
Unveiling Replication Timing-Dependent Mutational Biases: Mechanistic Insights from Gene Knockouts and Genotoxins Exposures


Abstract

1. Introduction
2. Results
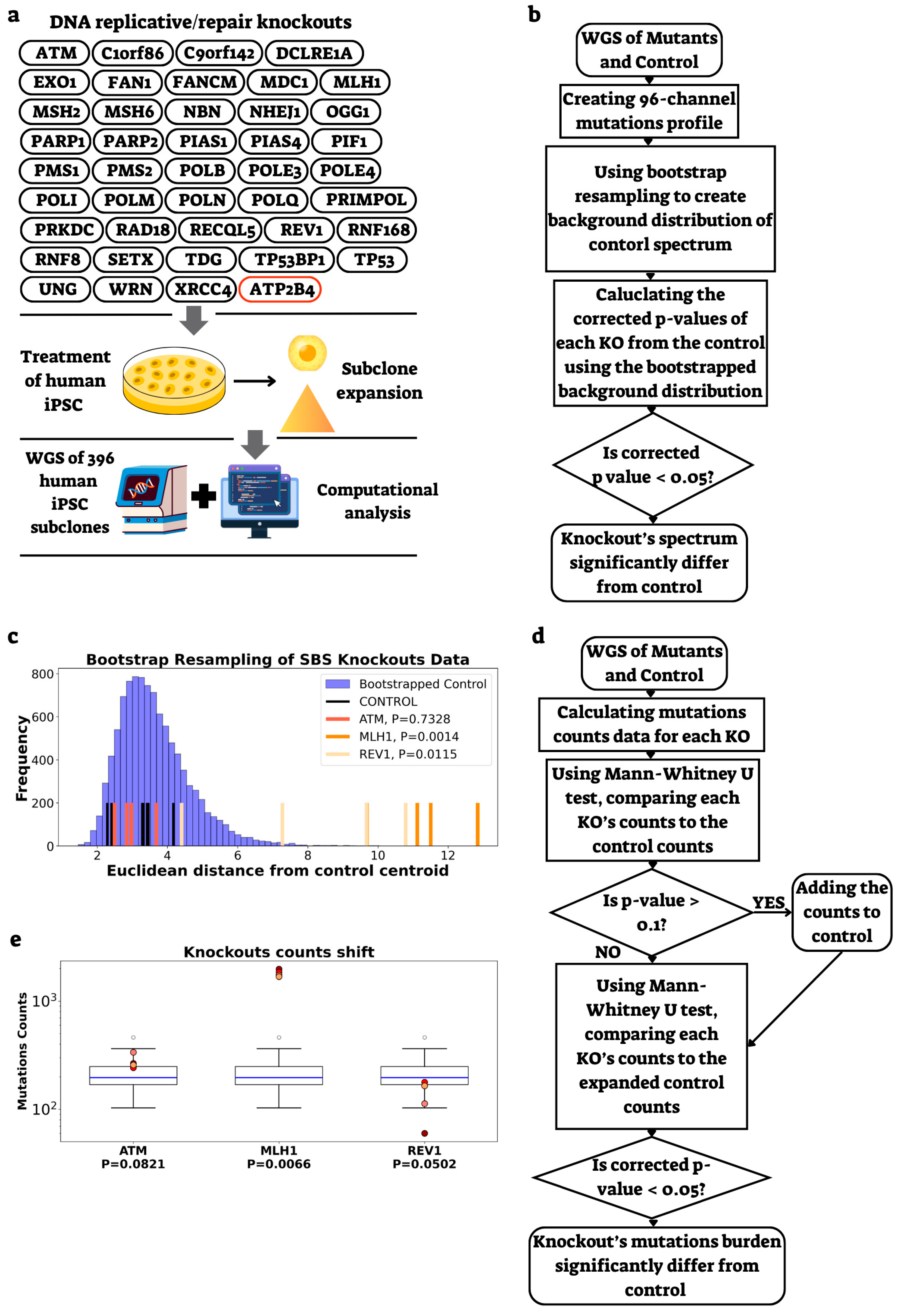
2.1. Development of a Methodology to Assess Gene Knockout Effects on Mutational Spectra and Burden
2.2. Identification of Genes Affecting Mutation Rate and Spectra in a Replication Timing-Dependent Manner
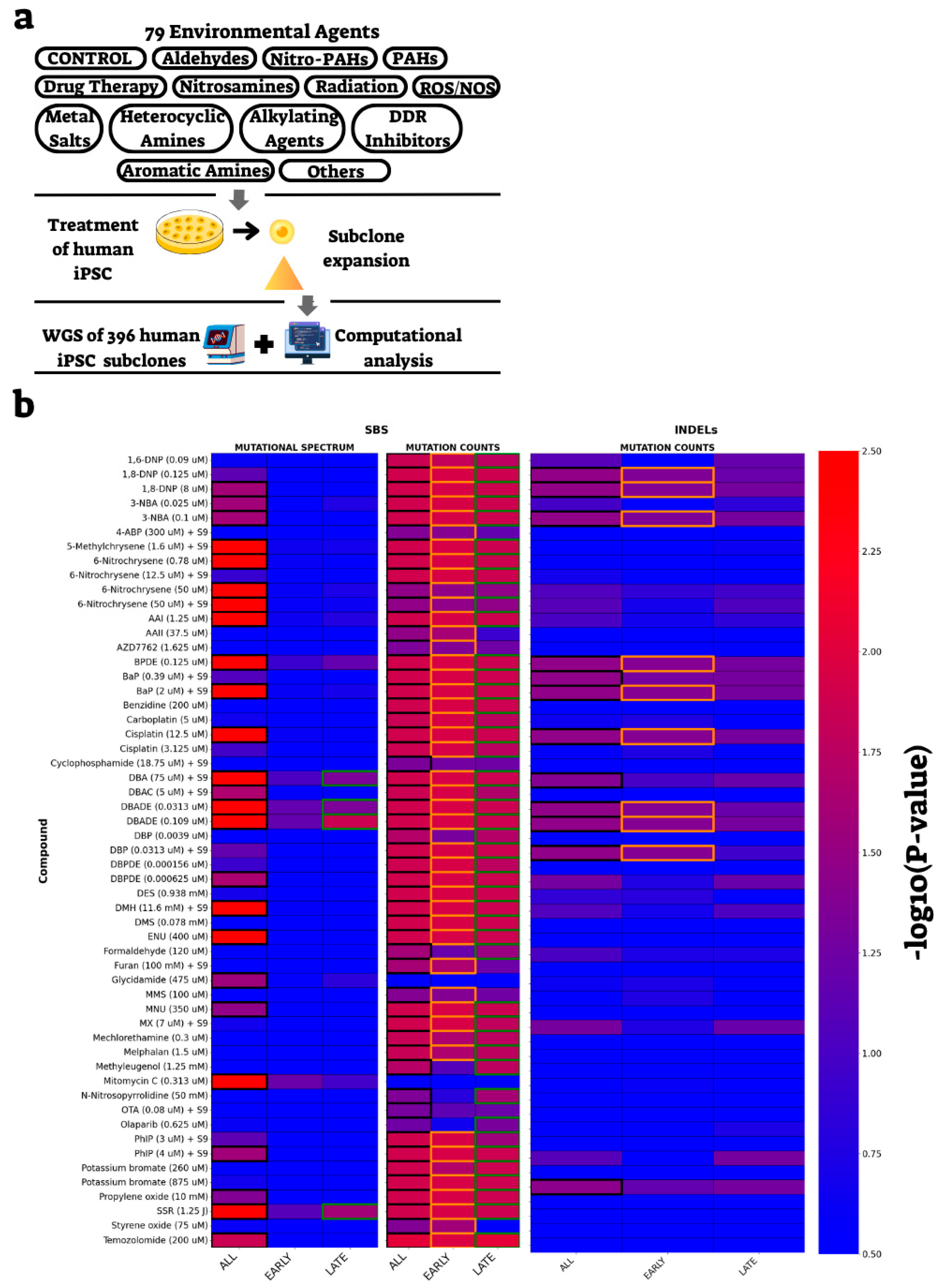
2.3. Identification of External Treatments Affecting Mutation Burden and Spectra in a Replication Timing-Dependent Manner
2.4. Power Analysis
3. Discussion
4. Materials and Methods
4.1. Data Sources
4.2. Mutational Profile Generation
4.3. Replication Timing Stratification
4.4. Trinucleotide Normalization
4.5. Bootstrapping Method for Qualitative Identification of Significant Differences in Mutational Spectra
4.6. Statistical Method for Quantitative Identification of Significant Differences in Mutation Counts
4.7. Power Analysis
4.8. Software and Code Availability
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BER | Base Excision Repair |
| DSB | Double-Strand Break |
| ERR | Early-Replicating Regions |
| FA | Fanconi Anemia |
| hiPSCs | Human-Induced Pluripotent Stem Cells |
| ICL | Inter-strand Cross-Link |
| INDEL | Insertions and Deletions |
| KO | Knockout |
| LRR | Late-Replicating Regions |
| MMEJ | Micro-Homology Mediated End Joining |
| MMR | Mismatch Repair |
| NER | Nucleotide Excision Repair |
| NHEJ | Non-Homologous End Joining |
| PAH | Polycyclic Aromatic Hydrocarbon |
| RT | Replication Timing |
| SBS | Single Base Substitution |
| TLS | Trans-lesion Synthesis |
| WGS | Whole-Genome Sequencing |
References
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The cancer genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef]
- Pfeifer, G.P. Environmental exposures and mutational patterns of cancer genomes. Genome Med. 2010, 2, 54. [Google Scholar] [CrossRef]
- Peña-Diaz, J.; Bregenhorn, S.; Ghodgaonkar, M.; Follonier, C.; Artola-Borán, M.; Castor, D.; Lopes, M.; Sartori, A.A.; Jiricny, J. Noncanonical mismatch repair as a source of genomic instability in human cells. Mol. Cell 2012, 47, 669–680. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef]
