
A 25K Wheat SNP Array Revealed the Genetic Diversity and Population Structure of Durum Wheat (Triticum turgidum subsp. durum) Landraces and Cultivars

 ,
,  , , and
, , and 
Abstract
1. Introduction
2. Results
2.1. Genomic-Wide Genetic Variation
2.2. SNP Loci Under Selection
2.3. Genetic Variation Between and Within Accessions and Their Groups
2.4. Analysis of Molecular Variance (AMOVA) and Population Differentiation
2.5. Cluster Analysis
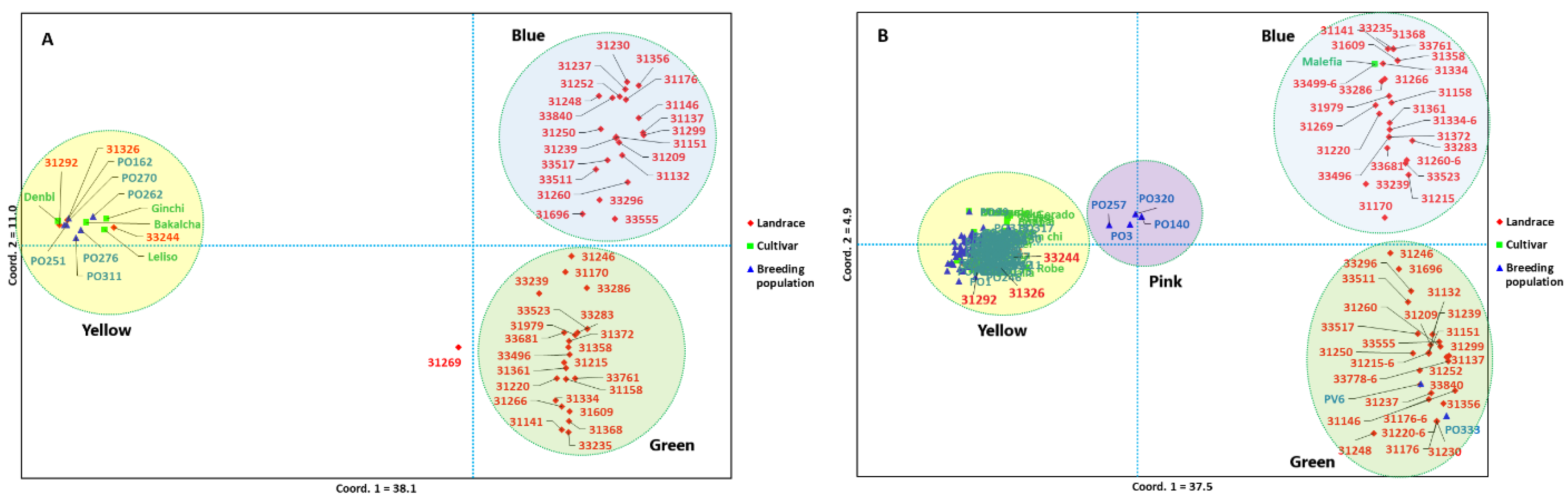
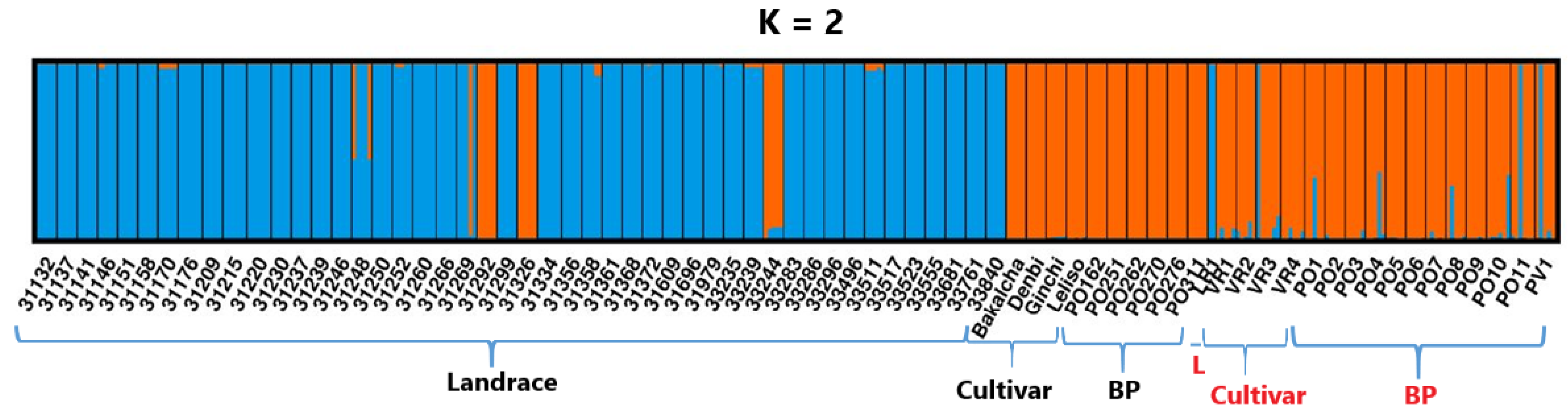
2.6. Principal Coordinate Analysis and Population Structure
3. Discussion
3.1. Genome-Wide SNP Distribution and Genetic Diversity
3.2. Selection Signatures and Functional Implications
3.3. Genetic Variation Between and Within Accessions and Their Groups
3.4. Geographic and Agronomic Group Comparisons
4. Materials and Methods
4.1. Plant Material
4.2. Planting, Leaf Tissue Sampling, and DNA Extraction
4.3. SNP Genotyping and Genotype Data Filtering
4.4. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Broccanello, C.; Bellin, D.; Dalcorso, G.; Furini, A.; Taranto, F. Genetic approaches to exploit landraces for improvement of Triticum turgidum ssp. durum in the age of climate change. Front. Plant Sci. 2023, 14, 1101271. [Google Scholar] [CrossRef] [PubMed]
- FAOSTAT. Food and Agriculture Organization of the United Nations. 2023. Available online: https://www.fao.org/faostat/en/#data (accessed on 1 July 2025).
