

The Role of Advanced Glycation End-Products in the Pathophysiology and Pharmacotherapy of Cardiovascular Disease


Abstract
1. Introduction
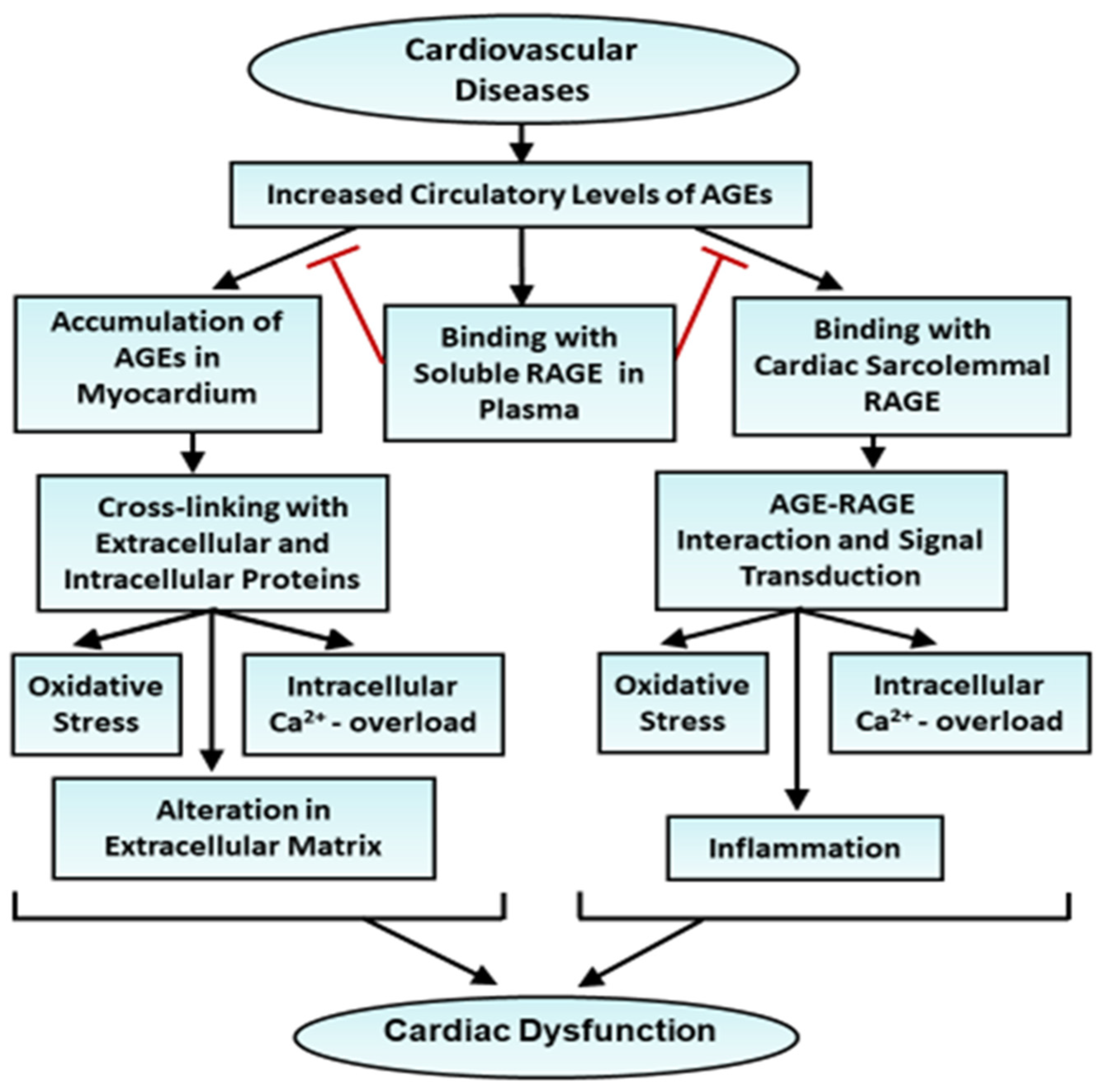
2. General Consideration of the Role of AGEs in Cardiovascular Diseases
3. AGE–RAGE Interaction and Signaling Pathways in Cardiovascular Dysfunction
- (i)
- NF-κB Pathway: The activation of RAGEs triggers the NF-κB signaling pathway, a critical mediator of inflammation. NF-κB transcription factors, such as p65 and p50, translocate to the nucleus and promote the expression of pro-inflammatory cytokines, adhesion molecules, and other inflammatory mediators. Chronic activation of NF-κB by AGEs leads to endothelial dysfunction, increased vascular permeability, and recruitment of inflammatory cells to sites of injury; these alterations contribute to the development of atherosclerosis [67,68,69].
- (ii)
- MAPK Pathway: RAGE activation also triggers the MAPK pathway, including p38 MAPK, extracellular signal-regulated kinase (ERK), and c-Jun N-terminal kinase (JNK). These kinases regulate gene expression and cellular responses such as cell proliferation, apoptosis, and inflammation. In the context of cardiovascular diseases, MAPK activation promotes the phenotypic switch of vascular smooth muscle cells, a key event in the development of atherosclerotic plaque [70,71,72].
- (iii)
- JAK/STAT Pathway: The JAK/STAT signaling pathway is also implicated in RAGE-mediated effects. Activation of JAK1 and JAK2 leads to the phosphorylation and activation of STAT3, which subsequently modulates the expression of genes involved in inflammation and cell survival. This pathway contributes to the endothelial dysfunction observed in cardiovascular diseases, promoting a pro-inflammatory and pro-thrombotic state [73,74].
