 536–548. [Google Scholar] [CrossRef]
- Kreso, A.; Dick, J.E. Evolution of the cancer stem cell model. Cell Stem Cell 2014, 14, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Liang, R.; Li, L.; Guan, J. Studies on the effect and mechanism of CD147 on melanoma stem cells. Allergol. Immunopathol. 2024, 52, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Fang, D.; Nguyen, T.K.; Leishear, K.; Finko, R.; Kulp, A.N.; Hotz, S.; Van Belle, P.A.; Xu, X.; Elder, D.E.; Herlyn, M. A tumorigenic subpopulation with stem cell properties in melanomas. Cancer Res. 2005, 65, 9328–9337. [Google Scholar] [CrossRef]
- Al Hmada, Y.; Brodell, R.T.; Kharouf, N.; Flanagan, T.W.; Alamodi, A.A.; Hassan, S.Y.; Shalaby, H.; Hassan, S.L.; Haikel, Y.; Megahed, M.; et al. Mechanisms of Melanoma Progression and Treatment Resistance: Role of Cancer Stem-like Cells. Cancers 2024, 16, 470. [Google Scholar] [CrossRef] [PubMed]
- Jamal, S.M.E.; Alamodi, A.; Wahl, R.U.; Grada, Z.; Shareef, M.A.; Hassan, S.Y.; Murad, F.; Hassan, S.L.; Santourlidis, S.; Gomez, C.R.; et al. Melanoma stem cell maintenance and chemo-resistance are mediated by CD133 signal to PI3K-dependent pathways. Oncogene 2020, 39, 5468–5478. [Google Scholar] [CrossRef] [PubMed]
- Sabău, A.H.; Niculescu, R.; Cocuz, I.G.; Tinca, A.C.; Szöke, A.R.; Lazar, B.A.; Chiorean, D.M.; Budin, C.E.; Tomuț, A.N.; Cotoi, O.S. Characterizing CD133 and NANOG Expression in Melanoma: Associations with Histological and Epidemiological Parameters. Medicina 2024, 60, 1658. [Google Scholar] [CrossRef] [PubMed]
- El-Khattouti, A.; Sheehan, N.T.; Monico, J.; Drummond, H.A.; Haikel, Y.; Brodell, R.T.; Megahed, M.; Hassan, M. CD133+ melanoma subpopulation acquired resistance to caffeic acid phenethyl ester-induced apoptosis. Cancer Lett. 2015, 357, 83–104. [Google Scholar] [CrossRef]
- Speigl, L.; Janssen, N.; Weide, B.; Sinnberg, T.; Pawelec, G.; Shipp, C. Putative Cancer Stem Cell Markers are Frequently Expressed by Melanoma Cells. Front. Biosci. 2023, 28, 193. [Google Scholar] [CrossRef]
- Beretti, F.; Gatti, M.; Zavatti, M.; Bassoli, S.; Pellacani, G.; Maraldi, T. Reactive Oxygen Species Regulation of Chemoresistance and Metastatic Capacity of Melanoma: Role of the Cancer Stem Cell Marker CD271. Biomedicines 2023, 11, 1229. [Google Scholar] [CrossRef]
- Gerard, L.; Duvivier, L.; Fourrez, M.; Salazar, P.; Sprimont, L.; Xia, D.; Ambudkar, S.V.; Gottesman, M.M.; Gillet, J.P. Identification of two novel heterodimeric ABC transporters in melanoma. J. Biol. Chem. 2024, 300, 105594. [Google Scholar] [CrossRef]
- Yoganandarajah, V.; Patel, J.; van Schaijik, B.; Bockett, N.; Brasch, H.D.; Paterson, E.; Sim, D.; Davis, P.F.; Roth, I.M.; Itinteang, T.; et al. Identification of Cancer Stem Cell Subpopulations in Melanoma. Cells 2020, 9, 324. [Google Scholar] [CrossRef]
- Louphrasitthiphol, P.; Chauhan, J.; Goding, C.R. ABCB5 is activated by MITF and β-catenin. Pigment. Cell Melanoma Res. 2020, 33, 112–118. [Google Scholar] [CrossRef]
- Abou-Hamad, J.; Hodgins, J.J.; de Souza, C.T.; Garland, B.; Labrèche, C.; Marotel, M.; Gibson, C.; Delisle, S.; Pascoal, J.; Auer, R.C.; et al. CEACAM1 is a direct SOX10 target and inhibits melanoma immune infiltration and stemness. iScience 2022, 25, 105524. [Google Scholar] [CrossRef]