- Sall, A.T.; Chiari, T.; Legesse, W.; Seid-Ahmed, K.; Ortiz, R.; Van Ginkel, M.; Bassi, F.M. Durum wheat (Triticum durum Desf): Origin, cultivation and potential expansion in Sub-Saharan Africa. Agronomy 2019, 9, 263. [Google Scholar] [CrossRef]
- Grosse-Heilmann, M.; Cristiano, E.; Deidda, R.; Viola, F. Durum wheat productivity today and tomorrow: A review of influencing factors and climate change effects. Resour. Environ. Sustain. 2024, 17, 100170. [Google Scholar] [CrossRef]
- Maccaferri, M.; Harris, N.S.; Twardziok, S.O.; Pasam, R.K.; Gundlach, H.; Spannagl, M.; Ormanbekova, D.; Lux, T.; Prade, V.M.; Milner, S.G.; et al. Durum wheat genome highlights past domestication signatures and future improvement targets. Nat. Genet. 2019, 51, 885–895. [Google Scholar] [CrossRef] [PubMed]
- Swarup, S.; Cargill, E.J.; Crosby, K.; Flagel, L.; Kniskern, J.; Glenn, K.C. Genetic diversity is indispensable for plant breeding to improve crops. Crop Sci. 2021, 61, 839–852. [Google Scholar] [CrossRef]
- Haudry, A.; Cenci, A.; Ravel, C.; Bataillon, T.; Brunel, D.; Poncet, C.; Hochu, I.; Poirier, S.; Santoni, S.; Glémin, S.; et al. Grinding up wheat: A massive loss of nucleotide diversity since domestication. Mol. Biol. Evol. 2007, 24, 1506–1517. [Google Scholar] [CrossRef] [PubMed]
- Vavilov, N.I. The Origin, Variation, Immunity and Breeding of Cultivated Plants; LWW: Philadelphia, PA, USA, 1951. [Google Scholar]
- Kabbaj, H.; Sall, A.T.; Al-Abdallat, A.; Geleta, M.; Amri, A.; Filali-Maltouf, A.; Belkadi, B.; Ortiz, R.; Balssi, F.M. Genetic diversity within a global panel of durum wheat (Triticum durum) landraces and modern germplasm reveals the history of alleles exchange. Front. Plant Sci. 2017, 8, 1277. [Google Scholar] [CrossRef] [PubMed]
- Tesemma, T.; Belay, G. Aspects of Ethiopian tetraploid wheat with emphasis on durum wheat genetics and breeding. In Wheat Research in Ethiopia; Tanner, D.G., Gebre-Mariam, H., Huluka, M., Eds.; Institute of Agricultural Research: Addis Ababa, Ethiopia, 1991; pp. 95–103. [Google Scholar]
- Kidane, Y.G.; Hailemariam, B.N.; Mengistu, D.K.; Fadda, C.; Pè, M.E.; Dell’Acqua, M. Genome-wide association study of Septoria tritici blotch resistance in Ethiopian durum wheat landraces. Front. Plant Sci. 2017, 8, 1586. [Google Scholar] [CrossRef] [PubMed]
- Negisho, K.; Shibru, S.; Matros, A.; Pillen, K.; Ordon, F.; Wehner, G. Association mapping of drought tolerance indices in Ethiopian durum wheat (Triticum turgidum ssp. durum). Front. Plant Sci. 2022, 13, 838088. [Google Scholar] [CrossRef] [PubMed]
- Mulugeta, B.; Tesfaye, K.; Geleta, M.; Johansson, E.; Hailesilassie, T.; Hammenhag, C.; Hailu, F.; Ortiz, R.; Alrzani, A. Multivariate analyses of Ethiopian durum wheat revealed stable and high yielding genotypes. PLoS ONE 2022, 17, e0273008. [Google Scholar] [CrossRef] [PubMed]
- Dagnaw, T.; Mulugeta, B.; Haileselassie, T.; Geleta, M.; Ortiz, R.; Tesfaye, K. Genetic diversity of durum wheat (Triticum turgidum L. ssp. durum, Desf) germplasm as revealed by morphological and SSR markers. Genes 2023, 14, 1155. [Google Scholar] [CrossRef] [PubMed]
- Mulugeta, B.; Tesfaye, K.; Ortiz, R.; Geleta, M.; Haileselassie, T.; Hammenhag, C.; Hailu, F.; Johansson, E. Unlocking the genetic potential of Ethiopian durum wheat landraces with high protein quality: Sources to be used in future breeding for pasta production. Food Energy Secur. 2023, 13, e511. [Google Scholar] [CrossRef]