4. The Role of AGEs in Ischemic Heart Disease
5. The Role of AGEs in Vascular Dysfunction
6. The Role of AGEs in Pathological Cardiac Hypertrophy and Heart Failure
7. Pathophysiological and Therapeutic Implications of the AGE–RAGE Axis
8. Conclusions and Perspectives
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chomistek, A.K.; Chiuve, S.E.; Eliassen, A.H.; Mukamal, K.J.; Willett, W.C.; Rimm, E.B. Healthy lifestyle in the primordial prevention of cardiovascular disease among young women. J. Am. Coll. Cardiol. 2015, 65, 43–51. [Google Scholar] [CrossRef]
- Sharifi-Rad, J.; Rodrigues, C.F.; Sharopov, F.; Docea, A.O.; Can Karaca, A.; Sharifi-Rad, M.; Kahveci Karıncaoglu, D.; Gülseren, G.; Şenol, E.; Demircan, E. Diet, lifestyle and cardiovascular diseases: Linking pathophysiology to cardioprotective effects of natural bioactive compounds. Int. J. Environ. Res. Public Health 2020, 17, 2326. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.; Matsui, T. Pathologic role of dietary advanced glycation end products in cardiometabolic disorders and therapeutic intervention. Nutrition 2016, 32, 157–165. [Google Scholar] [CrossRef]
- Prasad, K.; Dhar, I.; Caspar-Bell, G. Role of advanced glycation end products and its receptors in the pathogenesis of cigarette smoke-induced cardiovascular disease. Int. J. Angiol. 2015, 24, 75–80. [Google Scholar]
- Zieman, S.; Kass, D. Advanced glycation end product cross-linking: Pathophysiologic role and therapeutic target in cardiovascular disease. Congest. Heart Fail. 2004, 10, 144–149. [Google Scholar] [CrossRef]
- Thorpe, S.R.; Baynes, J.W. Maillard reaction products in tissue proteins: New products and new perspectives. Amino Acids 2003, 25, 275–281. [Google Scholar] [CrossRef]
- Twarda-Clapa, A.; Olczak, A.; Białkowska, A.M.; Koziołkiewicz, M. Advanced glycation end-products (AGEs): Formation, chemistry, classification, receptors, and diseases related to AGEs. Cells 2022, 11, 1312. [Google Scholar] [CrossRef] [PubMed]
- Perrone, A.; Giovino, A.; Benny, J.; Martinelli, F. Advanced glycation end products (AGEs): Biochemistry, signaling, analytical methods, and epigenetic effects. Oxid. Med. Cell. Longev. 2020, 2020, 3818196. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.I.; Ashfaq, F.; Alsayegh, A.A.; Hamouda, A.; Khatoon, F.; Altamimi, T.N.; Alhodieb, F.S.; Beg, M.M.A. Advanced glycation end product signaling and metabolic complications: Dietary approach. World J. Diabetes 2023, 14, 995–1012. [Google Scholar] [CrossRef]
- Onorato, J.M.; Thorpe, S.R.; Baynes, J.W. Immunohistochemical and ELISA assays for biomarkers of oxidative stress in aging and disease. Ann. N. Y. Acad. Sci. 1998, 854, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Deluyker, D.; Evens, L.; Bito, V. Advanced glycation end products (AGEs) and cardiovascular dysfunction: Focus on high molecular weight AGEs. Amino Acids 2017, 49, 1535–1541. [Google Scholar] [CrossRef]
- Gupta, J.K. The role of aldose reductase in polyol pathway: An emerging pharmacological target in diabetic complications and associated morbidities. Curr. Pharm. Biotechnol. 2024, 25, 1073–1081. [Google Scholar] [CrossRef]
- Lorenzi, M. The polyol pathway as a mechanism for diabetic retinopathy: Attractive, elusive, and resilient. Exp. Diabetes Res. 2007, 2007, 61038. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Randive, R.; Stewart, J.A. Molecular mechanisms of AGE/RAGE-mediated fibrosis in the diabetic heart. World J. Diabetes 2014, 5, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Bidasee, K.R.; Zhang, Y.; Shao, C.H.; Wang, M.; Patel, K.P.; Dincer, U.D.; Besch, H.R. Diabetes increases formation of advanced glycation end products on sarco (endo) plasmic reticulum Ca2+-ATPase. Diabetes 2004, 53, 463–473. [Google Scholar] [CrossRef]
- Ramasamy, R.; Schmidt, A.M. Receptor for advanced glycation end products (RAGE) and implications for the pathophysiology of heart failure. Curr. Heart Fail. Rep. 2012, 9, 107–116. [Google Scholar] [CrossRef]
- Suchal, K.; Malik, S.; Khan, S.I.; Malhotra, R.K.; Goyal, S.N.; Bhatia, J.; Kumari, S.; Ojha, S.; Arya, D.S. Protective effect of mangiferin on myocardial ischemia-reperfusion injury in streptozotocin-induced diabetic rats: Role of AGE-RAGE/MAPK pathways. Sci. Rep. 2017, 7, 42027. [Google Scholar] [CrossRef]
- Blackburn, N.J.R.; Vulesevic, B.; McNeill, B.; Cimenci, C.E.; Ahmadi, A.; Gonzalez-Gomez, M.; Ostojic, A.; Zhong, Z.; Brownlee, M.; Beisswenger, P.J.; et al. Methylglyoxal-derived advanced glycation end products contribute to negative cardiac remodeling and dysfunction post-myocardial infarction. Basic Res. Cardiol. 2017, 112, 57. [Google Scholar] [CrossRef]
- Gomes, R.; Sousa Silva, M.; Quintas, A.; Cordeiro, C.; Freire, A.; Pereira, P.; Martins, A.; Monteiro, E.; Barroso, E.; Ponces Freire, A. Argpyrimidine, a methylglyoxal-derived advanced glycation end-product in familial amyloidotic polyneuropathy. Biochem. J. 2005, 385 Pt 2, 339–345. [Google Scholar] [CrossRef]
- Gawlowski, T.; Stratman, B.; Stork, I.; Engelbrecht, B.; Brodehl, A.; Niehaus, K.; Körfer, R.; Tschoepe, D.; Milting, H. Heat shock protein 27 modification is increased in human diabetic failing heart. Horm. Metab. Res. 2009, 41, 594–599. [Google Scholar] [CrossRef]
- Prasad, K.; Bhanumathy, K.K. AGE-RAGE axis in the pathophysiology of chronic lower limb ischemia and a novel strategy for its treatment. Int. J. Angiol. 2020, 29, 156–167. [Google Scholar] [CrossRef]
- Prasad, K.; Dhar, I.; Zhou, Q.; Elmoselhi, H.; Shoker, M.; Shoker, A. AGEs/sRAGE, a novel risk factor in the pathogenesis of end-stage renal disease. Mol. Cell. Biochem. 2016, 423, 105–114. [Google Scholar] [CrossRef]