- Willis, B.C.; Johnson, G.; Wang, J.; Cohen, C. SOX10: A useful marker for identifying metastatic melanoma in sentinel lymph nodes. Appl. Immunohistochem. Mol. Morphol. 2015, 23, 109–112. [Google Scholar] [CrossRef]
- Kumar, D.; Gorain, M.; Kundu, G.; Kundu, G.C. Therapeutic implications of cellular and molecular biology of cancer stem cells in melanoma. Mol. Cancer 2017, 16, 7. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X. Targeting the Wnt/β-catenin signaling pathway in cancer. J. Hematol. Oncol. 2020, 13, 165. [Google Scholar] [CrossRef]
- Larue, L.; Delmas, V. The WNT/Beta-catenin pathway in melanoma. Front. Biosci. 2006, 11, 733–742. [Google Scholar] [CrossRef] [PubMed]
- Meisel, C.T.; Porcheri, C.; Mitsiadis, T.A. Cancer Stem Cells, Quo Vadis? The Notch Signaling Pathway in Tumor Initiation and Progression. Cells 2020, 9, 1879. [Google Scholar] [CrossRef]
- Muley, A.; Kim Uh, M.; Salazar-De Simone, G.; Swaminathan, B.; James, J.M.; Murtomaki, A.; Youn, S.W.; McCarron, J.D.; Kitajewski, C.; Gnarra Buethe, M.; et al. Unique functions for Notch4 in murine embryonic lymphangiogenesis. Angiogenesis 2022, 25, 205–224. [Google Scholar] [CrossRef]
- Fattahi, S.; Pilehchian Langroudi, M.; Akhavan-Niaki, H. Hedgehog signaling pathway: Epigenetic regulation and role in disease and cancer development. J. Cell. Physiol. 2018, 233, 5726–5735. [Google Scholar] [CrossRef] [PubMed]
- Marini, K.D.; Payne, B.J.; Watkins, D.N.; Martelotto, L.G. Mechanisms of Hedgehog signalling in cancer. Growth Factors 2011, 29, 221–234. [Google Scholar] [CrossRef]
- Giuntini, G.; Coppola, F.; Falsini, A.; Filippi, I.; Monaci, S.; Naldini, A.; Carraro, F. Role of the Hedgehog Pathway and CAXII in Controlling Melanoma Cell Migration and Invasion in Hypoxia. Cancers 2022, 14, 4776. [Google Scholar] [CrossRef] [PubMed]
- Adorno-Cruz, V.; Kibria, G.; Liu, X.; Doherty, M.; Junk, D.J.; Guan, D.; Hubert, C.; Venere, M.; Mulkearns-Hubert, E.; Sinyuk, M.; et al. Cancer stem cells: Targeting the roots of cancer, seeds of metastasis, and sources of therapy resistance. Cancer Res. 2015, 75, 924–929. [Google Scholar] [CrossRef]
- Dashti, A.; Ebrahimi, M.; Hadjati, J.; Memarnejadian, A.; Moazzeni, S.M. Dendritic cell based immunotherapy using tumor stem cells mediates potent antitumor immune responses. Cancer Lett. 2016, 374, 175–185. [Google Scholar] [CrossRef]
- Lobo, N.A.; Shimono, Y.; Qian, D.; Clarke, M.F. The biology of cancer stem cells. Annu. Rev. Cell Dev. Biol. 2007, 23, 675–699. [Google Scholar] [CrossRef]
- Huang, F.; Santinon, F.; Flores González, R.E.; Del Rincón, S.V. Melanoma Plasticity: Promoter of Metastasis and Resistance to Therapy. Front. Oncol. 2021, 11, 756001. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, F.; Haass, N.K. Microenvironment-Driven Dynamic Heterogeneity and Phenotypic Plasticity as a Mechanism of Melanoma Therapy Resistance. Front. Oncol. 2018, 8, 173. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Rebecca, V.W.; Kossenkov, A.V.; Connelly, T.; Liu, Q.; Gutierrez, A.; Xiao, M.; Li, L.; Zhang, G.; Samarkina, A.; et al. Neural Crest-Like Stem Cell Transcriptome Analysis Identifies LPAR1 in Melanoma Progression and Therapy Resistance. Cancer Res. 2021, 81, 5230–5241. [Google Scholar] [CrossRef] [PubMed]