- Legesse, W.; Tesfaye, H.; Bassi, F.M. Ancient and novel Ethiopian durum wheat cultivars: What is the future for their cultivation? Afr. J. Agric. Res. 2024, 20, 221–225. [Google Scholar] [CrossRef]
- Alemayehu, L.Z.; Mekbib, F.; Abebe, K.A.; Bishaw, Z. Estimation of genetic erosion on Ethiopian tetraploid wheat landraces using different approaches. J. Genet. Resour. 2021, 2, 66–71. [Google Scholar]
- Al-Khayri, J.M.; Jain, S.M.; Johnson, D.V. Advances in Plant Breeding Strategies: Breeding, Biotechnology and Molecular Tools; Springer International Publishing: Cham, Switzerland, 2016; p. 656. [Google Scholar]
- Geleta, M.; Ortiz, R. Molecular and genomic tools provide insights on crop domestication and evolution. Adv. Agron. 2016, 135, 181–223. [Google Scholar]
- Kumar, R.; Das, S.P.; Choudhury, B.U.; Kumar, A.; Prakash, N.R.; Verma, R.; Chakraborti, M.; Devi, A.G.; Bhattacharjee, B.; Das, R.; et al. Advances in genomic tools for plant breeding: Harnessing DNA molecular markers, genomic selection, and genome editing. Biol. Res. 2024, 57, 80. [Google Scholar] [CrossRef] [PubMed]
- Burridge, A.J.; Winfield, M.; Przewieslik-Allen, A.; Edwards, K.J.; Siddique, I.; Barral-Arca, R.; Griffiths, S.; Cheng, S.; Huang, Z.; Feng, C.; et al. Development of a next generation SNP genotyping array for wheat. Plant Biotechnol. J. 2024, 22, 2235–2247. [Google Scholar] [CrossRef] [PubMed]
- Turner-Hissong, S.D.; Mabry, M.E.; Beissinger, T.M.; Ross-Ibarra, J.; Pires, J.C. Evolutionary insights into plant breeding. Curr. Opin. Plant Biol. 2020, 54, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Bekele, E. Analysis of regional patterns of phenotypic diversity in the Ethiopian tetraploid and hexaploid wheats. Hereditas 1984, 100, 131–154. [Google Scholar] [CrossRef]
- Eticha, F.; Bekele, E.; Belay, G.; Börner, A. Phenotypic diversity in tetraploid wheats collected from Bale and Wello regions of Ethiopia. Plant Genet. Resour. 2005, 3, 35–43. [Google Scholar] [CrossRef]
- Mengistu, D.K.; Kiros, A.Y.; Pè, M.E. Phenotypic diversity in Ethiopian durum wheat (Triticum turgidum var. durum) landraces. Crop J. 2015, 3, 190–199. [Google Scholar] [CrossRef]
- Mengistu, D.K.; Kidane, Y.G.; Catellani, M.; Frascaroli, E.; Fadda, C.; Pè, M.E.; Dell’ACqua, M. High-density molecular characterization and association mapping in Ethiopian durum wheat landraces reveals high diversity and potential for wheat breeding. Plant Biotechnol. J. 2016, 14, 1800–1812. [Google Scholar] [CrossRef] [PubMed]
- Alemu, A.; Feyissa, T.; Letta, T.; Abeyo, B. Genetic diversity and population structure analysis based on the high-density SNP markers in Ethiopian durum wheat (Triticum turgidum ssp. durum). BMC Genet. 2020, 21, 18. [Google Scholar] [CrossRef] [PubMed]
- Negisho, K.; Shibru, S.; Pillen, K.; Ordon, F.; Wehner, G. Genetic diversity of Ethiopian durum wheat landraces. PLoS ONE 2021, 16, e0247016. [Google Scholar] [CrossRef] [PubMed]
- Mulugeta, B.; Ortiz, R.; Geleta, M.; Hailesilassie, T.; Hammenhag, C.; Hailu, F.; Tesfaye, K. Harnessing genome-wide genetic diversity, population structure and linkage disequilibrium in Ethiopian durum wheat gene pool. Front. Plant Sci. 2023, 14, 1192356. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.L.; Liu, J.X. STRUCTURESELECTOR: A web-based software to select and visualize the optimal number of clusters using multiple methods. Mol. Ecol. Resour. 2018, 18, 176–177. [Google Scholar] [CrossRef] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Mulugeta, B.; Tesfaye, K.; Johansson, E.; Ortiz, R.; Hailesilassie, T.; Hammenhag, C.; Hailu, F.; Geleta, M. Marker-trait association analyses revealed major novel QTLs for grain yield and related traits in durum wheat. Front. Plant Sci. 2023, 13, 1009244. [Google Scholar] [CrossRef] [PubMed]