- Fracasso, B.M.; Rangel, J.O.; Machado, A.G.; Curuja, F.S.; Lopes, A.; Olsen, V.; Clausell, N.; Biolo, A.; Rohde, L.E.; Andrades, M. Characterization of advanced glycation end products and their receptor (RAGE) in an animal model of myocardial infarction. PLoS ONE 2019, 14, e0209964. [Google Scholar] [CrossRef]
- Cao, X.; Li, B.; Han, X.; Zhang, X.; Dang, M.; Wang, H.; Du, F.; Zeng, X.; Guo, C. Soluble receptor for advanced glycation end-products promotes angiogenesis through activation of STAT3 in myocardial ischemia/reperfusion injury. Apoptosis 2020, 25, 341–353. [Google Scholar] [CrossRef]
- Yang, D.; Wang, Y.; Jiang, M.; Deng, X.; Pei, Z.; Li, F.; Xia, K.; Zhu, L.; Yang, T.; Chen, M. Downregulation of profilin-1 expression attenuates cardiomyocytes hypertrophy and apoptosis induced by advanced glycation end products in H9c2 cells. BioMed Res. Int. 2017, 2017, 9716087. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Liu, W.; Ma, L.; Wang, Y.; Ma, J.; Jiang, M.; Deng, X.; Huang, F.; Yang, T.; Chen, M. Profilin-1 contributes to cardiac injury induced by advanced glycation end-products in rats. Mol. Med. Rep. 2017, 16, 634–664. [Google Scholar] [CrossRef]
- Gao, W.; Zhou, Z.; Liang, B.; Huang, Y.; Yang, Z.; Chen, Y.; Zhang, L.; Yan, C.; Wang, J.; Lu, L.; et al. Inhibiting receptor of advanced glycation end products attenuates pressure overload-induced cardiac dysfunction by preventing excessive autophagy. Front. Physiol. 2018, 9, 1333. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Chen, J.; Chen, Y.; Chen, X.; Liu, P. Advanced glycation end products promote heart failure through inducing the immune maturation of dendritic cells. Appl. Biochem. Biotechnol. 2014, 172, 4062–4077. [Google Scholar] [CrossRef] [PubMed]
- Deluyker, D.; Evens, L.; Haesen, S. Glycolaldehyde-derived high-molecular-weight advanced glycation end-products induce cardiac dysfunction through structural and functional remodeling of cardiomyocytes. Cell. Physiol. Biochem. 2020, 54, 809–824. [Google Scholar]
- Bodiga, V.L.; Eda, S.R.; Bodiga, S. Advanced glycation end products: Role in pathology of diabetic cardiomyopathy. Heart Fail. Rev. 2014, 19, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Zhou, Q.Y.; Liu, D.; Yu, L.; Zhan, L.; Li, X.J.; Peng, H.Y.; Zhang, X.L.; Yuan, X.C. Advanced glycation end-products impair Na+/K+-ATPase activity in diabetic cardiomyopathy: Role of the adenosine monophosphate-activated protein kinase/sirtuin 1 pathway. Clin. Exp. Pharmacol. Physiol. 2014, 41, 127–133. [Google Scholar] [CrossRef]
- Wendt, T.; Harja, E.; Bucciarelli, L.; Qu, W.; Lu, Y.; Rong, L.L.; Jenkins, D.G.; Stein, G.; Schmidt, A.M.; Yan, S.F. RAGE modulates vascular inflammation and atherosclerosis in a murine model of type 2 diabetes. Atherosclerosis 2006, 185, 70–77. [Google Scholar] [CrossRef]
- Soro-Paavonen, A.; Watson, A.M.D.; Li, J.; Paavonen, K.; Koitka, A.; Calkin, A.C.; Barit, D.; Coughlan, M.T.; Drew, B.G.; Lancaster, G.I.; et al. Receptor for advanced glycation end products (RAGE) deficiency attenuates the development of atherosclerosis in diabetes. Diabetes 2008, 57, 2461–2469. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, X.; Zhou, W.; Men, H.; Bao, T.; Sun, Y.; Wang, Q.; Tan, Y.; Keller, B.B.; Tong, Q.; et al. Ferroptosis is essential for diabetic cardiomyopathy and is prevented by sulforaphane via AMPK/NRF2 pathways. Acta Pharm. Sin. B 2022, 12, 708–722. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K.; Tiwari, S. Therapeutic interventions for advanced glycation-end products and its receptor- mediated cardiovascular disease. Curr. Pharm. Des. 2017, 23, 937–943. [Google Scholar] [CrossRef] [PubMed]
- McNair, E.; Qureshi, M.; Prasad, K.; Pearce, C. Atherosclerosis and the hypercholesterolemic AGE-RAGE axis. Int. J. Angiol. 2016, 25, 110–116. [Google Scholar]
- Prasad, K. AGE-RAGE stress in the pathophysiology of atrial fibrillation and its treatment. Int. J. Angiol. 2020, 29, 72–80. [Google Scholar] [CrossRef]
- Prasad, K.; Mishra, M. AGE-RAGE stress, stressors, and antistressors in health and disease. Int. J. Angiol. 2018, 27, 1–12. [Google Scholar]
- Prasad, K. Is there any evidence that AGE/sRAGE is a universal biomarker/risk marker for diseases? Mol. Cell. Biochem. 2019, 451, 139–144. [Google Scholar] [CrossRef]
- Prasad, K. Low levels of serum soluble receptors for advanced glycation end products, biomarkers for disease state: Myth or reality. Int. J. Angiol. 2014, 23, 11–16. [Google Scholar] [CrossRef]
- Singh, V.P.; Bali, A.; Singh, N.; Jaggi, A.S. Advanced glycation end products and diabetic complications. Korean J. Physiol. Pharmacol. 2014, 18, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Semba, R.D.; Nicklett, E.J.; Ferrucci, L. Does accumulation of advanced glycation end products contribute to the aging phenotype? J. Gerontol. A Biol. Sci. Med. Sci. 2010, 65, 963–975. [Google Scholar] [CrossRef]
- Shen, C.Y.; Lu, C.H.; Wu, C.H.; Li, K.J.; Kuo, Y.M.; Hsieh, S.C.; Yu, C.L. The development of maillard reaction, and advanced glycation end product (AGE)-receptor for AGE (RAGE) signaling inhibitors as novel therapeutic strategies for patients with AGE-related diseases. Molecules 2020, 25, 5591. [Google Scholar] [CrossRef]
- Uceda, A.B.; Mariño, L.; Casasnovas, R.; Adrover, M. An overview on glycation: Molecular mechanisms, impact on proteins, pathogenesis, and inhibition. Biophys. Rev. 2024, 16, 189–218. [Google Scholar] [CrossRef]
- Sakai-Sakasai, A.; Takeda, K.; Suzuki, H.; Takeuchi, M. Structures of toxic advanced glycation end-products derived from glyceraldehyde, a sugar metabolite. Biomolecules 2024, 14, 202. [Google Scholar] [CrossRef]