- Diazzi, S.; Tartare-Deckert, S.; Deckert, M. The mechanical phenotypic plasticity of melanoma cell: An emerging driver of therapy cross-resistance. Oncogenesis 2023, 12, 7. [Google Scholar] [CrossRef]
- Hossain, S.M.; Eccles, M.R. Phenotype Switching and the Melanoma Microenvironment; Impact on Immunotherapy and Drug Resistance. Int. J. Mol. Sci. 2023, 24, 1601. [Google Scholar] [CrossRef]
- Nallasamy, P.; Nimmakayala, R.K.; Parte, S.; Are, A.C.; Batra, S.K.; Ponnusamy, M.P. Tumor microenvironment enriches the stemness features: The architectural event of therapy resistance and metastasis. Mol. Cancer 2022, 21, 225. [Google Scholar] [CrossRef]
- Kyriakou, G.; Melachrinou, M. Cancer stem cells, epigenetics, tumor microenvironment and future therapeutics in cutaneous malignant melanoma: A review. Future Oncol. 2020, 16, 1549–1567. [Google Scholar] [CrossRef]
- Tanabe, S. Microenvironment of Cancer Stem Cells. Adv. Exp. Med. Biol. 2022, 1393, 103–124. [Google Scholar] [CrossRef]
- Guo, Q.; Zhou, Y.; Xie, T.; Yuan, Y.; Li, H.; Shi, W.; Zheng, L.; Li, X.; Zhang, W. Tumor microenvironment of cancer stem cells: Perspectives on cancer stem cell targeting. Genes Dis. 2023, 11, 101043. [Google Scholar] [CrossRef]
- Maccalli, C.; Volontè, A.; Cimminiello, C.; Parmiani, G. Immunology of cancer stem cells in solid tumours. A review. Eur. J. Cancer 2014, 50, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Tinca, A.C.; Cocuz, I.G.; Șincu, M.C.; Niculescu, R.; Sabău, A.H.; Chiorean, D.M.; Szőke, A.R.; Cotoi, O.S. VISTA, PDL-L1, and BRAF—A Review of New and Old Markers in the Prognosis of Melanoma. Medicina 2022, 58, 74. [Google Scholar] [CrossRef] [PubMed]
- Friedmann-Morvinski, D.; Verma, I.M. Dedifferentiation and reprogramming: Origins of cancer stem cells. EMBO Rep. 2014, 15, 244–253. [Google Scholar] [CrossRef]
- Marzagalli, M.; Raimondi, M.; Fontana, F.; Montagnani Marelli, M.; Moretti, R.M.; Limonta, P. Cellular and molecular biology of cancer stem cells in melanoma: Possible therapeutic implications. Semin. Cancer Biol. 2019, 59, 221–235. [Google Scholar] [CrossRef] [PubMed]
- Bogunovic, D.; O’Neill, D.W.; Belitskaya-Levy, I.; Vacic, V.; Yu, Y.L.; Adams, S.; Darvishian, F.; Berman, R.; Shapiro, R.; Pavlick, A.C.; et al. Immune profile and mitotic index of metastatic melanoma lesions enhance clinical staging in predicting patient survival. Proc. Natl. Acad. Sci. USA 2009, 106, 20429–20434. [Google Scholar] [CrossRef]
- Schlaak, M.; Schmidt, P.; Bangard, C.; Kurschat, P.; Mauch, C.; Abken, H. Regression of metastatic melanoma in a patient by antibody targeting of cancer stem cells. Oncotarget 2012, 3, 22–30. [Google Scholar] [CrossRef]
- Madjd, Z.; Erfani, E.; Gheytanchi, E.; Moradi-Lakeh, M.; Shariftabrizi, A.; Asadi-Lari, M. Expression of CD133 cancer stem cell marker in melanoma: A systematic review and meta-analysis. Int. J. Biol. Markers 2016, 31, e118–e125. [Google Scholar] [CrossRef]
- Welte, Y.; Davies, C.; Schäfer, R.; Regenbrecht, C.R. Patient derived cell culture and isolation of CD133+ putative cancer stem cells from melanoma. J. Vis. Exp. 2013, 73, e50200. [Google Scholar] [CrossRef]
