
Genetic and Metabolic Factors of Familial Dysbetalipoproteinemia Phenotype: Insights from a Cross-Sectional Study

 ,
,  , , , , ,
, , , , ,  and
and
Abstract
1. Introduction
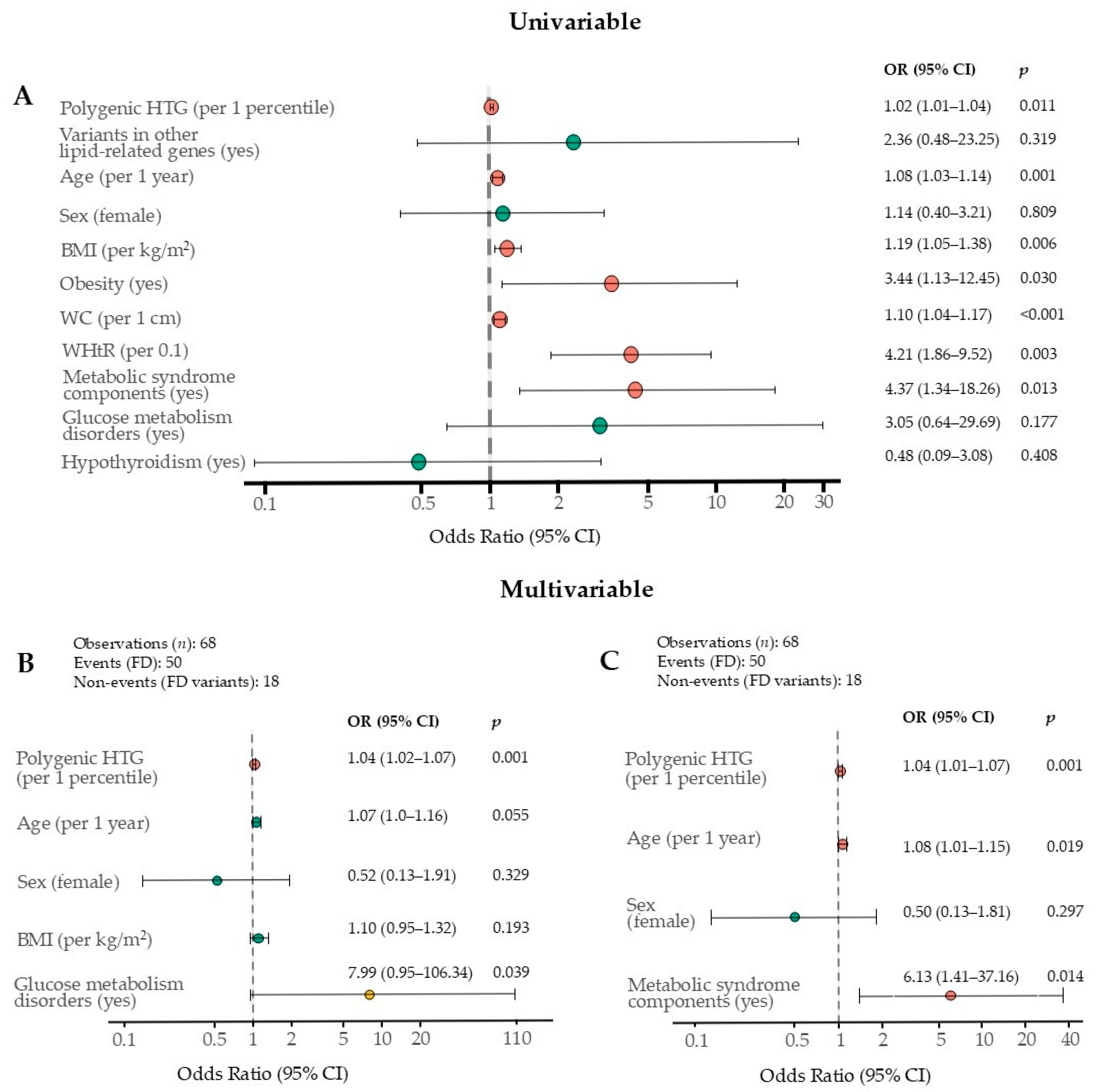
2. Results
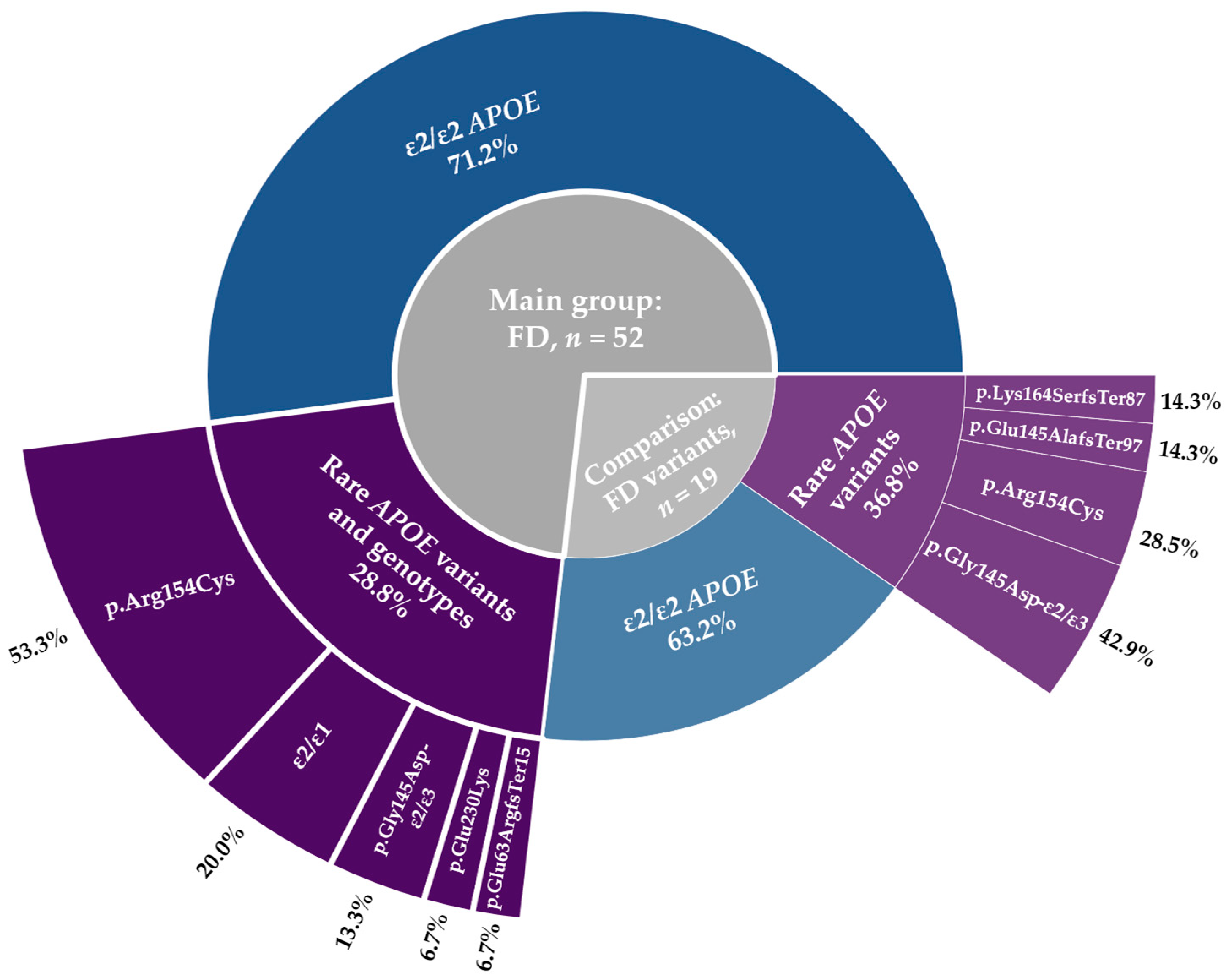
2.1. Genetic Factors
2.1.1. Pathogenic Variants in Lipid-Related Genes Beyond APOE
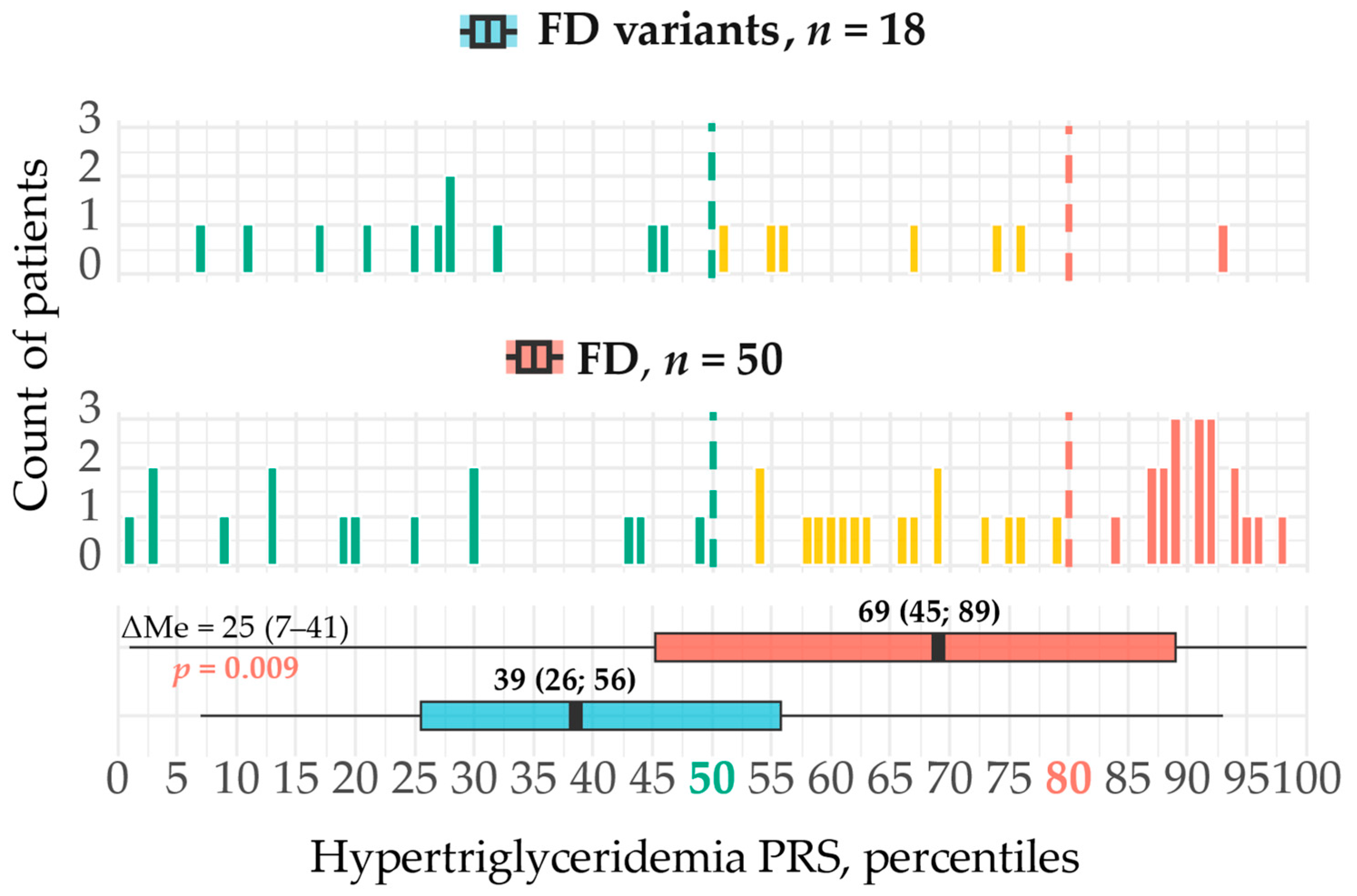
2.1.2. Polygenic Risk
2.2. Clinical Factors
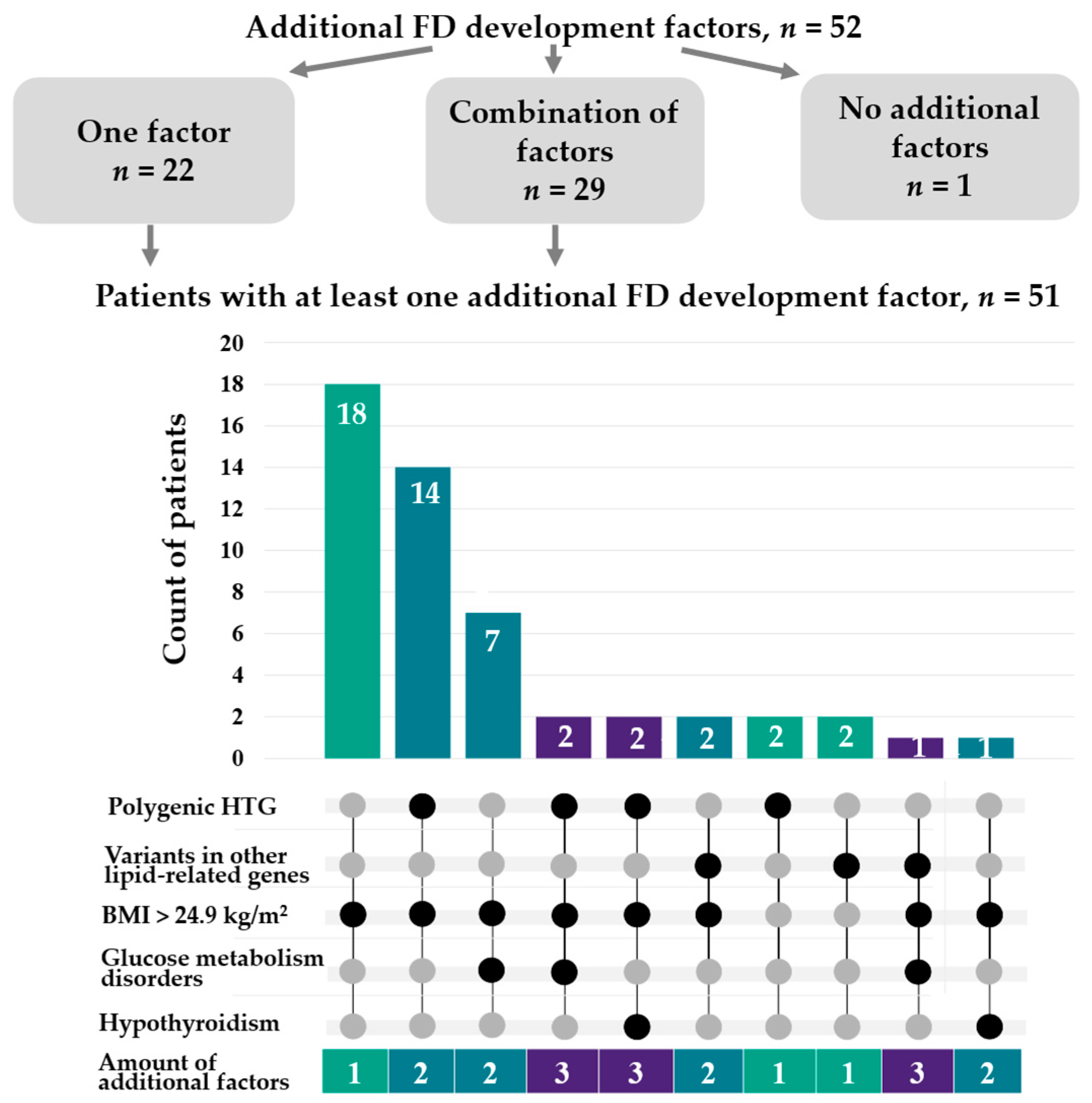
2.3. Independent FD Phenotype Factors
2.4. Treatment Targets in FD Patients
3. Discussion
3.1. Polygenic HTG
3.2. Rare Additional Genetic Variants Beyond APOE
3.3. Metabolic Factors
4. Materials and Methods
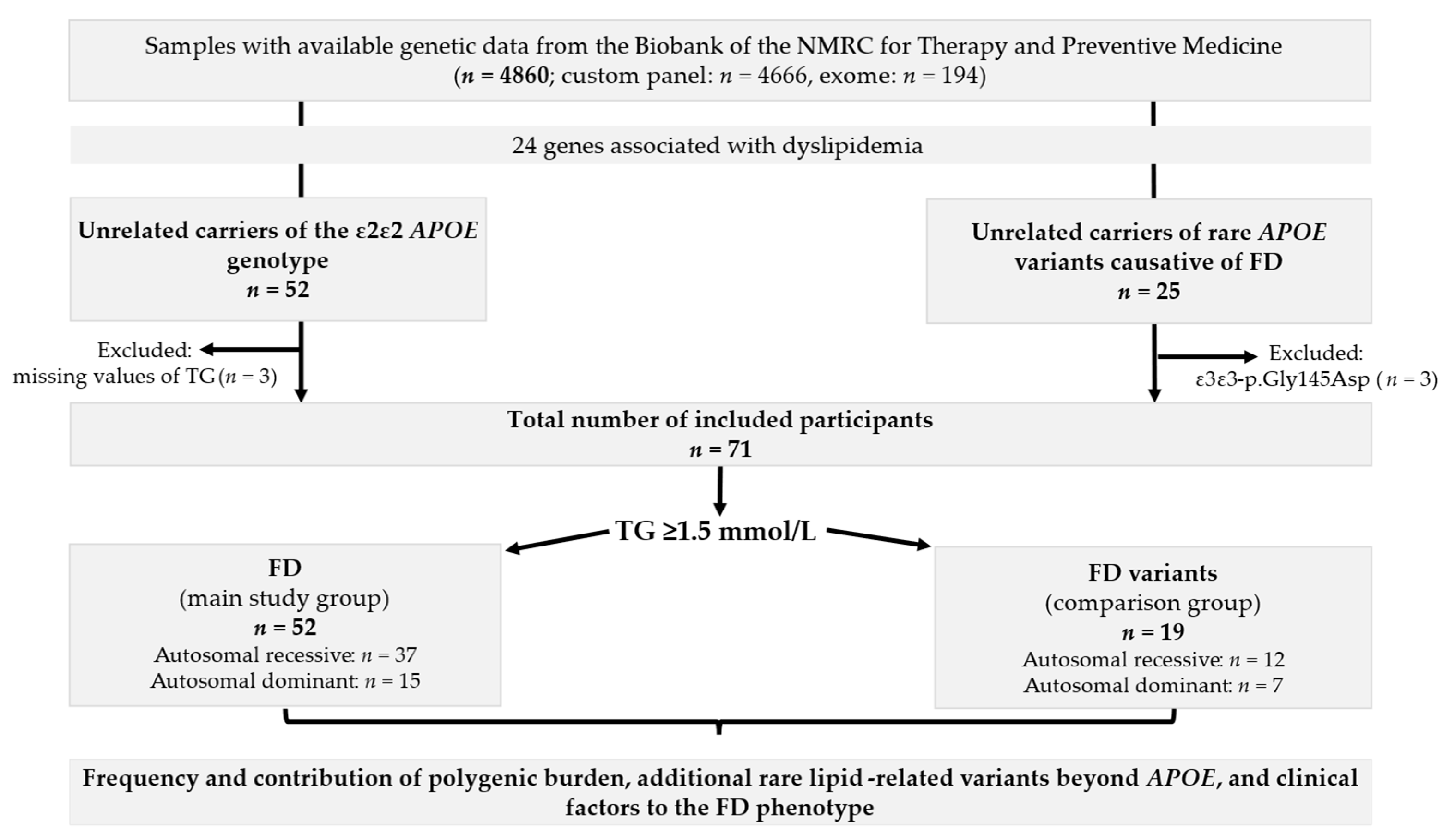
4.1. Sampling
- Population samples from the “Epidemiology of Cardiovascular Diseases and Risk Factors in Regions of the Russian Federation” (ESSE-RF) study, a cross-sectional study conducted from 2012 to 2013 across 13 regions of Russia [28]. The current study included representative samples from the Ivanovo region (n = 1858) [1] and the Vologda region (n = 880) [29], as well as single subjects from other regions (n = 167), and participants from the ESSE-FH-RF study with a clinical diagnosis of definite or probable heterozygous FH (n = 119) [30];
- Subjects (the Russian patient sample (RPS)) with diverse chronic noncommunicable diseases, including lipid metabolism disorders, whose blood samples were collected at the NMRC for Therapy and Preventive Medicine Biobank (n = 1836) [27].
4.2. Clinical and Biochemical Data
4.3. Genetic Analysis
4.3.1. NGS
4.3.2. Bioinformatic Analysis and Clinical Interpretation
4.4. Polygenic Risk Score
4.5. Ethical Statement
4.6. Statistical Analysis
Statistical Power Analysis
4.7. Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| FD | Familial dysbetalipoproteinemia |
| HTG | Hypertriglyceridemia |
| TG | Triglyceride |
| NGS | Next-generation sequencing |
| AR | Autosomal recessive |
| AD | Autosomal dominant |
| FH | Familial hypercholesterolemia |
| CV | Cardiovascular |
| OR | Odds ratio |
| CI | Confidence interval |
| PRS | Polygenic risk score |
| ΔMe | Median difference |
| Lp(a) | Lipoprotein (a) |
| BMI | Body mass index |
| WHtR | Waist-to-height ratio |
| CMI | Cardiometabolic index |