- Tsegaye, S. Estimation of outcrossing rate in landraces of tetraploid wheat (Triticum turgidum L.). Plant Breed. 1996, 115, 195–197. [Google Scholar] [CrossRef]
- Hucl, P.; Matus-Cádiz, M. Isolation distances for minimizing outcrossing in spring wheat. Crop Sci. 2001, 41, 1348–1351. [Google Scholar] [CrossRef]
- Wright, S. Isolation by distance. Genetics 1943, 28, 114–138. [Google Scholar] [CrossRef] [PubMed]
- Wright, S. The genetical structure of populations. Ann. Eugen. 1949, 15, 323–354. [Google Scholar] [CrossRef] [PubMed]
- Wright, S. The interpretation of population structure by F-statistics with special regard to systems of mating. Evolution 1965, 19, 395–420. [Google Scholar] [CrossRef]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Glémin, S.; Bazin, E.; Charlesworth, D. Impact of mating systems on patterns of sequence polymorphism in flowering plants. Proc. R. Soc. B 2006, 273, 3011–3019. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhao, X.; Li, Y.; Xu, J.; Bi, A.; Kang, L.; Xu, D.; Chen, H.; Wang, Y.; Wang, Y.-G.; et al. Triticum population sequencing provides insights into wheat adaptation. Nat. Genet. 2020, 52, 1412–1422. [Google Scholar] [CrossRef] [PubMed]
- Jordan, K.W.; Wang, S.; He, F.; Chao, S.; Lun, Y.; Paux, E.; Sourdille, P.; Sherman, J.; Akhunova, A.; Blake, N.K.; et al. The genetic architecture of genome-wide recombination rate variation in allopolyploid wheat revealed by nested association mapping. Plant J. 2018, 95, 1039–1054. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, C.M.; Emons, A.M.C.; Ketelaar, T. The Plant Exocyst. J. Integr. Plant Biol. 2010, 52, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhang, X.; Wan, W.; Zhang, H.; Liu, J.; Li, M.; Wang, H.; Xiao, J.; Wang, X. Identification and characterization of the EXO70 gene family in polyploid wheat and related species. Int. J. Mol. Sci. 2018, 20, 60. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, P.; Yang, Y.; Chi, Y.; Fan, B.; Chen, Z. Expression and functional analysis of a novel group of legume-specific WRKY and Exo70 protein variants from soybean. Sci. Rep. 2016, 6, 32090. [Google Scholar] [CrossRef] [PubMed]
- Tuteja, N.; Tuteja, R. Prokaryotic and eukaryotic DNA helicases: Essential molecular motor proteins for cellular machinery. Eur. J. Biochem. 2004, 271, 1835–1848. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Jin, X.; Chen, J.; Xu, H.; Zhang, M.; Lu, X.; Wu, R.; Zhao, Y.; Guo, Y.; An, Y.; et al. ATP-dependent DNA helicase (TaDHL), a novel reduced-height (Rht) gene in wheat. Genes 2022, 13, 979. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, J.; Gautam, T.; Gahlaut, V.; Singh, K.; Kumar, S.; Batra, R.; Gupta, P.K. Identification and characterization of RuvBL DNA helicase genes for tolerance against abiotic stresses in bread wheat (Triticum aestivum L.) and related species. Funct. Integr. Genom. 2023, 23, 255. [Google Scholar] [CrossRef] [PubMed]