- Chen, Y.; Meng, Z.; Li, Y.; Liu, S.; Hu, P.; Luo, E. Advanced glycation end products and reactive oxygen species: Uncovering the potential role of ferroptosis in diabetic complications. Mol. Med. 2024, 30, 141. [Google Scholar] [CrossRef]
- Rungratanawanich, W.; Qu, Y.; Wang, X.; Essa, M.M.; Song, B.J. Advanced glycation end products (AGEs) and other adducts in aging-related diseases and alcohol-mediated tissue injury. Exp. Mol. Med. 2021, 53, 168–188. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Elimban, V.; Bartekova, M.; Adameova, A. Involvement of oxidative stress in the development of subcellular defects and heart disease. Biomedicines 2022, 10, 393. [Google Scholar] [CrossRef]
- Li, H.; Ping, Y.; Niranjan, K.; Dong, H. Structure, antioxidant properties and AGEs (advanced glycation end products) formation of modified wheat gluten protein after enzymatic hydrolysis and Maillard reaction. J. Food Compos. Anal. 2024, 136, 106795. [Google Scholar] [CrossRef]
- Brings, S.; Mier, W.; Beijer, B.; Müller, P.C.; Herzig, S.; Szendroedi, J.; Nawroth, P.P.; Fleming, T. Non-cross-linking advanced glycation end products affect prohormone processing. Biochem. J. 2024, 481, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Nash, A.; Notou, M.; Lopez-Clavijo, A.F.; Bozec, L.; de Leeuw, N.H.; Birch, H.L. Glucosepane is associated with changes to structural and physical properties of collagen fibrils. Matrix Biol. Plus 2019, 4, 100013. [Google Scholar] [CrossRef] [PubMed]
- Fedintsev, A.; Moskalev, A. Stochastic non-enzymatic modification of long-lived macromolecules—A missing hallmark of aging. Ageing Res. Rev. 2020, 62, 101097. [Google Scholar] [CrossRef]
- Bordeleau, F.; Mason, B.N.; Lollis, E.M.; Mazzola, M.; Zanotelli, M.R.; Somasegar, S.; Califano, J.P.; Montague, C.; LaValley, D.J.; Huynh, J.; et al. Matrix stiffening promotes a tumor vasculature phenotype. Proc. Natl. Acad. Sci. USA 2017, 114, 492–497. [Google Scholar] [CrossRef]
- Sroga, G.E.; Vashishth, D. In vivo glycation—Interplay between oxidant and carbonyl stress in bone. JBMR Plus 2024, 8, e110. [Google Scholar] [CrossRef] [PubMed]
- Hegab, Z.; Gibbons, S.; Neyses, L.; Mamas, M.A. Role of advanced glycation end products in cardiovascular disease. World J. Cardiol. 2012, 4, 90–102. [Google Scholar] [CrossRef]
- Chuah, Y.K.; Basir, R.; Talib, H.; Tie, T.H.; Nordin, N. Receptor for advanced glycation end products and its involvement in inflammatory diseases. Int. J. Inflam. 2013, 2013, 403460. [Google Scholar] [CrossRef] [PubMed]
- Kosmopoulos, M.; Drekolias, D.; Zavras, P.D.; Piperi, C.; Papavassiliou, A.G. Impact of advanced glycation end products (AGEs) signaling in coronary artery disease. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 611–619. [Google Scholar] [CrossRef]
- Casper-Bell, G.; Dhar, I.; Prasad, K. Advanced glycation products (AGEs) and its receptors in the pathogenesis of hyperthyroidism. Mol. Cell. Biochem. 2016, 414, 171–178. [Google Scholar] [CrossRef]
- Harvey, A.; Montezano, A.C.; Lopes, R.A.; Rios, F.J.; Touyz, R.M. Vascular fibrosis in aging and hypertension: Molecular mechanisms and clinical implications. Can. J. Cardiol. 2016, 32, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Cardiac fibrosis. Cardiovasc. Res. 2021, 117, 1450–1488. [Google Scholar] [CrossRef]
- Bierhaus, A.; Humpert, P.M.; Morcos, M.; Wendt, T.; Chavakis, T.; Arnold, B.; Stern, D.M.; Nawroth, P.P. Understanding RAGE, the receptor for advanced glycation end products. J. Mol. Med. 2005, 83, 876–886. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.M.; Yan, S.D.; Yan, S.F.; Stern, D.M. The biology of the receptor for advanced glycation end products and its ligands. Biochim. Biophys. Acta 2000, 1498, 99–111. [Google Scholar] [CrossRef]
- Vegivinti, C.T.R.; Keesari, P.R.; Veeraballi, S.; Martins Maia, C.M.P.; Mehta, A.K.; Lavu, R.R.; Thakur, R.K.; Tella, S.H.; Patel, R.; Kakumani, V.K.; et al. Role of innate immunological/inflammatory pathways in myelodysplastic syndromes and AML: A narrative review. Exp. Hematol. Oncol. 2023, 12, 60. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.D.; Schmidt, A.M.; Anderson, G.M.; Zhang, J.; Brett, J.; Zou, Y.S.; Pinsky, D.; Stern, D. Enhanced cellular oxidant stress by the interaction of advanced glycation end products with their receptors/binding proteins. J. Biol. Chem. 1994, 269, 9889–9897. [Google Scholar] [CrossRef]
- Feng, Y.; Zou, L.; Zhang, M.; Li, Y.; Chen, C.; Chao, W. MyD88 and Trif signaling play distinct roles in cardiac dysfunction and mortality during endotoxin shock and polymicrobial sepsis. Anesthesiology 2011, 115, 555–567. [Google Scholar] [CrossRef]
- Hoebe, K.; Du, X.; Georgel, P.; Janssen, K.; Tabeta, K.; Kim, S.O.; Goode, J.; Lin, P.; Mann, N.; Mudd, S.; et al. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature 2003, 424, 743–748. [Google Scholar] [CrossRef]
- Byun, K.; Yoo, Y.; Son, M.; Lee, J.; Jeong, G.B.; Park, Y.M.; Salekdeh, G.H.; Lee, B. Advanced glycation end-products produced systemically and by macrophages: A common contributor to inflammation and degenerative diseases. Pharmacol. Ther. 2017, 177, 44–55. [Google Scholar] [CrossRef]
- Schmidt, A.M.; Yan, S.D.; Wautier, J.L.; Stern, D. Activation of receptor for advanced glycation end products: A mechanism for chronic vascular dysfunction in diabetic vasculopathy and atherosclerosis. Circ. Res. 1999, 84, 489–497. [Google Scholar] [CrossRef]
- Chavakis, T.; Bierhaus, A.; Nawroth, P.P. RAGE receptor for advanced glycation end products): A central player in the inflammatory response. Microbes Infect. 2004, 6, 1219–1225. [Google Scholar] [CrossRef]