| LDL-C | Low-density lipoprotein cholesterol |
| HDL-C | High-density lipoprotein cholesterol |
| WC | Waist circumference |
| LLT | Lipid-lowering therapy |
| NMRC | National Medical Research Center |
| ESSE-RF | Epidemiology of Cardiovascular Diseases and Risk Factors in Regions of the Russian Federation |
| TC | Total cholesterol |
| RPS | Russian patient sample |
| PCA | Principal component analysis |
References
- Blokhina, A.V.; Ershova, A.I.; Kiseleva, A.V.; Sotnikova, E.A.; Zharikova, A.A.; Zaicenoka, M.; Vyatkin, Y.V.; Ramensky, V.E.; Kutsenko, V.A.; Garbuzova, E.V.; et al. Spectrum and Prevalence of Rare APOE Variants and Their Association with Familial Dysbetalipoproteinemia. Int. J. Mol. Sci. 2024, 25, 12651. [Google Scholar] [CrossRef]
- Bea, A.M.; Larrea-Sebal, A.; Marco-Benedi, V.; Uribe, K.B.; Galicia-Garcia, U.; Lamiquiz-Moneo, I.; Laclaustra, M.; Moreno-Franco, B.; Fernandez-Corredoira, P.; Olmos, S.; et al. Contribution of APOE genetic variants to dyslipidemia. Arterioscler. Thromb. Vasc. Biol. 2023, 43, 1066–1077. [Google Scholar] [CrossRef] [PubMed]
- Koopal, C.; Marais, A.D.; Visseren, F.L.J. Familial dysbetalipoproteinemia: An underdiagnosed lipid disorder. Curr. Opin. Endocrinol. Diabetes Obes. 2017, 24, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Blokhina, A.V.; Ershova, A.I.; Meshkov, A.N.; Drapkina, O.M. Familial dysbetalipoproteinemia: Highly atherogenic and underdiagnosed disorder. Cardiovasc. Ther. Prev. 2021, 20, 2893. (In Russian) [Google Scholar] [CrossRef]
- Marais, A.D.; Solomon, G.A.; Blom, D.J. Dysbetalipoproteinaemia: A mixed hyperlipidaemia of remnant lipoproteins due to mutations in apolipoprotein E. Crit. Rev. Clin. Lab. Sci. 2014, 51, 46–62. [Google Scholar] [CrossRef] [PubMed]
- Smelt, A.H.M.; De Beer, F. Apolipoprotein E and familial dysbetalipoproteinemia: Clinical, biochemical, and genetic aspects. Semin. Vasc. Med. 2004, 4, 249–257. [Google Scholar] [CrossRef]
- Heidemann, B.E.; Koopal, C.; Baass, A.; Defesche, J.C.; Zuurbier, L.; Mulder, M.T.; Roeters van Lennep, J.E.; Riksen, N.P.; Boot, C.; Marais, A.D.; et al. Establishing the relationship between familial dysbetalipoproteinemia and genetic variants in the APOE gene. Clin. Genet. 2022, 102, 253–261. [Google Scholar] [CrossRef]