- Tomasetti, C.; Vogelstein, B. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 2015, 347, 78–81. [Google Scholar] [CrossRef]
- Singh, V.K.; Rastogi, A.; Hu, X.; Wang, Y.; De, S. Mutational signature SBS8 predominantly arises due to late replication errors in cancer. Commun. Biol. 2020, 3, 421. [Google Scholar] [CrossRef]
- Tomkova, M.; Tomek, J.; Kriaucionis, S.; Schuster-Böckler, B. Mutational signature distribution varies with DNA replication timing and strand asymmetry. Genome Biol. 2018, 19, 129. [Google Scholar] [CrossRef]
- Blumenfeld, B.; Ben-Zimra, M.; Simon, I. Perturbations in the Replication Program Contribute to Genomic Instability in Cancer. Int. J. Mol. Sci. 2017, 18, 1138. [Google Scholar] [CrossRef]
- Greenberg, A.; Blumenfeld, B.; Simon, I. The delicate relationship between DNA replication timing and gene expression. Curr. Opin. Syst. Biol. 2020, 19, 8–15. [Google Scholar] [CrossRef]
- Farkash-Amar, S.; Simon, I. Genome-wide analysis of the replication program in mammals. Chromosome Res. 2010, 18, 115–125. [Google Scholar] [CrossRef]
- Yaacov, A.; Vardi, O.; Blumenfeld, B.; Greenberg, A.; Massey, D.J.; Koren, A.; Adar, S.; Simon, I.; Rosenberg, S. Cancer Mutational Processes Vary in Their Association with Replication Timing and Chromatin Accessibility. Cancer Res. 2021, 81, 6106–6116. [Google Scholar] [CrossRef]
- Supek, F.; Lehner, B. Differential DNA mismatch repair underlies mutation rate variation across the human genome. Nature 2015, 521, 81–84. [Google Scholar] [CrossRef]
- Zheng, C.L.; Wang, N.J.; Chung, J.; Moslehi, H.; Sanborn, J.Z.; Hur, J.S.; Collisson, E.A.; Vemula, S.S.; Naujokas, A.; Chiotti, K.E.; et al. Transcription Restores DNA Repair to Heterochromatin, Determining Regional Mutation Rates in Cancer Genomes. Cell Rep. 2014, 9, 1228–1234. [Google Scholar] [CrossRef]
- Zou, X.; Koh, G.C.C.; Nanda, A.S.; Degasperi, A.; Urgo, K.; Roumeliotis, T.I.; Agu, C.A.; Badja, C.; Momen, S.; Young, J.; et al. A systematic CRISPR screen defines mutational mechanisms underpinning signatures caused by replication errors and endogenous DNA damage. Nat. Cancer 2021, 2, 643–657. [Google Scholar] [CrossRef]
- Kucab, J.E.; Zou, X.; Morganella, S.; Joel, M.; Nanda, A.S.; Nagy, E.; Gomez, C.; Degasperi, A.; Harris, R.; Jackson, S.P.; et al. A Compendium of Mutational Signatures of Environmental Agents. Cell 2019, 177, 821–836.e16. [Google Scholar] [CrossRef]