- Mudgil, Y.; Shiu, S.H.; Stone, S.L.; Salt, J.N.; Goring, D.R. A large complement of the predicted Arabidopsis ARM repeat proteins are members of the U-box E3 ubiquitin ligase family. Plant Physiol. 2004, 134, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Yee, D.; Goring, D.R. The diversity of plant U-box E3 ubiquitin ligases: From upstream activators to downstream target substrates. J. Exp. Bot. 2009, 60, 1109–1121. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, C.; Qin, A.; Deng, W.; Chen, R.; Yu, H.; Wang, Y.; Song, J.; Zeng, L. Genome-wide identification and transcriptome profiling expression analysis of the U-box E3 ubiquitin ligase gene family related to abiotic stress in maize (Zea mays L.). BMC Genom. 2024, 25, 132. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, M.; Gao, Y.; Jia, C.; Pan, X.; Cao, P.; Zhao, X.; Zhang, J.; Chang, W. Structural implications of Dpy30 oligomerization for MLL/SET1 COMPASS H3K4 trimethylation. Protein Cell 2015, 6, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Wang, S.; Jiang, H.; Cheng, B.; Wu, K.; Ding, Y. The COMPASS-like complex promotes flowering and panicle branching in rice. Plant Physiol. 2018, 176, 2761–2771. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Huang, F.; Zhai, H.; Zheng, Y.; Yu, J.; Chen, Z.; Fan, Y.; Zhao, H.; Sun, Q.; Liang, R.; et al. The Tetratricopeptide repeat protein TaTPR-B1 regulates spike compactness in bread wheat. Plant Physiol. 2024, 197, kiae546. [Google Scholar] [CrossRef] [PubMed]
- Toledo-Ortiz, G.; Huq, E.; Quail, P.H. The Arabidopsis basic/helix-loop-helix transcription factor family. Plant Cell 2003, 15, 1749–1770. [Google Scholar] [CrossRef] [PubMed]
- Carretero-Paulet, L.; Galstyan, A.; Roig-Villanova, I.; Martínez-García, J.F.; Bilbao-Castro, J.R. Genome-wide classification and evolutionary analysis of the bHLH family of transcription factors in Arabidopsis, poplar, rice, moss, and algae. Plant Physiol. 2010, 153, 1398–1412. [Google Scholar] [CrossRef] [PubMed]
- Pires, N.; Dolan, L. Origin and diversification of basic-helix-loop-helix proteins in plants. Mol. Biol. Evol. 2010, 27, 862–874. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xiang, L.; Hong, J.; Xie, Z.; Li, B. Genome-wide analysis of bHLH transcription factor family reveals their involvement in biotic and abiotic stress responses in wheat (Triticum aestivum L.). 3 Biotech 2019, 9, 236. [Google Scholar] [CrossRef] [PubMed]
- Ellis, J.; Dodds, P.; Pryor, T. Structure, function and evolution of plant disease resistance genes. Curr. Opin. Plant Biol. 2000, 3, 278–284. [Google Scholar] [CrossRef] [PubMed]
- McHale, L.; Tan, X.; Koehl, P.; Michelmore, R.W. Plant NBS-LRR proteins: Adaptable guards. Genome Biol. 2006, 7, 212. [Google Scholar] [CrossRef] [PubMed]
- Krattinger, S.G.; Lagudah, E.S.; Spielmeyer, W.; Singh, R.P.; Huerta-Espino, J.; McFadden, H.; Bossolini, E.; Selter, L.L.; Keller, B. A putative ABC transporter confers durable resistance to multiple fungal pathogens in wheat. Science 2009, 323, 1360–1363. [Google Scholar] [CrossRef] [PubMed]
- Periyannan, S.; Moore, J.; Ayliffe, M.; Bansal, U.; Wang, X.; Huang, L.; Deal, K.; Luo, M.; Kong, X.; Bariana, H.; et al. The gene Sr33, an ortholog of barley Mla genes, encodes resistance to wheat stem rust race Ug99. Science 2013, 341, 786–788. [Google Scholar] [CrossRef] [PubMed]
- Zielinski, R.E. Calmodulin and calmodulin-binding proteins in plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1998, 49, 697–725. [Google Scholar] [CrossRef] [PubMed]
- Dugasa, M.T.; Feng, X.; Wang, N.H.; Wang, J.; Wu, F. Comparative transcriptome and tolerance mechanism analysis in the two contrasting wheat (Triticum aestivum L.) cultivars in response to drought and salinity stresses. Plant Growth Regul. 2021, 94, 101–114. [Google Scholar] [CrossRef]