- Koopal, C.; Marais, A.D.; Westerink, J.; Visseren, F.L.J. Autosomal dominant familial dysbetalipoproteinemia: A pathophysiological framework and practical approach to diagnosis and therapy. J. Clin. Lipidol. 2017, 11, 12–23. [Google Scholar] [CrossRef]
- Blokhina, A.V.; Ershova, A.I.; Kiseleva, A.V.; Sotnikova, E.A.; Zharikova, A.A.; Zaicenoka, M.; Vyatkin, Y.V.; Ramensky, V.E.; Kutsenko, V.A.; Litinskaya, O.A.; et al. Clinical and biochemical features of atherogenic hyperlipidemias with different genetic basis: A comprehensive comparative study. PLoS ONE 2024, 19, e0315693. [Google Scholar] [CrossRef]
- Hopkins, P.N.; Wu, L.L.; Hunt, S.C.; Brinton, E.A. Plasma triglycerides and type III hyperlipidemia are independently associated with premature familial coronary artery disease. J. Am. Coll. Cardiol. 2005, 45, 1003–1012. [Google Scholar] [CrossRef]
- Koopal, C.; Retterstøl, K.; Sjouke, B.; Hovingh, G.K.; Ros, E.; de Graaf, J.; Dullaart, R.P.; Bertolini, S.; Visseren, F.L. Vascular risk factors, vascular disease, lipids and lipid targets in patients with familial dysbetalipoproteinemia: A European cross-sectional study. Atherosclerosis 2015, 240, 90–97. [Google Scholar] [CrossRef]
- Paquette, M.; Trinder, M.; Guay, S.P.; Brunham, L.R.; Baass, A. Predictors of Cardiovascular Disease in Individuals with Dysbetalipoproteinemia: A Prospective Study in the UK Biobank. J. Clin. Endocrinol. Metab. 2025, 110, e1959–e1965. [Google Scholar] [CrossRef] [PubMed]
- Bea, A.M.; Civeira, F.; Jarauta, E.; Lamiquiz-Moneo, I.; Pérez-Calahorra, S.; Marco-Benedí, V.; Cenarro, A.; Mateo-Gallego, R. Association Between the Presence of Carotid Artery Plaque and Cardiovascular Events in Patients with Genetic Hypercholesterolemia. Rev. Esp. Cardiol. 2017, 70, 551–558. [Google Scholar] [CrossRef]
- Blokhina, A.V.; Ershova, A.I.; Kiseleva, A.V.; Sotnikova, E.A.; Zharikova, A.A.; Zaicenoka, M.; Vyatkin, Y.V.; Ramensky, V.E.; Kutsenko, V.A.; Shalnova, S.A.; et al. Applicability of Diagnostic Criteria and High Prevalence of Familial Dysbetalipoproteinemia in Russia: A Pilot Study. Int. J. Mol. Sci. 2023, 24, 13159. [Google Scholar] [CrossRef]