- Song, J.S.; Kang, C.M.; Park, C.K.; Yoon, H.K.; Lee, S.Y.; Ahn, J.H.; Moon, H.S. Inhibitory effect of receptor for advanced glycation end products (RAGE) on the TGF-β-induced alveolar epithelial to mesenchymal transition. Exp. Mol. Med. 2011, 43, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Reustle, A.; Torzewski, M. Role of p38 MAPK in atherosclerosis and aortic valve sclerosis. Int. J. Mol. Sci. 2018, 19, 3761. [Google Scholar] [CrossRef] [PubMed]
- Rajamäki, K.; Mäyränpää, M.I.; Risco, A.; Tuimala, J.; Nurmi, K.; Cuenda, A.; Eklund, K.K.; Öörni, K.; Kovanen, P.T. p38δ MAPK: A novel regulator of NLRP3 inflammasome activation with increased expression in coronary atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1937–1946. [Google Scholar] [CrossRef]
- Aleshin, A.; Ananthakrishnan, R.; Li, Q.; Rosario, R.; Lu, Y.; Qu, W.; Song, F.; Bakr, S.; Szabolcs, M.; D’Agati, V.; et al. RAGE modulates myocardial injury consequent to LAD infarction via impact on JNK and STAT signaling in a murine model. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H1823–H1832. [Google Scholar] [CrossRef]
- Shang, L.; Ananthakrishnan, R.; Li, Q.; Quadri, N.; Abdillahi, M.; Zhu, Z.; Qu, W.; Rosario, R.; Touré, F.; Yan, S.F.; et al. RAGE modulates hypoxia/reoxygenation injury in adult murine cardiomyocytes via JNK and GSK-3beta signaling pathways. PLoS ONE 2010, 5, e10092. [Google Scholar] [CrossRef]
- Seryogina, E.S.; Kamynina, A.V.; Koroev, D.O.; Volpina, O.M.; Vinokurov, A.Y.; Abramov, A.Y. RAGE induces physiological activation of NADPH oxidase in neurons and astrocytes and neuroprotection. FEBS J. 2024, 291, 1944–1957. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.P. Oxidative stress in health and disease. Biomedicines 2023, 11, 2925. [Google Scholar] [CrossRef]
- Goh, S.Y.; Cooper, M.E. The role of advanced glycation end products in progression and complications of diabetes. J. Clin. Endocrinol. Metab. 2008, 93, 1143–1152. [Google Scholar] [CrossRef]
- Yashima, H.; Terasaki, M.; Sotokawauchi, A.; Matsui, T.; Mori, Y.; Saito, T.; Osaka, N.; Kushima, H.; Hiromura, M.; Ohara, M.; et al. AGE-RAGE axis stimulates oxidized LDL uptake into macrophages through cyclin-dependent kinase 5-CD36 pathway via oxidative stress generation. Int. J. Mol. Sci. 2020, 21, 9263. [Google Scholar] [CrossRef]
- Olejarz, W.; Łacheta, D.; Kubiak-Tomaszewska, G. Matrix metalloproteinases as biomarkers of atherosclerotic plaque instability. Int. J. Mol. Sci. 2020, 21, 3946. [Google Scholar] [CrossRef]
- Dong, H.; Zhang, Y.; Huang, Y.; Deng, H. Pathophysiology of RAGE in inflammatory diseases. Front. Immunol. 2022, 13, 931473. [Google Scholar] [CrossRef] [PubMed]
- Theofilis, P.; Sagris, M.; Oikonomou, E.; Antonopoulos, A.S.; Siasos, G.; Tsioufis, C.; Tousoulis, D. Inflammatory mechanisms contributing to endothelial dysfunction. Biomedicines 2021, 9, 781. [Google Scholar] [CrossRef]
- Yu, Y.; Liu, S.; Yang, L.; Song, P.; Liu, Z.; Liu, X.; Yan, X.; Dong, Q. Roles of reactive oxygen species in inflammation and cancer. MedComm 2020, 5, e519. [Google Scholar] [CrossRef]
- Maiolino, G.; Rossitto, G.; Caielli, P.; Bisogni, V.; Rossi, G.P.; Calò, L.A. The role of oxidized low-density lipoproteins in atherosclerosis: The myths and the facts. Mediat. Inflamm. 2013, 2013, 714653. [Google Scholar] [CrossRef]
- Stirban, A.; Gawlowski, T.; Roden, M. Vascular effects of advanced glycation endproducts: Clinical effects and molecular mechanisms. Mol. Metab. 2013, 3, 94–108. [Google Scholar] [CrossRef]
- Yamagishi, S.; Takeuchi, M.; Matsui, T.; Nakamura, K.; Imaizumi, T.; Inoue, H. Angiotensin II augments advanced glycation end product-induced pericyte apoptosis through RAGE overexpression. FEBS Lett. 2005, 579, 4265–4270. [Google Scholar] [CrossRef]
- Nakamura, K.; Yamagishi, S.; Adachi, H.; Kurita-Nakamura, Y.; Matsui, T.; Yoshida, T.; Imaizumi, T. Serum levels of sRAGE, the soluble form of receptor for advanced glycation end products, are associated with inflammatory markers in patients with type 2 diabetes. Mol. Med. 2007, 13, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Vlassara, H.; Striker, G.E. AGE restriction in diabetes mellitus: A paradigm shift. Nat. Rev. Endocrinol. 2011, 7, 526–539. [Google Scholar] [CrossRef]
- Ott, C.; Jacobs, K.; Haucke, E.; Navarrete Santos, A.; Grune, T.; Simm, A. Role of advanced glycation end products in cellular signaling. Redox Biol. 2014, 2, 411–429. [Google Scholar] [CrossRef] [PubMed]
- Hamasaki, S.; Kobori, T.; Yamazaki, Y.; Kitaura, A.; Niwa, A.; Nishinaka, T.; Nishibori, M.; Mori, S.; Nakao, S.; Takahashi, H. Effects of scavenger receptors-1 class A stimulation on macrophage morphology and highly modified advanced glycation end product-protein phagocytosis. Sci. Rep. 2018, 8, 5901. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Shah, A.K.; Tappia, P.S. Role of oxidation stress in metabolic and subcellular abnormalities in diabetic cardiomyopathy. Int. J. Mol. Sci. 2020, 21, 2413. [Google Scholar] [CrossRef] [PubMed]
- Wasim, R.; Mahmood, T.; Siddiqui, M.H.; Ahsan, F.; Shamim, A.; Singh, A.; Shariq, M.; Parveen, S. Aftermath of AGE-RAGE Cascade in the pathophysiology of cardiovascular ailments. Life Sci. 2022, 307, 120860. [Google Scholar] [CrossRef]
- Yan, S.F.; Ramasamy, R.; Schmidt, A.M. The receptor for advanced glycation endproducts (RAGE) and cardiovascular disease. Expert Rev. Mol. Med. 2009, 11, e9. [Google Scholar]
- Ramasamy, R.; Shekhtman, A.; Schmidt, A.M. RAGE/DIAPH1 and atherosclerosis through an evolving lens: Viewing the cell from the “Inside–Out”. Atherosclerosis 2024, 394, 117304. [Google Scholar] [CrossRef] [PubMed]