- Roudi, R.; Korourian, A.; Shariftabrizi, A.; Madjd, Z. Differential Expression of Cancer Stem Cell Markers ALDH1 and CD133 in Various Lung Cancer Subtypes. Cancer Investig. 2015, 33, 294–302. [Google Scholar] [CrossRef]
- Akil, A.; Gutiérrez-García, A.K.; Guenter, R.; Rose, J.B.; Beck, A.W.; Chen, H.; Ren, B. Notch Signaling in Vascular Endothelial Cells, Angiogenesis, and Tumor Progression: An Update and Prospective. Front. Cell Dev. Biol. 2021, 9, 642352. [Google Scholar] [CrossRef]
- Monzani, E.; Facchetti, F.; Galmozzi, E.; Corsini, E.; Benetti, A.; Cavazzin, C.; Gritti, A.; Piccinini, A.; Porro, D.; Santinami, M.; et al. Melanoma contains CD133 and ABCG2 positive cells with enhanced tumourigenic potential. Eur. J. Cancer 2007, 43, 935–946. [Google Scholar] [CrossRef] [PubMed]
- Klein, W.M.; Wu, B.P.; Zhao, S.; Wu, H.; Klein-Szanto, A.J.; Tahan, S.R. Increased expression of stem cell markers in malignant melanoma. Mod. Pathol. 2007, 20, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Thomas-Pfaab, M.; Annereau, J.P.; Munsch, C.; Guilbaud, N.; Garrido, I.; Paul, C.; Brousset, P.; Lamant, L.; Meyer, N. CD10 expression by melanoma cells is associated with aggressive behavior in vitro and predicts rapid metastatic progression in humans. J. Dermatol. Sci. 2013, 69, 105–113. [Google Scholar] [CrossRef]
- Yin, Q.; Shi, X.; Lan, S.; Jin, H.; Wu, D. Effect of melanoma stem cells on melanoma metastasis. Oncol. Lett. 2021, 22, 566. [Google Scholar] [CrossRef]
- Boiko, A.D.; Razorenova, O.V.; van de Rijn, M.; Swetter, S.M.; Johnson, D.L.; Ly, D.P.; Butler, P.D.; Yang, G.P.; Joshua, B.; Kaplan, M.J.; et al. Human melanoma-initiating cells express neural crest nerve growth factor receptor CD271. Nature 2010, 466, 133–137. [Google Scholar] [CrossRef]
- Vidal, A.; Redmer, T. Decoding the Role of CD271 in Melanoma. Cancers 2020, 12, 2460. [Google Scholar] [CrossRef] [PubMed]
- Cheli, Y.; Bonnazi, V.F.; Jacquel, A.; Allegra, M.; De Donatis, G.M.; Bahadoran, P.; Bertolotto, C.; Ballotti, R. CD271 Is an Imperfect Marker for Melanoma Initiating Cells. Oncotarget 2014, 5, 5272–5283. [Google Scholar] [CrossRef]
- Frank, N.Y.; Margaryan, A.; Huang, Y.; Schatton, T.; Waaga-Gasser, A.M.; Gasser, M.; Sayegh, M.H.; Sadee, W.; Frank, M.H. ABCB5-Mediated Doxorubicin Transport and Chemoresistance in Human Malignant Melanoma. Cancer Res. 2005, 65, 4320–4333. [Google Scholar] [CrossRef]
- Huang, Y.; Anderle, P.; Bussey, K.J.; Barbacioru, C.; Shankavaram, U.; Dai, Z.; Reinhold, W.C.; Papp, A.; Weinstein, J.N.; Sadée, W. Membrane Transporters and Channels: Role of the Transportome in Cancer Chemosensitivity and Chemoresistance. Cancer Res. 2004, 64, 4294–4301. [Google Scholar] [CrossRef] [PubMed]
- Murphy, G.F.; Wilson, B.J.; Girouard, S.D.; Frank, N.Y.; Frank, M.H. Stem Cells and Targeted Approaches to Melanoma Cure. Mol. Asp. Med. 2014, 39, 33–49. [Google Scholar] [CrossRef]
- Wang, S.; Tang, L.; Lin, J.; Shen, Z.; Yao, Y.; Wang, W.; Tao, S.; Gu, C.; Ma, J.; Xie, Y.; et al. ABCB5 Promotes Melanoma Metastasis through Enhancing NF-κB p65 Protein Stability. Biochem. Biophys. Res. Commun. 2017, 492, 18–26. [Google Scholar] [CrossRef]