- Paquette, M.; Trinder, M.; Guay, S.P.; Brunham, L.R.; Baass, A. Prevalence of Dysbetalipoproteinemia in the UK Biobank According to Different Diagnostic Criteria. J. Clin. Endocrinol. Metab. 2024, 110, 703–709. [Google Scholar] [CrossRef]
- Pallazola, V.A.; Sathiyakumar, V.; Park, J.; Vakil, R.M.; Toth, P.P.; Lazo-Elizondo, M.; Brown, E.; Quispe, R.; Guallar, E.; Banach, M.; et al. Modern prevalence of dysbetalipoproteinemia (Fredrickson-Levy-Lees type III hyperlipoproteinemia). Arch. Med. Sci. 2019, 16, 993–1003. [Google Scholar] [CrossRef]
- Bea, A.M.; Cenarro, A.; Marco-Bened, V.; Laclaustra, M.; Martn, C.; Ibarretxe, D.; Pint, X.; Arrobas, T.; Vials, C.; Civeira, F.; et al. Diagnosis of Familial Dysbetalipoproteinemia Based on the Lipid Abnormalities Driven by APOE2/E2 Genotype. Clin. Chem. 2023, 69, 140–148. [Google Scholar] [CrossRef]
- Heidemann, B.E.; Wolters, F.J.; Kavousi, M.; Gruppen, E.G.; Dullaart, R.P.; Marais, A.D.; Visseren, F.L.; Koopal, C. Adiposity and the development of dyslipidemia in APOE ε2 homozygous subjects: A longitudinal analysis in two population-based cohorts. Atherosclerosis 2021, 325, 57–62. [Google Scholar] [CrossRef] [PubMed]
- De Beer, F.; Stalenhoef, A.F.H.; Hoogerbrugge, N.; Kastelein, J.J.P.; Gevers Leuven, J.A.; van Duijn, C.M.; Havekes, L.M.; Smelt, A.H. Expression of type III hyperlipoproteinemia in apolipoprotein E2 (Arg158 → Cys) homozygotes is associated with hyperinsulinemia. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 294–299. [Google Scholar] [CrossRef]
- Corsetti, J.P.; Love, T.M.; Sparks, C.E.; Bakker, S.J.L.; Dullaart, R.P.F. Insulin resistance involvement in prevalence of familial dysbetalipoproteinemia in ε2ε2 subjects by Bayesian network modeling. Clin. Biochem. 2018, 59, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.; Seedorf, U.; Beil, F.U. Polymorphisms in the apolipoprotein A5 (APOA5) gene and type III hyperlipidemia. Clin. Genet. 2005, 68, 369–372. [Google Scholar] [CrossRef]
- Henneman, P.; van der Sman-de Beer, F.; Moghaddam, P.H.; Huijts, P.; Stalenhoef, A.F.; Kastelein, J.J.; van Duijn, C.M.; Havekes, L.M.; Frants, R.R.; van Dijk, K.W.; et al. The expression of type III hyperlipoproteinemia: Involvement of lipolysis genes. Eur. J. Hum. Genet. 2009, 17, 620–628. [Google Scholar] [CrossRef]