- Zeng, H.; Zhu, Q.; Yuan, P.; Yan, Y.; Yi, K.; Du, L. Calmodulin and calmodulin-like protein-mediated plant responses to biotic stresses. Plant Cell Environ. 2023, 46, 3680–3703. [Google Scholar] [CrossRef] [PubMed]
- Santo, T.; Rashkova, M.; Alabaça, C.; Leitão, J. The ENU-induced powdery mildew resistant mutant pea (Pisum sativum L.) lines S (er1mut1) and F (er1mut2) harbour early stop codons in the PsMLO1 gene. Mol. Breed. 2013, 32, 723–727. [Google Scholar] [CrossRef]
- Amiteye, S. Genetic mutations and molecular detection techniques in plant breeding. In Advanced Crop Improvement; Volume 1: Theory and Practice; Springer International Publishing: Cham, Switzerland, 2023; pp. 277–308. [Google Scholar]
- Mazzucotelli, E.; Sciara, G.; Mastrangelo, A.M.; Desiderio, F.; Xu, S.S.; Faris, J.; Hayden, M.J.; Tricker, P.J.; Ozkan, H.; Echenique, V.; et al. The global durum wheat panel (GDP): An international platform to identify and exchange beneficial alleles. Front. Plant Sci. 2020, 11, 569905. [Google Scholar] [CrossRef] [PubMed]
- Sansaloni, C.; Franco, J.; Santos, B.; Percival-Alwyn, L.; Singh, S.; Petroli, C.; Campos, J.; Dreher, K.; Payne, T.; Marshall, D.; et al. Diversity analysis of 80,000 wheat accessions reveals consequences and opportunities of selection footprints. Nat. Commun. 2020, 11, 4572. [Google Scholar] [CrossRef] [PubMed]
- Teklu, Y.; Hammer, K.; Huang, X.Q.; Röder, M.S. Analysis of microsatellite diversity in Ethiopian tetraploid wheat landraces. Genet. Resour. Crop Evol. 2006, 53, 1115–1126. [Google Scholar] [CrossRef]
- Mangini, G.; Gadaleta, A.; Colasuonno, P.; Marcotuli, I.; Signorile, A.M.; Simeone, R.; De Vita, P.; Mastrangelo, A.M.; Laidò, G.; Pecchioni, N.; et al. Genetic dissection of the relationships between grain yield components by genome-wide association mapping in a collection of tetraploid wheats. PLoS ONE 2018, 13, e0190162. [Google Scholar] [CrossRef] [PubMed]
- Frichot, E.; Schoville, S.D.; Bouchard, G.; François, O. Testing for associations between loci and environmental gradients using latent factor mixed models. Mol. Biol. Evol. 2013, 30, 1687–1699. [Google Scholar] [CrossRef] [PubMed]
- Schlötterer, C.; Tobler, R.; Kofler, R.; Nolte, V. Sequencing pools of individuals—Mining genome-wide polymorphism data without big funding. Nat. Rev. Genet. 2014, 15, 749–763. [Google Scholar] [CrossRef] [PubMed]
- Lopes, M.S.; Dreisigacker, S.; Peña, R.J.; Sukumaran, S.; Reynolds, M.P. Genetic characterization of the wheat association mapping initiative (WAMI) panel for dissection of complex traits in spring wheat. Theor. Appl. Genet. 2015, 128, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Soriano, J.M.; Colasuonno, P.; Marcotuli, I.; Gadaleta, A. Meta-QTL analysis and identification of candidate genes for quality, abiotic and biotic stress in durum wheat. Sci. Rep. 2021, 11, 11877. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Pressoir, G.; Briggs, W.H.; Vroh Bi, I.; Yamasaki, M.; Doebley, J.F.; McMullen, M.D.; Gaut, B.S.; Nielsen, D.M.; Holland, J.B.; et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 2006, 38, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.M.; Sul, J.H.; Service, S.K.; Zaitlen, N.A.; Kong, S.Y.; Freimer, N.B.; Sabatti, C.; Etskin, E. Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 2010, 42, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Mengistu, D.K.; Kidane, Y.G.; Fadda, C.; Pè, M.E. Genetic diversity in Ethiopian durum wheat (Triticum turgidum var. durum) inferred from phenotypic variations. Plant Genet. Resour. 2018, 16, 39–49. [Google Scholar] [CrossRef]