- Parmiani, G. Melanoma Cancer Stem Cells: Markers and Functions. Cancers 2016, 8, 34. [Google Scholar] [CrossRef]
- Douville, J.; Beaulieu, R.; Balicki, D. ALDH1 as a Functional Marker of Cancer Stem and Progenitor Cells. Stem Cells Dev. 2009, 18, 17–25. [Google Scholar] [CrossRef]
- Toledo-Guzmán, M.E.; Hernández, M.I.; Gómez-Gallegos, Á.A.; Ortiz-Sánchez, E. ALDH as a Stem Cell Marker in Solid Tumors. Curr. Stem Cell Res. Ther. 2019, 14, 375–388. [Google Scholar] [CrossRef]
- Burger, P.E.; Gupta, R.; Xiong, X.; Ontiveros, C.S.; Salm, S.N.; Moscatelli, D.; Wilson, E.L. High Aldehyde Dehydrogenase Activity: A Novel Functional Marker of Murine Prostate Stem/Progenitor Cells. Stem Cells 2009, 27, 2220–2228. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Chai, S.; Wang, P.; Zhang, C.; Yang, Y.; Yang, Y.; Wang, K. Aldehyde Dehydrogenases and Cancer Stem Cells. Cancer Lett. 2015, 369, 50–57. [Google Scholar] [CrossRef]
- Luo, Y.; Dallaglio, K.; Chen, Y.; Robinson, W.A.; Robinson, S.E.; McCarter, M.D.; Wang, J.; Gonzalez, R.; Thompson, D.C.; Norris, D.A.; et al. ALDH1A Isozymes Are Markers of Human Melanoma Stem Cells and Potential Therapeutic Targets. Stem Cells 2012, 30, 2100–2113. [Google Scholar] [CrossRef] [PubMed]
- Prasmickaite, L.; Engesaeter, B.Ø.; Skrbo, N.; Hellenes, T.; Kristian, A.; Oliver, N.K.; Suo, Z.; Maelandsmo, G.M. Aldehyde Dehydrogenase (ALDH) Activity Does Not Select for Cells with Enhanced Aggressive Properties in Malignant Melanoma. PLoS ONE 2010, 5, e10731. [Google Scholar] [CrossRef]
- Tinca, A.C.; Raicea, A.; Szőke, A.R.; Cocuz, I.G.; Şincu, M.C.; Niculescu, R.; Sabău, A.H.; Popelea, M.C.; Fruntelată, R.F.; Cotoi, O.S. Morphological Aspects and Therapeutic Options in Melanoma: A Narrative Review of the Past Decade. Rom. J. Morphol. Embryol. 2023, 64, 135–141. [Google Scholar] [CrossRef]
- Sun, C.; Wang, L.; Huang, S.; Heynen, G.J.; Prahallad, A.; Robert, C.; Haanen, J.; Blank, C.; Wesseling, J.; Willems, S.M.; et al. Reversible and Adaptive Resistance to BRAF(V600E) Inhibition in Melanoma. Nature 2014, 508, 118–122. [Google Scholar] [CrossRef]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; et al. Targeting Cancer Stem Cell Pathways for Cancer Therapy. Signal Transduct. Target Ther. 2020, 5, 8. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, G.; Stromhaug, K.; Klaeger, S.; Kula, T.; Frederick, D.T.; Le, P.M.; Forman, J.; Huang, T.; Li, S.; Zhang, W.; et al. Phenotype, Specificity and Avidity of Antitumour CD8+ T Cells in Melanoma. Nature 2021, 596, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Bradley, E.W.; Drissi, M.H. Wnt5b Regulates Mesenchymal Cell Aggregation and Chondrocyte Differentiation through the Planar Cell Polarity Pathway. J. Cell. Physiol. 2011, 226, 1683–1693. [Google Scholar] [CrossRef]
- Radaszkiewicz, T.; Nosková, M.; Gömöryová, K.; Vondálová Blanářová, O.; Radaszkiewicz, K.A.; Picková, M.; Víchová, R.; Gybeľ, T.; Kaiser, K.; Demková, L.; et al. RNF43 Inhibits WNT5A-driven Signaling and Suppresses Melanoma Invasion and Resistance to the Targeted Therapy. eLife 2021, 10, e65759. [Google Scholar] [CrossRef]