- Šatný, M.; Todorovová, V.; Altschmiedová, T.; Hubáček, J.A.; Dlouhá, L.; Laňská, V.; Soška, V.; Kyselák, O.; Freiberger, T.; Bobák, M.; et al. Genetic risk score in patients with the APOE2/E2 genotype as a predictor of familial dysbetalipopro-teinemia. J. Clin. Lipidol. 2024, 18, e230–e237. [Google Scholar] [CrossRef]
- Pieri, K.; Trichia, E.; Neville, M.J.; Taylor, H.; Bennett, D.; Karpe, F.; Koivula, R.W. Polygenic risk in Type III hyperlipidaemia and risk of cardiovascular disease: An epidemiological study in UK Biobank and Oxford Biobank. Int. J. Cardiol. 2023, 373, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Marmontel, O.; Abou-Khalil, Y.; Bluteau, O.; Cariou, B.; Carreau, V.; Charrière, S.; Divry, E.; Gallo, A.; Moulin, P.; Paillard, F.; et al. Additive effect of APOE Rare Variants on the phenotype of familial hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 2023, 43, e270–e278. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, I.; Daimon, T. The “cardiometabolic index” as a new marker determined by adiposity and blood lipids for discrimination of diabetes mellitus. Clin. Chim. Acta. 2015, 438, 274–278. [Google Scholar] [CrossRef] [PubMed]
- Kopylova, O.V.; Ershova, A.I.; Pokrovskaya, M.S.; Meshkov, A.N.; Efimova, I.A.; Serebryanskaya, Z.Z.; Blokhina, A.V.; Borisova, A.L.; Kondratskaya, V.A.; Limonova, A.S.; et al. Population-nosological research biobank of the National Medical Research Center for Therapy and Preventive Medicine: Analysis of biosamples, principles of collecting and storing information. Cardiovasc. Ther. Prev. 2021, 20, 3119. (In Russian) [Google Scholar] [CrossRef]
- Boytsov, S.A.; Drapkina, O.M.; Shlyakhto, E.V.; Konradi, A.O.; Balanova, Y.A.; Zhernakova, Y.V.; Metelskaya, V.A.; Oshchepkova, E.V.; Rotar, O.P.; Shalnova, S.A. Epidemiology of cardiovascular diseases and their risk factors in regions of Russian Federation (ESSE-RF) study. Ten years later. Cardiovasc. Ther. Prev. 2021, 20, 3007. (In Russian) [Google Scholar] [CrossRef]
- Zaicenoka, M.; Ershova, A.I.; Kiseleva, A.V.; Blokhina, A.V.; Kutsenko, V.A.; Sotnikova, E.A.; Zharikova, A.A.; Vyatkin, Y.V.; Pokrovskaya, M.S.; Shalnova, S.A.; et al. Blood Lipid Polygenic Risk Score Development and Application for Atherosclerosis Ultrasound Parameters. Biomedicines 2024, 12, 2798. [Google Scholar] [CrossRef]