- Bucciarelli, L.G.; Wendt, T.; Qu, W.; Lu, Y.; Lalla, E.; Rong, L.L.; Goova, M.T.; Moser, B.; Kislinger, T.; Lee, D.C.; et al. RAGE blockade stabilizes established atherosclerosis in diabetic apolipoprotein E-null mice. Circulation 2002, 106, 2827–2835. [Google Scholar] [CrossRef] [PubMed]
- Sawada, N.; Liao, J.K. Rho/Rho-associated coiled-coil forming kinase pathway as therapeutic targets for statins in atherosclerosis. Antioxid. Redox Signal. 2014, 20, 1251–1267. [Google Scholar] [CrossRef]
- Wang, X.L.; Lau, W.B.; Yuan, Y.X.; Wang, Y.J.; Yi, W.; Christopher, T.A.; Lopez, B.L.; Liu, H.R.; Ma, X.L. Methylglyoxal increases cardiomyocyte ischemia-reperfusion injury via glycative inhibition of thioredoxin activity. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E207–E214. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Shah, A.K.; Adameova, A.; Elimban, V. Role of oxidative stress in cardiac dysfunction and subcellular defects due to ischemia-reperfusion injury. Biomedicines 2022, 10, 1473. [Google Scholar] [CrossRef]
- Zhang, J.; Han, X.; Chang, J.; Liu, J.; Liu, Y.; Wang, H.; Du, F.; Zeng, X.; Guo, C. Soluble RAGE attenuates myocardial I/R injuries via FoxO3-Bnip3 pathway. Cell. Mol. Life Sci. 2022, 79, 269. [Google Scholar] [CrossRef]
- Zhang, J.; Han, X.; Chang, J.; Liu, J.; Liu, Y.; Wang, H.; Du, F.; Zeng, X.; Guo, C. Soluble RAGE attenuates myocardial I/R injury by suppressing interleukin-6. Am. J. Med. Sci. 2025, 369, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ma, Y.; Wang, R.; Xia, C.; Zhang, R.; Lian, K.; Luan, R.; Sun, L.; Yang, L.; Lau, W.B.; et al. Advanced glycation end products accelerate ischemia/reperfusion injury through receptor of advanced end product/nitrative thioredoxin inactivation in cardiac microvascular endothelial cells. Antioxid. Redox Signal. 2011, 15, 1769–1778. [Google Scholar] [CrossRef]
- Aronson, D. Cross-linking of glycated collagen in the pathogenesis of arterial and myocardial stiffening of aging and diabetes. J. Hypertens. 2003, 21, 3–12. [Google Scholar] [CrossRef]
- Ma, Z.; Mao, C.; Jia, Y.; Fu, Y.; Kong, W. Extracellular matrix dynamics in vascular remodeling. Am. J. Physiol. Cell Physiol. 2020, 319, C481–C499. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.; Maharana, K.C.; Meenakshi, S.; Baisakh, S.S. Endothelial dysfunction and its relation in different disorders: Recent update. Health Sci. Rev. 2023, 7, 100084. [Google Scholar] [CrossRef]
- Kranstuber, A.L.; Del Rio, C.; Biesiadecki, B.J.; Wold, L.E. Advanced glycation end product cross-link breaker attenuates diabetes-induced cardiac dysfunction by improving sarcoplasmic reticulum calcium handling. Front. Physiol. 2012, 3, 292. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhao, X.; Wu, H. Arterial stiffness: A focus on vascular calcification and its link to bone mineralization. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1078–1093. [Google Scholar] [CrossRef] [PubMed]
- Shu, M.; Cheng, W.; Jia, X.; Bai, X.; Zhao, Y.; Lu, Y.; Zhu, L.; Zhu, Y.; Wang, L.; Shu, Y.; et al. AGEs promote atherosclerosis by increasing LDL transcytosis across endothelial cells via RAGE/NF-κB/Caveolin-1 pathway. Mol. Med. 2023, 29, 113. [Google Scholar] [CrossRef]
- Zhu, W.; Li, W.; Silverstein, R.L. Advanced glycation end products induce a prothrombotic phenotype in mice via interaction with platelet CD36. Blood 2012, 119, 6136–6144. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.X.; Srinivasan, S.; O’Neill, K.; Nickolas, T.L.; Wallace, J.M.; Allen, M.R.; Metzger, C.E.; Creecy, A.; Avin, K.G.; Moe, S.M. Effect of advanced glycation end-products (AGE) lowering drug ALT-711 on biochemical, vascular, and bone parameters in a rat model of CKD-MBD. J. Bone Miner. Res. 2020, 35, 608–617. [Google Scholar] [CrossRef]
- Freidja, M.L.; Tarhouni, K.; Toutain, B.; Fassot, C.; Loufrani, L.; Henrion, D. The AGE-breaker ALT-711 restores high blood flow-dependent remodeling in mesenteric resistance arteries in a rat model of type 2 diabetes. Diabetes 2012, 61, 1562–1572. [Google Scholar] [CrossRef]
- Lu, Y.; Qin, W.; Shen, T.; Zhou, L.; Dou, L.; Man, Y.; Wang, S.; Xiao, C.; Li, J. The antioxidant N-acetylcysteine promotes atherosclerotic plaque stabilization through suppression of RAGE, MMPs and NF-κB in ApoE-deficient mice. J. Atheroscler. Thromb. 2011, 18, 998–1008. [Google Scholar] [CrossRef]
- Zhou, Z.; Tang, Y.; Jin, X.; Chen, C.; Lu, Y.; Liu, L.; Shen, C. Metformin inhibits advanced glycation end products-induced inflammatory response in murine macrophages partly through AMPK activation and RAGE/NFκB pathway suppression. J. Diabetes Res. 2016, 2016, 4847812. [Google Scholar] [CrossRef]
- Ko, S.Y.; Lin, I.H.; Shieh, T.M.; Ko, H.A.; Chen, H.I.; Chi, T.C.; Chang, S.S.; Hsu, Y.C. Cell hypertrophy and MEK/ERK phosphorylation are regulated by glyceraldehyde-derived AGEs in cardiomyocyte H9c2 cells. Cell Biochem. Biophys. 2013, 67, 537–544. [Google Scholar] [CrossRef]
- Trial, J.; Cieslik, K.A. Changes in cardiac resident fibroblast physiology and phenotype in aging. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H745–H755. [Google Scholar] [CrossRef]
- Nelson, M.B.; Swensen, A.C.; Winden, D.R.; Bodine, J.S.; Bikman, B.T.; Reynolds, P.R. Cardiomyocyte mitochondrial respiration is reduced by receptor for advanced glycation end-product signaling in a ceramide-dependent manner. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H63–H69. [Google Scholar] [CrossRef]
- Herrmann, K.L.; McCulloch, A.D.; Omens, J.H. Glycated collagen cross-linking alters cardiac mechanics in volume-overload hypertrophy. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H1277–H1284. [Google Scholar] [CrossRef] [PubMed]