- Ding, Q.; Edwards, M.M.; Wang, N.; Zhu, X.; Bracci, A.N.; Hulke, M.L.; Hu, Y.; Tong, Y.; Hsiao, J.; Charvet, C.J.; et al. The genetic architecture of DNA replication timing in human pluripotent stem cells. Nat. Commun. 2021, 12, 6746. [Google Scholar] [CrossRef]
- Secrier, M.; Li, X.; De Silva, N.; Eldridge, M.D.; Contino, G.; Bornschein, J.; MacRae, S.; Grehan, N.; O’Donovan, M.; Miremadi, A.; et al. Mutational signatures in esophageal adenocarcinoma define etiologically distinct subgroups with therapeutic relevance. Nat. Genet. 2016, 48, 1131–1141. [Google Scholar] [CrossRef]
- Stenzinger, A.; Pfarr, N.; Endris, V.; Penzel, R.; Jansen, L.; Wolf, T.; Herpel, E.; Warth, A.; Klauschen, F.; Kloor, M.; et al. Mutations in POLE and survival of colorectal cancer patients--link to disease stage and treatment. Cancer Med. 2014, 3, 1527–1538. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Zou, X.; Owusu, M.; Harris, R.; Jackson, S.P.; Loizou, J.I.; Nik-Zainal, S. Validating the concept of mutational signatures with isogenic cell models. Nat. Commun. 2018, 9, 1744. [Google Scholar] [CrossRef] [PubMed]
- Bergstrom, E.N.; Huang, M.N.; Mahto, U.; Barnes, M.; Stratton, M.R.; Rozen, S.G.; Alexandrov, L.B. SigProfilerMatrixGenerator: A tool for visualizing and exploring patterns of small mutational events. BMC Genom. 2019, 20, 685. [Google Scholar] [CrossRef] [PubMed]
- Bellelli, R.; Belan, O.; Pye, V.E.; Clement, C.; Maslen, S.L.; Skehel, J.M.; Cherepanov, P.; Almouzni, G.; Boulton, S.J. POLE3-POLE4 Is a Histone H3-H4 Chaperone that Maintains Chromatin Integrity during DNA Replication. Mol. Cell 2018, 72, 112–126.e5. [Google Scholar] [CrossRef] [PubMed]
- Grollman, A.P.; Shibutani, S.; Moriya, M.; Miller, F.; Wu, L.; Moll, U.; Suzuki, N.; Fernandes, A.; Rosenquist, T.; Medverec, Z.; et al. Aristolochic acid and the etiology of endemic (Balkan) nephropathy. Proc. Natl. Acad. Sci. USA 2007, 104, 12129–12134. [Google Scholar] [CrossRef]
- Bastlová, T.; Podlutsky, A. Molecular analysis of styrene oxide-induced hprt mutation in human T-lymphocytes. Mutagenesis 1996, 11, 581–591. [Google Scholar] [CrossRef]