- Regad, T. Molecular and Cellular Pathogenesis of Melanoma Initiation and Progression. Cell. Mol. Life Sci. 2013, 70, 4055–4065. [Google Scholar] [CrossRef]
- Santini, R.; Vinci, M.C.; Pandolfi, S.; Penachioni, J.Y.; Montagnani, V.; Olivito, B.; Gattai, R.; Pimpinelli, N.; Gerlini, G.; Borgognoni, L.; et al. Hedgehog-GLI Signaling Drives Self-renewal and Tumorigenicity of Human Melanoma-initiating Cells. Stem Cells 2012, 30, 1808–1818. [Google Scholar] [CrossRef]
- Li, C.; Nguyen, V.; Clark, K.N.; Zahed, T.; Sharkas, S.; Filipp, F.V.; Boiko, A.D. Down-regulation of FZD3 Receptor Suppresses Growth and Metastasis of Human Melanoma Independently of Canonical WNT Signaling. Proc. Natl. Acad. Sci. USA 2019, 116, 4548–4557. [Google Scholar] [CrossRef]
- Skoda, A.M.; Simovic, D.; Karin, V.; Kardum, V.; Vranic, S.; Serman, L. The Role of the Hedgehog Signaling Pathway in Cancer: A Comprehensive Review. Bosn. J. Basic Med. Sci. 2018, 18, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Stecca, B.; Mas, C.; Clement, V.; Zbinden, M.; Correa, R.; Piguet, V.; Beermann, F.; Ruiz i Altaba, A. Melanomas Require HEDGEHOG-GLI Signaling Regulated by Interactions between GLI1 and the RAS-MEK/AKT Pathways. Proc. Natl. Acad. Sci. USA 2007, 104, 5895–5900. [Google Scholar] [CrossRef] [PubMed]
- Faião-Flores, F.; Alves-Fernandes, D.K.; Pennacchi, P.C.; Sandri, S.; Vicente, A.L.; Scapulatempo-Neto, C.; Vazquez, V.L.; Reis, R.M.; Chauhan, J.; Goding, C.R.; et al. Targeting the Hedgehog Transcription Factors GLI1 and GLI2 Restores Sensitivity to Vemurafenib-resistant Human Melanoma Cells. Oncogene 2017, 36, 1849–1861. [Google Scholar] [CrossRef] [PubMed]
- Barbato, L.; Bocchetti, M.; Di Biase, A.; Regad, T. Cancer Stem Cells and Targeting Strategies. Cells 2019, 8, 926. [Google Scholar] [CrossRef]
- Valdez-Salazar, F.; Jiménez-Del Rio, L.A.; Padilla-Gutiérrez, J.R.; Valle, Y.; Muñoz-Valle, J.F.; Valdés-Alvarado, E. Advances in Melanoma: From Genetic Insights to Therapeutic Innovations. Biomedicines 2024, 12, 1851. [Google Scholar] [CrossRef] [PubMed]
- Hazarika, M.; Chuk, M.K.; Theoret, M.R.; Mushti, S.; He, K.; Weis, S.L.; Putman, A.H.; Helms, W.S.; Cao, X.; Li, H.; et al. U.S. FDA Approval Summary: Nivolumab for Treatment of Unresectable or Metastatic Melanoma Following Progression on Ipilimumab. Clin. Cancer Res. 2017, 23, 3484–3488. [Google Scholar] [CrossRef]
- Ertas, Y.N.; Abedi Dorcheh, K.; Akbari, A.; Jabbari, E. Nanoparticles for Targeted Drug Delivery to Cancer Stem Cells: A Review of Recent Advances. Nanomaterials 2021, 11, 1755. [Google Scholar] [CrossRef]
- Tang, Y.; Chen, Y.; Zhang, Z.; Tang, B.; Zhou, Z.; Chen, H. Nanoparticle-Based RNAi Therapeutics Targeting Cancer Stem Cells: Update and Prospective. Pharmaceutics 2021, 13, 2116. [Google Scholar] [CrossRef]
- Cheng, Z.; Li, M.; Dey, R.; Chen, Y. Nanomaterials for Cancer Therapy: Current Progress and Perspectives. J. Hematol. Oncol. 2021, 14, 85. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| MARKER | TYPE | PROPOSED ROLE | SPECIFICITY AND OBSERVATIONS |
|---|---|---|---|
| CD133 (PROMININ-1) | Membrane marker | Self-renewal and tumorigenesis | Limited to a small fraction of cells, but also present in non-stem cells |