- Tran, D.H.; Wang, Z.V. Glucose metabolism in cardiac hypertrophy and heart failure. J. Am. Heart Assoc. 2019, 8, e012673. [Google Scholar] [CrossRef]
- Tikellis, C.; Thomas, M.C.; Harcourt, B.E.; Coughlan, M.T.; Pete, J.; Bialkowski, K.; Tan, A.; Bierhaus, A.; Cooper, M.E.; Forbes, J.M. Cardiac inflammation associated with a Western diet is mediated via activation of RAGE by AGEs. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E323–E330. [Google Scholar] [CrossRef]
- Hartog, J.W.; Voors, A.A.; Bakker, S.J.; Smit, A.J.; van Veldhuisen, D.J. Advanced glycation end-products (AGEs) and heart failure: Pathophysiology and clinical implications. Eur. J. Heart Fail. 2007, 9, 1146–1155. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Bhullar, S.K.; Adameova, A.; Elimban, V. Status of β1-adrenoceptor signal transduction system in cardiac hypertrophy and heart failure. Rev. Cardiovasc. Med. 2023, 24, 264. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Hastings, M.H.; Rhee, J.; Trager, L.E.; Roh, J.D.; Rosenzweig, A. Targeting age-related pathways in heart failure. Circ. Res. 2020, 126, 533–551. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Hirayama, E. Age-related changes of wall composition and collagen cross-linking in the rat carotid artery—In relation with arterial mechanics. J. Mech. Behav. Biomed. Mater. 2017, 65, 881–889. [Google Scholar] [CrossRef]
- Meagher, P.; Adam, M.; Civitarese, R.; Bugyei-Twum, A.; Connelly, K.A. Heart failure with preserved ejection fraction in diabetes: Mechanisms and management. Can. J. Cardiol. 2018, 34, 632–643. [Google Scholar] [CrossRef]
- Kilfoil, P.J.; Lotteau, S.; Zhang, R.; Yue, X.; Aynaszyan, S.; Solymani, R.E.; Cingolani, E.; Marbán, E.; Goldhaber, J.I. Distinct features of calcium handling and β-adrenergic sensitivity in heart failure with preserved versus reduced ejection fraction. J. Physiol. 2020, 598, 5091–5108. [Google Scholar] [CrossRef]
- Tuleta, I.; Frangogiannis, N.G. Fibrosis of the diabetic heart: Clinical significance, molecular mechanisms, and therapeutic opportunities. Adv. Drug Deliv. Rev. 2021, 176, 113904. [Google Scholar] [CrossRef]
- Taguchi, K.; Fukami, K. RAGE signaling regulates the progression of diabetic complications. Front. Pharmacol. 2023, 14, 1128872. [Google Scholar] [CrossRef]
- Linssen, P.B.; Henry, R.M.; Schalkwijk, C.G.; van der Kallen, C.J. Serum advanced glycation endproducts are associated with left ventricular dysfunction in normal glucose metabolism but not in type 2 diabetes: The Hoorn Study. Diab. Vasc. Dis. Res. 2016, 13, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Norton, G.R.; Candy, G.; Woodiwiss, A.J. Aminoguanidine prevents the decreased myocardial compliance produced by streptozotocin-induced diabetes mellitus in rats. Circulation 1996, 93, 1905–1912. [Google Scholar] [CrossRef]
- Avendano, G.F.; Agarwal, R.K.; Bashey, R.I.; Lyons, M.M.; Soni, B.J.; Jyothirmayi, G.N.; Regan, T.J. Effects of glucose intolerance on myocardial function and collagen-linked glycation. Diabetes 1999, 48, 1443–1447. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, S.; Huber, J.; Wihler, C.; Rütten, H.; Busch, A.E.; Linz, W. Impaired left ventricular relaxation in type 2 diabetic rats is related to myocardial accumulation of N(epsilon)-(carboxymethyl) lysine. Eur. J. Heart Fail. 2006, 8, 2–6. [Google Scholar] [CrossRef]
- Asif, M.; Egan, J.; Vasan, S.; Jyothirmayi, G.N.; Masurekar, M.R.; Lopez, S.; Williams, C.; Torres, R.L.; Wagle, D.; Ulrich, P.; et al. An advanced glycation endproduct cross-link breaker can reverse age-related increases in myocardial stiffness. Proc. Natl. Acad. Sci. USA 2000, 97, 2809–2813. [Google Scholar] [CrossRef] [PubMed]
- Vaitkevicius, P.V.; Lane, M.; Spurgeon, H.; Ingram, D.K.; Roth, G.S.; Egan, J.J.; Vasan, S.; Wagle, D.R.; Ulrich, P.; Brines, M.; et al. A cross-link breaker has sustained effects on arterial and ventricular properties in older rhesus monkeys. Proc. Natl. Acad. Sci. USA 2001, 98, 1171–1175. [Google Scholar] [CrossRef]
- Liu, J.; Masurekar, M.R.; Vatner, D.E.; Vatner, S.F. Glycation end-product cross-link breaker reduces collagen and improves cardiac function in aging diabetic heart. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H2587–H2591. [Google Scholar] [CrossRef]
- Brown, D.A.; Perry, J.B.; Allen, M.E.; Sabbah, H.N.; Stauffer, B.L.; Shaikh, S.R.; Cleland, J.G.; Colucci, W.S.; Butler, J.; Voors, A.A.; et al. Mitochondrial function as a therapeutic target in heart failure. Nat. Rev. Cardiol. 2017, 14, 238–250. [Google Scholar] [CrossRef]
- Hinton, A., Jr.; Claypool, S.M.; Neikirk, K.; Senoo, N.; Wanjalla, C.N.; Kirabo, A.; Williams, C.R. Mitochondrial structure and function in human heart failure. Circ. Res. 2024, 135, 372–396. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Elimban, V.; Adameova, A.D.; Goyal, R.K. Molecular mechanisms for pathophysiology and therapy of cardiac dysfunction in heart failure. Scr. Med. 2025, 56, 117–136. [Google Scholar] [CrossRef]
- Coirault, C.; Guellich, A.; Barbry, T.; Samuel, J.L.; Riou, B.; Lecarpentier, Y. Oxidative stress of myosin contributes to skeletal muscle dysfunction in rats with chronic heart failure. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1009–H1017. [Google Scholar] [CrossRef]
- Franssen, C.; González Miqueo, A. The role of titin and extracellular matrix remodelling in heart failure with preserved ejection fraction. Neth. Heart J. 2016, 24, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Horn, M.A.; Graham, H.K.; Richards, M.A.; Clarke, J.D.; Greensmith, D.J.; Briston, S.J.; Hall, M.C.; Dibb, K.M.; Trafford, A.W. Age-related divergent remodeling of the cardiac extracellular matrix in heart failure: Collagen accumulation in the young and loss in the aged. J. Mol. Cell. Cardiol. 2012, 53, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K. AGE-RAGE stress in the pathophysiology of pulmonary hypertension and its treatment. Int. J. Angiol. 2019, 28, 71–79. [Google Scholar] [CrossRef]