- Yalcin, E.; de la Monte, S. Tobacco nitrosamines as culprits in disease: Mechanisms reviewed. J. Physiol. Biochem. 2016, 72, 107–120. [Google Scholar] [CrossRef]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef]
- Talhout, R.; Schulz, T.; Florek, E.; Van Benthem, J.; Wester, P.; Opperhuizen, A. Hazardous compounds in tobacco smoke. Int. J. Environ. Res. Public Health 2011, 8, 613–628. [Google Scholar] [CrossRef]
- Koren, A.; Massey, D.J.; Bracci, A.N. TIGER: Inferring DNA replication timing from whole-genome sequence data. Bioinformatics 2021, 37, 4001–4005. [Google Scholar] [CrossRef]
- Zhu, N.; Zhao, Y.; Mi, M.; Lu, Y.; Tan, Y.; Fang, X.; Weng, S.; Yuan, Y. REV1: A novel biomarker and potential therapeutic target for various cancers. Front. Genet. 2022, 13, 997970. [Google Scholar] [CrossRef]
- Waters, L.S.; Walker, G.C. The critical mutagenic translesion DNA polymerase Rev1 is highly expressed during G(2)/M phase rather than S phase. Proc. Natl. Acad. Sci. USA 2006, 103, 8971–8976. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Richie, C.; Legerski, R.J. Translation of hSNM1 is mediated by an internal ribosome entry site that upregulates expression during mitosis. DNA Repair 2002, 1, 379–390. [Google Scholar] [CrossRef]
- Heilbrun, E.E.; Tseitline, D.; Wasserman, H.; Kirshenbaum, A.; Cohen, Y.; Gordan, R.; Adar, S. The epigenetic landscape shapes smoking-induced mutagenesis by modulating DNA damage susceptibility and repair efficiency. Nucleic Acids Res. 2025, 53, gkaf048. [Google Scholar] [CrossRef]
- Adar, S.; Hu, J.; Lieb, J.D.; Sancar, A. Genome-wide kinetics of DNA excision repair in relation to chromatin state and mutagenesis. Proc. Natl. Acad. Sci. USA 2016, 113, E2124–E2133. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene KO | Pathway | Spectrum RT-Differentiation | Counts RT-Differentiation |
|---|---|---|---|
| SBS | |||
| DCLRE1A | FA and ICL repair | LRR | LRR—LESS |
| MLH1 | MMR | LRR | NA 1 |
| MSH2 | MMR | LRR | NA 1 |
| MSH6 | MMR | LRR | NA 1 |
| PMS1 | MMR | LRR | NA |
| PMS2 | MMR | LRR | NA 1 |
| POLE3 | DNA replication | LRR | NA 2 |
| PRKDC | NHEJ and MMEJ | LRR | NA |
| REV1 | TLS | LRR | LRR—LESS |
| TP53 | Checkpoint/DSB repair | LRR | NA |
| INDELs | |||
| PARP1 | BER/DSB repair/NER | - | ERR *—MORE |
| PMS1 | MMR | - | LRR—MORE |
| POLB | BER | - | ERR—MORE |
| POLE4 | DNA replication | - | LRR—LESS |