| CD271 (P75 NGFR) | Membrane marker | Associated with invasion and survival | Present in CSCs, but also in other malignant cell populations and normal melanocytes |
| ABCB5/ ABCG2 | Functional marker | Therapeutic resistance through toxic efflux | Expressed in CSCs and associated with resistance, but not specific to the CSC subpopulation |
| ALDH1 | Enzymatic marker (high metabolic activity) | Self-renewal and therapeutic resistance | Associated with the CSC phenotype, but also detected in non-CSC cell populations in melanoma |
| OCT4, SOX2, NANOG | Transcription factor | Maintenance of pluripotency | Variable expression in CSCs and in tumor cells with increased plasticity |
| MOLECULAR PATHWAY | KEY COMPONENTS | EFFECTS ON CSCS | THERAPEUTIC IMPLICATIONS |
|---|---|---|---|
| WNT/β-CATENIN | Nuclear β-catenin, FZD3 | Self-renewal, survival specific | β-catenin inhibitors |
| NOTCH | Notch receptors, Jagged/Delta ligands | Maintenance of undifferentiation and self-renewal | γ-secretase inhibitors |
| HEDGEHOG (HH) | SHH-GLI | Proliferation, invasion, therapeutic resistance | Vismodegib-like inhibitors |
| HIPOXIA/HIF-1 | HIF-1α, TWIST, SNAIL, ZEB1 | EMT, survival, and metabolic adaptation | Agents that block HIF activity |
| MICROENVIRONMENT COMPONENT | SECRETED FACTORS | ROLE IN INTERACTION WITH CSC | POTENTIAL THERAPEUTIC TARGET |
|---|---|---|---|
| CAF SITES | miR-214, pro-tumor cytokines | Support of CSC invasion and self-renewal | Inhibitors of CAF activation |
| M2 MACROPHAGES (TAM) | TGF-β, IL-10 | Immunosuppression and protection of CSCs | TAM repolarization toward the M1 phenotype |
| MYELOID SUPPRESSOR CELLS (MDSCS) | IL-6 | CSC maintenance via STAT3 activation | IL-6/JAK-STAT inhibitors |
| NEUTROPHILS N2 | ROS, MMP-9 | Promotion of invasion and potentiation of CSCs | Blocking chemotactic factors (IL-8) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sabău, A.-H.; Tinca, A.-C.; Niculescu, R.; Cocuz, I.G.; Cozac-Szöke, A.R.; Lazar, B.A.; Chiorean, D.M.; Budin, C.E.; Cotoi, O.S. Cancer Stem Cells in Melanoma: Drivers of Tumor Plasticity and Emerging Therapeutic Strategies. Int. J. Mol. Sci. 2025, 26, 7419. https://doi.org/10.3390/ijms26157419
Sabău A-H, Tinca A-C, Niculescu R, Cocuz IG, Cozac-Szöke AR, Lazar BA, Chiorean DM, Budin CE, Cotoi OS. Cancer Stem Cells in Melanoma: Drivers of Tumor Plasticity and Emerging Therapeutic Strategies. International Journal of Molecular Sciences. 2025; 26(15):7419. https://doi.org/10.3390/ijms26157419
Chicago/Turabian StyleSabău, Adrian-Horațiu, Andreea-Cătălina Tinca, Raluca Niculescu, Iuliu Gabriel Cocuz, Andreea Raluca Cozac-Szöke, Bianca Andreea Lazar, Diana Maria Chiorean, Corina Eugenia Budin, and Ovidiu Simion Cotoi. 2025. "Cancer Stem Cells in Melanoma: Drivers of Tumor Plasticity and Emerging Therapeutic Strategies" International Journal of Molecular Sciences 26, no. 15: 7419. https://doi.org/10.3390/ijms26157419
APA StyleSabău, A.-H., Tinca, A.-C., Niculescu, R., Cocuz, I. G., Cozac-Szöke, A. R., Lazar, B. A., Chiorean, D. M., Budin, C. E., & Cotoi, O. S. (2025). Cancer Stem Cells in Melanoma: Drivers of Tumor Plasticity and Emerging Therapeutic Strategies. International Journal of Molecular Sciences, 26(15), 7419. https://doi.org/10.3390/ijms26157419