- Meshkov, A.N.; Ershova, A.I.; Kiseleva, A.V.; Shalnova, S.A.; Drapkina, O.M.; Boytsov, S.A. The prevalence of heterozygous familial hypercholesterolemia in selected regions of the Russian Federation: The FH-ESSE-RF study. J. Pers. Med. 2021, 11, 464. [Google Scholar] [CrossRef]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk: The Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS). Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef]
- Ashwell, M.; Gunn, P.; Gibson, S. Waist-to-height ratio is a better screening tool than waist circumference and BMI for adult cardiometabolic risk factors: Systematic review and meta-analysis. Obes. Rev. 2012, 13, 275–286. [Google Scholar] [CrossRef]
- Blokhina, A.V.; Ershova, A.I.; Meshkov, A.N.; Limonova, A.S.; Mikhailina, V.I.; Drapkina, O.M. Lipid Clinic is an Efficacious Model of Preventive Medicine. Ration. Pharmacother. Cardiol. 2021, 17, 4–10. (In Russian) [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for 321 the interpretation of sequence variants: A joint consensus recommendation of the American 322 College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Chora, J.R.; Iacocca, M.A.; Tichý, L.; Wand, H.; Kurtz, C.L.; Zimmermann, H.; Leon, A.; Williams, M.; Humphries, S.E.; Hooper, A.J.; et al. The Clinical Genome Resource (ClinGen) Familial Hypercholesterolemia Variant Curation Expert Panel consensus guidelines for LDLR variant classification. Genet. Med. 2022, 24, 293–306. [Google Scholar] [CrossRef]
- Willer, C.J.; Schmidt, E.M.; Sengupta, S.; Peloso, G.M.; Gustafsson, S.; Kanoni, S.; Ganna, A.; Chen, J.; Buchkovich, M.L.; Mora, S. Discovery and refinement of loci associated with lipid levels. Nat. Genet. 2013, 45, 1274–1283. [Google Scholar] [CrossRef] [PubMed]
- Futema, M.; Shah, S.; Cooper, J.A.; Li, K.; Whittall, R.A.; Sharifi, M.; Goldberg, O.; Drogari, E.; Mollaki, V.; Wiegman, A.; et al. Refinement of variant selection for the LDL cholesterol genetic risk score in the diagnosis of the polygenic form of clinical familial hypercholesterolemia and replication in samples from 6 countries. Clin. Chem. 2015, 61, 231–238. [Google Scholar] [CrossRef]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013; Available online: https://www.R-project.org/ (accessed on 17 January 2025).