| Mutagen | Group | Spectrum RT-Differentiation | Counts RT-Differentiation |
|---|---|---|---|
| SBS | |||
| Formaldehyde (120 μM) | Aldehydes | NA | LRR *—MORE |
| MMS (100 μM) | Alkylating Agents | NA | ERR *—MORE |
| 4-ABP (300 μM) + S9 | Aromatic Amines | NA | ERR *—MORE |
| AZD7762 (1.625 μM) | DNA Damage Response Inhibitors | NA | ERR *—MORE |
| Olaparib (0.625 μM) | Drug Therapy | NA | LRR—LESS |
| N-Nitrosopyrrolidine (50 mM) | Nitrosamine | NA | LRR—MORE |
| DBA (75 μM) + S9 | PAHs | LRR * | NA 1 |
| DBADE (0.0313 μM) | PAHs | ERR * | NA 1 |
| DBP (0.0039 μM) | PAHs | NA | LRR *—MORE |
| SSR (1.25 J) | Radiation | LRR * | NA 1 |
| AAII (3.75 μM) | Others | NA | ERR—MORE |
| Furan (100 mM) + S9 | Others | NA | ERR *—MORE |
| Methyleugenol (1.25 mM) | Others | NA | LRR—MORE |
| Styrene oxide (75 μM) | Others | NA | ERR—MORE |
| INDELs | |||
| Cisplatin (12.5 μM) | Drug Therapy | - | ERR *—MORE |
| BPDE (0.125 μM) | PAHs | - | ERR *—MORE |
| BaP (2 μM) + S9 | PAHs | - | ERR *—MORE |
| DBADE (0.0313 μM) | PAHs | - | ERR *—MORE |
| DBADE (0.109 μM) | PAHs | - | ERR *—MORE |
| DBP (0.0313 μM) + S9 | PAHs | - | ERR—MORE |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gross-Samuels, H.; Koren, A.; Simon, I. Unveiling Replication Timing-Dependent Mutational Biases: Mechanistic Insights from Gene Knockouts and Genotoxins Exposures. Int. J. Mol. Sci. 2025, 26, 7307. https://doi.org/10.3390/ijms26157307
Gross-Samuels H, Koren A, Simon I. Unveiling Replication Timing-Dependent Mutational Biases: Mechanistic Insights from Gene Knockouts and Genotoxins Exposures. International Journal of Molecular Sciences. 2025; 26(15):7307. https://doi.org/10.3390/ijms26157307
Chicago/Turabian StyleGross-Samuels, Hadas, Amnon Koren, and Itamar Simon. 2025. "Unveiling Replication Timing-Dependent Mutational Biases: Mechanistic Insights from Gene Knockouts and Genotoxins Exposures" International Journal of Molecular Sciences 26, no. 15: 7307. https://doi.org/10.3390/ijms26157307
APA StyleGross-Samuels, H., Koren, A., & Simon, I. (2025). Unveiling Replication Timing-Dependent Mutational Biases: Mechanistic Insights from Gene Knockouts and Genotoxins Exposures. International Journal of Molecular Sciences, 26(15), 7307. https://doi.org/10.3390/ijms26157307