- Hadado, T.T.; Rau, D.; Bitocchi, E.; Papa, R. Adaptation and diversity along an altitudinal gradient in Ethiopian barley (Hordeum vulgare L.) landraces revealed by molecular analysis. BMC Plant Biol. 2010, 10, 121. [Google Scholar] [CrossRef] [PubMed]
- Robbana, C.; Kehel, Z.; Ben Naceur, M.B.; Sansaloni, C.; Bassi, F.; Amri, A. Genome-wide genetic diversity and population structure of Tunisian durum wheat landraces based on DArTseq technology. Int. J. Mol. Sci. 2019, 20, 1352. [Google Scholar] [CrossRef] [PubMed]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- Nei, M. Molecular Evolutionary Genetics; Columbia University Press: New York, NY, USA, 1987. [Google Scholar]
- Pfeifer, B.; Wittelsbu, U.; Ramos-onsins, S.E.; Lercher, M.J. PopGenome: An Efficient Swiss Army Knife for Population Genomic Analyses in R. Mol. Biol. Evol. 2014, 31, 1929–1936. [Google Scholar] [CrossRef] [PubMed]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Muse, S.V. PowerMarker: An integrated analysis environment for genetic marker analysis. Bioinformatics 2005, 21, 2128–2129. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A.; Mayrose, I. Clumpak: A program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Resour. 2015, 15, 1179–1191. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
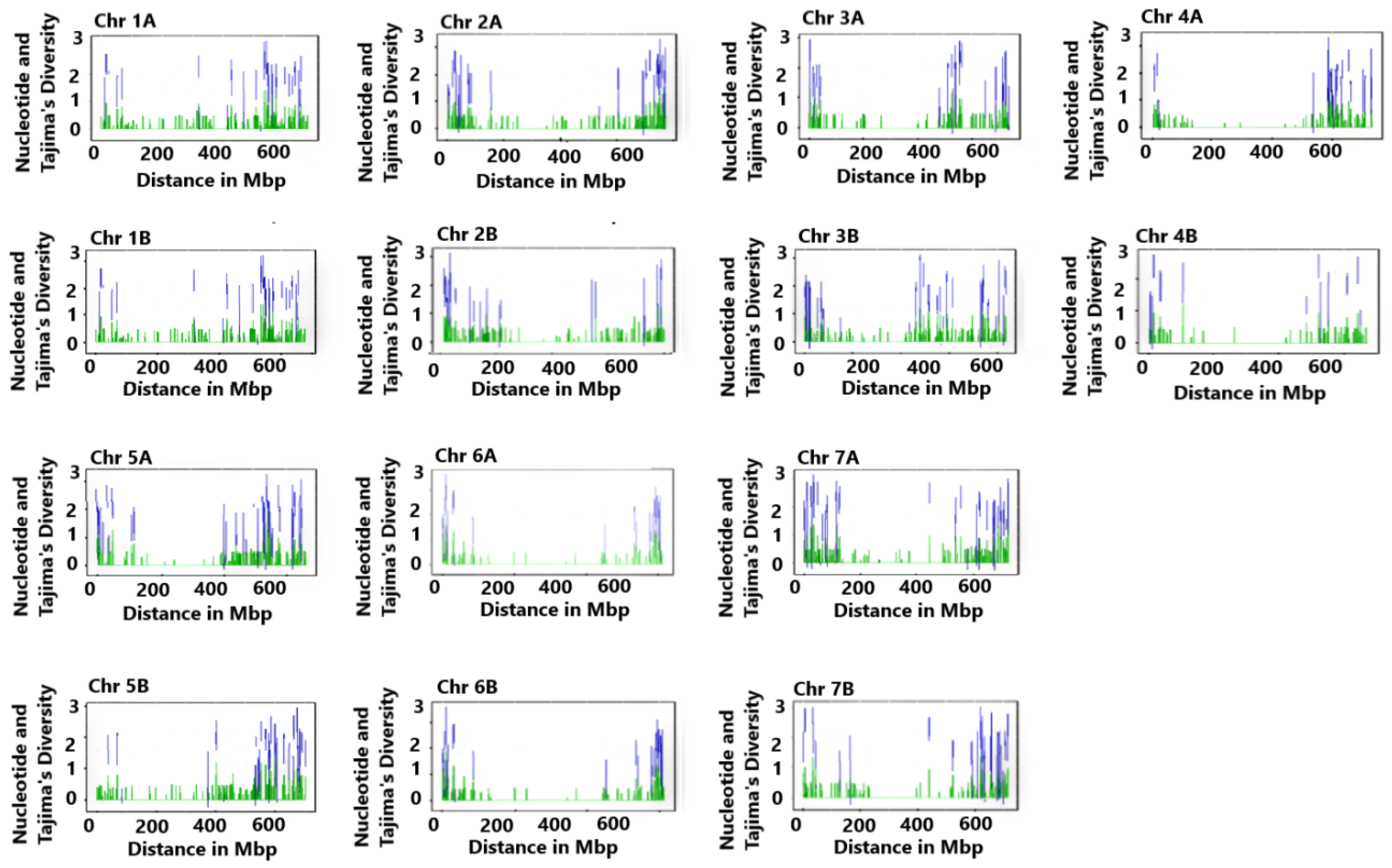
| Chr | NM | GCR (bp) | CGRS (Mbp) | ND | TD (p > 0.10) |
|---|---|---|---|---|---|
| 1A | 487 | 1,159,612–584,771,671 | 583.612 | 0.209 | 1.499 |
| 2A | 524 | 295,475–774,814,125 | 774.519 | 0.211 | 1.530 |
| 3A | 453 | 304,055–746,465,146 | 746.161 | 0.200 | 1.295 |
| 4A | 345 | 698,412–735,809,633 | 735.111 | 0.197 | 1.230 |
| 5A | 539 | 27,537–667,289,264 | 667.262 | 0.211 | 1.540 |
| 6A | 381 | 770,173–615,260,837 | 614.491 | 0.204 | 1.391 |
| 7A | 593 | 173,256–727,310,461 | 727.137 | 0.205 | 1.417 |
| 1B | 537 | 313,555–681,099,620 | 680.786 | 0.203 | 1.368 |
| 2B | 575 | 406,084–789,416,853 | 789.376 | 0.194 | 1.164 |
| 3B | 582 | 306,806–836,443,340 | 836.137 | 0.196 | 1.225 |
| 4B | 270 | 1,400,884–675,805,446 | 674.405 | 0.221 | 1.724 |
| 5B | 563 | 2,555,603–701,346,725 | 698.760 | 0.196 | 1.217 |
| 6B | 477 | 2,064,505–698,590,527 | 696.526 | 0.210 | 1.507 |
| 7B | 418 | 113,839–719,907,662 | 719.794 | 0.206 | 1.425 |
| A genome | 3322 a | - | 4176.5 a | 0.21 b | 1.41 b |
| B genome | 3422 a | - | 5095.8 a | 0.20 b | 1.38 b |
| Whole genome | 6744 a | - | 9272.3 a | 0.20 b | 1.40 b |
| Marker | Chr | SNP Position | Obs. Het. | Obs FST | FST p-Value | SNP | Mutation Type | Impact | AA Change | Codons | SIFT Score | Gene | Gene Description |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AX-108886825 | 2A | 749161581 | 0.33 | 0.95 | 0.00245 | A/T | Stop-lost | High | */L | tAa/tTa | - | TRITD2Av1G282370a | Exocyst complex component, putative |
| AX-158531685 | 6A | 99013566 | 0.31 | 0.95 | 0.00419 | A/G | missense | Moderate | Q/R | cAg/cGg | 0.01 | TRITD6Av1G042660a | Leucine-rich repeat receptor-like protein kinase family protein |
| AX-158543425 | 6B | 694134398 | 0.3 | 0.98 | 0.00004 | A/C | missense | Moderate | K/Q | Aaa/Caa | 0.03 | TRITD6Bv1G227240a | Seed maturation-like protein |
| AX-158544944 | 1B | 616467524 | 0.39 | 0.68 | 0.00591 | G/T | missense | Moderate | G/W | Ggg/Tgg | 0 | TRITD1Bv1G201940a | ABC transporter B family protein |
| AX-158554628 | 7A | 713342696 | 0.33 | 0.95 | 0.00245 | A/C | Stop-lost | High | */S | tAg/tCg | - | TRITD7Av1G274730a | DNA helicase |