- Gross, J.; Ligges, U. nortest: Tests for Normality. R package version 1.0-4. 2015. Available online: https://CRAN.R-project.org/package=nortest (accessed on 22 July 2025).
- Heinze, G.; Ploner, M.; Jiricka, L.; Steiner, G.; Dunkler, D.; Southworth, H. logistf: Firth’s Bias-Reduced Logistic Regression; R Package Version 1.26.1. 2025. Available online: https://CRAN.R-project.org/package=logistf (accessed on 16 June 2025).
- Kuhn, M.; Wing, J.; Weston, S.; Williams, A.; Keefer, C.; Engelhardt, A.; Cooper, T.; Mayer, Z.; Kenkel, B.; R Core Team; et al. caret: Classification and Regression Training, R package Version 7.0-1. 2024. Available online: https://CRAN.R-project.org/package=caret (accessed on 22 July 2025).
- Wickham, H. GGPLOT2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; p. 3. [Google Scholar]
- Sievert, C.; Parmer, C.; Hocking, T.; Chamberlain, S.; Ram, K.; Corvellec, M.; Despouy, P.; Brüggemann, S.; Plotly Technologies Inc. plotly: Create Interactive Web Graphics via ‘plotly.js’; R Package Version 4.11.0. 2025. Available online: https://CRAN.R-project.org/package=plotly (accessed on 19 June 2025).
- Chen, H.; Boutros, P.C. VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef] [PubMed]
- Krassowski, M. ComplexUpset: Create Complex UpSet plots using ggplot2. Bioinformatics 2021, 37, 4350–4352. [Google Scholar] [CrossRef]
- Champely, S.; Ekstrom, C.; Dalgaard, P.; Gill, J.; Weibelzahl, S.; Anandkumar, A.; Ford, C.; Volcic, R.; De Rosario, H. pwr: Basic Functions for Power Analysis; R Package Version 1.3-0. 2020. Available online: https://CRAN.R-project.org/package=pwr (accessed on 16 June 2025).
- Sawilowsky, S.S. New effect size rules of thumb. J. Mod. Appl. Stat. Methods 2009, 8, 597–599. [Google Scholar] [CrossRef]
- Varghese, B.; Park, J.; Chew, E.; Sajja, A.; Brownstein, A.; Pallazola, V.A.; Sathiyakumar, V.; Jones, S.R.; Sniderman, A.D.; Martin, S.S. Importance of the triglyceride level in identifying patients with a Type III Hyperlipoproteinemia phenotype using the ApoB algorithm. J. Clin. Lipidol. 2021, 15, 104–115. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study Group | Carriers No. | Additional Variant 1, Gene/HGVSp/Zygosity | Additional Variant 2, Gene/HGVSp/Zygosity | APOE Genotype | Rare APOE Variant HGVSp/Zygosity |
|---|---|---|---|---|---|
| FD | 1 | LDLR/p.Ser554Leu/het | - | ε2/ε2 | - |
| 2 | LDLR/p.Ser586Pro/het | - | ε2/ε2 | - | |
| 3 | LPL/p.Asp202Asn/het | - | ε2/ε2 | - | |
| 4 | LPL/p.Asp202Asn/het | LPL/p.Tyr233Cys/het | ε3/ε3 | p.Arg154Cys/het | |
| 5 | LMF1/p.Tyr439Cys/homo | - | ε2/ε2 | - | |
| Comparison | 1 | LDLR/p.Gly592Glu/het | - | ε2/ε3 | p.Gly145Asp/het |
| Parameter | All Subjects (n = 71) | Carriers of the ε2/ε2 Genotype and Rare APOE Variants (n = 19) | Patients with FD (n = 52) | p-Value 1 |
OR (95% CI) 2/ ΔMe (95% CI) |
|---|---|---|---|---|---|
| Men, n (%) | 32 (45.1) | 9 (47.4) | 23 (44.2) | 1.0 | 1.1 (0.3–3.7) |
| Age, years, Me (Q1; Q3) | 50 (40; 56) | 40 (31; 52) | 51 (46; 58) | 0.004 | 11 (4–18) |
| Smoking (current or former smokers), n (%) | 32 (45.1) | 8 (42.1) | 24 (46.2) | 0.794 | 1.2 (0.4–4.0) |
| Hypertension, n (%) | 42 (59.2) | 8 (42.1) | 34 (65.4) | 0.104 | 2.6 (0.8–8.8) |
| Metabolic Factors | |||||
| BMI, kg/m2, Me (Q1; Q3) | 28.7 (25.4; 31.7) | 24.9 (22.8; 28.8) | 29.5 (26.9; 31.9) | 0.001 | 4.1 (1.7–6.4) |
| WC, cm, Me (Q1; Q3) | 94.0 (84.2; 100.2) n = 58 | 81.0 (75.4; 95.5) n = 16 | 95.0 (90.0; 104.0) n = 42 | 0.002 | 13.0 (6.0–21.0) |
| WHtR, Me (Q1; Q3) | 0.6 (0.5; 0.6) n = 58 | 0.5 (0.4; 0.6) n = 16 | 0.6 (0.6; 0.6) n = 42 | 0.001 | 0.1 (0.04–12.7) |
| CMI, Me (Q1; Q3) | 1.300 (0.622; 3.410) n = 58 | 0.337 (0.176; 0.523) n = 16 | 2.160 (1.050; 5.150) n = 42 | <0.001 | 1.823 (1.030–3.670) |
| Glucose metabolism disorders, n (%) 4 | 11 (15.5) | 1 (5.3) | 10 (19.2) | 0.267 | 4.2 (0.5–196.0) |