- Prasad, K. Involvement of AGE and its receptors in the pathogenesis of hypertension in elderly people and its treatment. Int. J. Angiol. 2022, 31, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K. AGE-RAGE stress play a role in aortic aneurysm: A comprehensive review and novel potential therapeutic target. Rev. Cardiovasc. Med. 2019, 20, 201–208. [Google Scholar] [CrossRef]
- Sarkar, A.; Prasad, K.; Ziganshin, B.A.; Elefteriades, J.A. Reasons to investigate the soluble receptor for advanced glycation end-product (sRAGE) pathway in aortic disease. Aorta 2013, 1, 210–217. [Google Scholar] [CrossRef]
- Prasad, K. AGE-RAGE stress and coronary artery disease. Int. J. Angiol. 2021, 30, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K.; Khan, A.S.; Bhanumathy, K.K. Does AGE-RAGE stress play a role in the development of coronary artery disease in obesity? Int. J. Angiol. 2022, 31, 1–9. [Google Scholar] [CrossRef] [PubMed]
- McNair, E.D.; Wells, C.R.; Qureshi, A.M.; Basran, R.S.; Pearce, C.; Orvold, J.; Devilliers, J.; Prasad, K. Low levels of soluble receptor for advanced glycation end products in non-ST elevation myocardial infarction patients. Int. J. Angiol. 2009, 18, 187–192. [Google Scholar] [CrossRef]
- McNair, E.D.; Wells, C.R.; Qureshi, A.M.; Pearce, C.; Caspar-Bell, G.; Prasad, K. Inverse association between cardiac troponin-I and soluble receptor for advanced glycation end products in patients with non-ST-segment elevation myocardial infarction. Int. J. Angiol. 2011, 20, 49–54. [Google Scholar] [CrossRef]
- Azevedo, R.F.L.; Varzino, M.; Steinman, E.; Rogers, W.A. Evaluating effectiveness of mHealth apps for older adults with diabetes: Meta-analysis of randomized controlled trials. J. Med. Internet Res. 2025, 27, e65855. [Google Scholar] [CrossRef]
- Barton, A.B.; Okorodudu, D.E.; Bosworth, H.B.; Crowley, M.J. Clinical inertia in a randomized trial of telemedicine-based chronic disease management: Lessons learned. Telemed. J. E-Health 2018, 24, 742–748. [Google Scholar] [CrossRef] [PubMed]
- Popp, C.J.; Hu, L.; Kharmats, A.Y.; Curran, M.; Berube, L.; Wang, C.; Pompeii, M.L.; Illiano, P.; St-Jules, D.E.; Mottern, M.; et al. Effect of a personalized diet to reduce postprandial glycemic response vs a low-fat diet on weight loss in adults with abnormal glucose metabolism and obesity: A randomized clinical trial. JAMA Netw. Open 2022, 5, e2233760. [Google Scholar] [CrossRef] [PubMed]
- Victoria-Montesinos, D.; Cerdá Martínez-Pujalte, B.; Zafrilla, P.; Ballester, P.; García-Muñoz, A.M. Effect of the consumption of species from the zingiberaceae or berberidaceae family on glycemic profile parameters: A systematic review and meta-analysis. Int. J. Mol. Sci. 2025, 26, 5565. [Google Scholar] [CrossRef]
- Yamagishi, S. Role of advanced glycation end products (AGEs) and receptor for AGEs (RAGE) in vascular damage in diabetes. Exp. Gerontol. 2011, 46, 217–224. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| AGE Groups | Composition | Targets |
|---|---|---|
| A. Fluorescent, cross-linked AGEs | a. Targets for cross-linked AGEs in groups A and B: | |
| 1 Pentosidine: | Arginine and lysine residue cross-linked with ribose | (i) Cross-linking with extracellular matrix proteins: collagen, vitronectin and lamin. |
| 2 Pentodilysine: | Lysine and lysine residues cross-linked with ribose | (ii) Cross-linking with cardiomyocyte and vascular proteins: collagen, sarcoplasmic reticulum Ca2+-ATPase and ryanodine receptors. |
| B. Non-fluorescent, cross-linked AGEs | ||
| 1 GOLD: | Glyoxal-lysine dimer | |
| 2 MOLD: | Methylglycoxal-lysine dimer | |
| C. Non-fluorescent, non-cross-linked AGEs | b. Targets for non-cross-linked AGEs in groups C and D: | |
| 1 CEL: | Carboxyethyl-lysine | |
| 2 CML: | Condensation of glucose with lysine of amino group | (i) Binding with RAGEs in macrophages, fibroblasts, endothelial cells, and Cardiomyocytes. |
| 3 Pyrraline: | Reaction between glucose and lysine residues | (ii) Binding with sRAGEs and esRAGEs as well as AGE receptors such as AGE-R1, AGE-R2, and AGE-R3. |
| D. Fluorescent, non-cross-linked AGEs | ||
| 1 Argpyrimidine: | Formed from arginine and methylglycoxal |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mota, K.O.; de Vasconcelos, C.M.L.; Kirshenbaum, L.A.; Dhalla, N.S. The Role of Advanced Glycation End-Products in the Pathophysiology and Pharmacotherapy of Cardiovascular Disease. Int. J. Mol. Sci. 2025, 26, 7311. https://doi.org/10.3390/ijms26157311
Mota KO, de Vasconcelos CML, Kirshenbaum LA, Dhalla NS. The Role of Advanced Glycation End-Products in the Pathophysiology and Pharmacotherapy of Cardiovascular Disease. International Journal of Molecular Sciences. 2025; 26(15):7311. https://doi.org/10.3390/ijms26157311
Chicago/Turabian StyleMota, Karina O., Carla M. L. de Vasconcelos, Lorrie A. Kirshenbaum, and Naranjan S. Dhalla. 2025. "The Role of Advanced Glycation End-Products in the Pathophysiology and Pharmacotherapy of Cardiovascular Disease" International Journal of Molecular Sciences 26, no. 15: 7311. https://doi.org/10.3390/ijms26157311
APA StyleMota, K. O., de Vasconcelos, C. M. L., Kirshenbaum, L. A., & Dhalla, N. S. (2025). The Role of Advanced Glycation End-Products in the Pathophysiology and Pharmacotherapy of Cardiovascular Disease. International Journal of Molecular Sciences, 26(15), 7311. https://doi.org/10.3390/ijms26157311