| Metabolic syndrome components, n (%) 5 | 40 (56.3) | 3 (15.8) | 37 (71.2) | <0.001 | 13 (3–77) |
| Hypothyroidism, n (%) | 5 (7.2) n = 69 | 2 (11.1) n = 18 | 3 (5.9) n = 51 | 0.600 | 0.5 (0.1–6.6) |
| CHD | |||||
| CHD, n (%) | 14 (19.7) | 0 | 14 (26.9) | 0.015 | 14.7 (0.8–259.4) 3 |
| Age at onset of CHD, years, Me (Q1; Q3) | 42 (37; 49) | — | 42 (37; 49) | — | — |
| Pretreatment Lipid Levels | |||||
| TC, mmol/L, Me (Q1; Q3) | 5.63 (4.23; 9.67) n = 67 | 4.15 (2.87; 4.65) | 6.92 (4.99; 10.83) n = 48 | <0.001 | 2.93 (1.67–5.25) |
| LDL-C, mmol/L, Me (Q1; Q3) | 2.39 (1,66; 3.31) n = 61 | 1.66 (0.99; 2.32) | 2.95 (2.11; 3.55) n = 42 | 0.001 | 1.13 (0.55–1.74) |
| HDL-C, mmol/L, Me (Q1; Q3) | 1.09 (0.89; 1.44) | 1.36 (1.17; 1.69) | 1.03 (0.84; 1.31) | 0.002 | −0.33 (−0.57; −0.14) |
| TG, mmol/L, Me (Q1; Q3) | 3.01 (1,47; 6.15) | 0.9 (0.70; 1.29) | 4.47 (2.65; 9.67) | <0.001 | 3.52 (2.24–5.05) |
| Non-HDL-C, mmol/L, Me (Q1; Q3) | 4.28 (2.69; 8.33) n = 57 | 2.43 (1.65; 3.27) | 5.69 (4.01; 9.98) n = 38 | <0.001 | 3.26 (1.92–6.08) |
| Remnant cholesterol, mmol/L, Me (Q1; Q3) | 1.34 (0.71; 2.02) n = 50 | 0.65 (0.46; 0.91) | 1.87 (1.25; 2.59) n = 31 | <0.001 | 1.17 (0.70–1.70) |
| Predictor | β Coefficient (SE) | 95% CI | p-Value |
|---|---|---|---|
| Polygenic HTG, per 1 percentile | 0.05 (0.02) | 0.01–0.09 | 0.011 |
| Variants in other lipid-related genes (yes) | 7.0 (1.70) | 3.60–10.41 | < 0.001 |
| Glucose metabolism disorders (yes) | 3.62 (1.52) | 0.58–6.66 | 0.020 |
| Initial model statistics: R2 = 0.30, Adjusted R2 = 0.27, Residual SE = 4.50 (df = 64), p < 0.001, observations number = 68 | |||
| Fivefold cross-validation model statistics 1: R2 = 0.30, Residual SE = 4.74, MAE = 3.48, observations number = 68 | |||
| Parameter | Standard Deviation in ESSE-Ivanovo (n = 1652) | Minimum Detectable Effect Sizes | Cohen’s d |
|---|---|---|---|
| HTG polygenic risk, score | 0.21 | 0.16 1 | 0.76 |
| Hypercholesterolemia polygenic risk, score | 0.34 | 0.26 2 | 0.76 |
| BMI, kg/m2 | 5.90 | 4.49 | 0.76 |
| TG, mmol/L | 1.04 | 0.79 | 0.76 |
| LDL-C, mmol/L | 1.15 | 0.88 | 0.77 |
| HDL-C, mmol/L | 0.34 | 0.26 | 0.76 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blokhina, A.V.; Ershova, A.I.; Kiseleva, A.V.; Sotnikova, E.A.; Zaicenoka, M.; Zharikova, A.A.; Vyatkin, Y.V.; Ramensky, V.E.; Novokhatskaya, E.A.; Borisova, A.L.; et al. Genetic and Metabolic Factors of Familial Dysbetalipoproteinemia Phenotype: Insights from a Cross-Sectional Study. Int. J. Mol. Sci. 2025, 26, 7376. https://doi.org/10.3390/ijms26157376
Blokhina AV, Ershova AI, Kiseleva AV, Sotnikova EA, Zaicenoka M, Zharikova AA, Vyatkin YV, Ramensky VE, Novokhatskaya EA, Borisova AL, et al. Genetic and Metabolic Factors of Familial Dysbetalipoproteinemia Phenotype: Insights from a Cross-Sectional Study. International Journal of Molecular Sciences. 2025; 26(15):7376. https://doi.org/10.3390/ijms26157376
Chicago/Turabian StyleBlokhina, Anastasia V., Alexandra I. Ershova, Anna V. Kiseleva, Evgeniia A. Sotnikova, Marija Zaicenoka, Anastasia A. Zharikova, Yuri V. Vyatkin, Vasily E. Ramensky, Elizaveta A. Novokhatskaya, Anna L. Borisova, and et al. 2025. "Genetic and Metabolic Factors of Familial Dysbetalipoproteinemia Phenotype: Insights from a Cross-Sectional Study" International Journal of Molecular Sciences 26, no. 15: 7376. https://doi.org/10.3390/ijms26157376
APA StyleBlokhina, A. V., Ershova, A. I., Kiseleva, A. V., Sotnikova, E. A., Zaicenoka, M., Zharikova, A. A., Vyatkin, Y. V., Ramensky, V. E., Novokhatskaya, E. A., Borisova, A. L., Shalnova, S. A., Meshkov, A. N., & Drapkina, O. M. (2025). Genetic and Metabolic Factors of Familial Dysbetalipoproteinemia Phenotype: Insights from a Cross-Sectional Study. International Journal of Molecular Sciences, 26(15), 7376. https://doi.org/10.3390/ijms26157376