| AX-94416225 | 6B | 539466257 | 0.32 | 0.98 | 0.00002 | C/T | missense | Moderate | R/K | aGa/aAa | 0.02 | TRITD6Bv1G168530b | Enhancer of mRNA-decapping protein 4 |
| AX-94439358 | 3A | 200789462 | 0.32 | 0.98 | 0.00002 | C/T | missense | Moderate | T/M | aCg/aTg | 0.04 | TRITD3Av1G082030a | Epoxide hydrolase 2 |
| AX-94458766 | 3A | 481186192 | 0.32 | 0.98 | 0.00002 | A/G | missense | Moderate | M/T | aTg/aCg | 0 | TRITD3Av1G171840b | SWAP (Suppressor-of-White-APricot)/surp domain-containing protein |
| AX-94463985 | 7B | 578955005 | 0.38 | 0.94 | 0.00718 | C/T | missense | Moderate | S/N | aGc/aAc | 0.04 | TRITD7Bv1G185250b | Glycosyltransferases |
| AX-94603856 | 1B | 600672886 | 0.18 | 0.63 | 0.00964 | G/T | missense | Moderate | S/I | aGc/aTc | 0.01 | TRITD1Bv1G196100a | 60 kDa chaperonin |
| AX-94639471 | 2A | 51194356 | 0.36 | 0.68 | 0.00952 | G/T | missense | Moderate | G/V | gGc/gTc | 0 | TRITD2Av1G025670a | CAP-gly domain linker G |
| AX-94646444 | 4B | 633082630 | 0.31 | 0.95 | 0.00419 | C/T | Stop-gained | High | Q/* | Cag/Tag | - | TRITD4Bv1G190600a | Tetratricopeptide repeat protein 7A |
| AX-94969179 | 1A | 537772342 | 0.35 | 0.67 | 0.00794 | C/T | missense | Moderate | P/L | cCc/cTc | 0 | TRITD1Av1G206330a | Pentatricopeptide repeat-containing protein |
| AX-95006148 | 2B | 611276157 | 0.32 | 0.98 | 0.00002 | C/T | Stop-gained | High | Q/* | Cag/Tag | - | TRITD2Bv1G204550a | Basic Helix-Loop-Helix (bHLH) DNA-binding superfamily protein G |
| AX-95073999 | 2B | 674106642 | 0.32 | 0.98 | 0.00002 | C/T | Stop-gained | High | Q/* | Caa/Taa | - | TRITD2Bv1G223490a | NBS-LRR disease resistance protein-like protein |
| BS00009789_51 | 5B | 410632320 | 0.32 | 0.98 | 0.00002 | G/T | missense | Moderate | P/Q | cCg/cAg | 0 | TRITD5Bv1G137170b | Processing peptidase |
| BS00046963_51 | 6B | 145924311 | 0.34 | 0.98 | 0.00005 | A/C | Stop-lost | High | */E | Tag/Gag | - | TRITD6Bv1G052050b | Plant calmodulin-binding protein-like protein |
| CAP8_c2210_103 | 6B | 679394894 | 0.2 | 0.58 | 0.00004 | C/T | missense | Moderate | V/I | Gtc/Atc | 0.02 | TRITD6Bv1G221320b | DNL-type zinc finger protein |
| Excalibur_rep_c111629_239 | 7B | 538297853 | 0.25 | 0.64 | 0.00217 | A/C | missense | Moderate | K/N | aaA/aaC | 0.03 | TRITD7Bv1G170020a | ATP-citrate synthase, putative |
| Ra_c56305_1946 | 7B | 168560355 | 0.3 | 0.98 | 0.00004 | C/T | Stop-gained | High | Q/* | Caa/Taa | - | TRITD7Bv1G059650a | U-box domain-containing family protein |
| RAC875_c65710_156 | 6B | 679642547 | 0.23 | 0.61 | 0.0011 | C/T | missense | Moderate | P/L | cCg/cTg | 0.02 | TRITD6Bv1G221530a | n/a |
| Tdurum_contig15512_429 | 2B | 138766219 | 0.28 | 0.98 | 0.00032 | A/G | missense | Moderate | V/A | gTa/gCa | 0.03 | TRITD2Bv1G053850b | Dihydrolipoamide acetyltransferase component of pyruvate dehydrogenase complex |
| Tdurum_contig97611_150 | 6A | 6813755 | 0.29 | 0.96 | 0.00401 | A/G | missense | Moderate | V/A | gTg/gCg | 0 | TRITD6Av1G002940b | Glycosyltransferase |
| wsnp_Ex_c12818_20334501 | 4A | 101554190 | 0.32 | 0.98 | 0.00002 | G/T | missense | Moderate | P/Q | cCa/cAa | 0 | TRITD4Av1G042900b | Serine/arginine repetitive matrix protein 1 G |
| wsnp_Ex_c55245_57821568 | 4A | 40245812 | 0.29 | 0.63 | 0.00018 | A/C | Stop-gained | High | L/* | tTa/tGa | - | TRITD4Av1G018270b | Retinoblastoma-binding protein 5 |
| wsnp_Ku_c3081_5776947 | 4A | 588463325 | 0.32 | 0.98 | 0.00002 | C/T | missense | Moderate | R/C | Cgc/Tgc | 0.01 | TRITD4Av1G201710a | DWNN domain, A CCHC-type zinc finger protein |
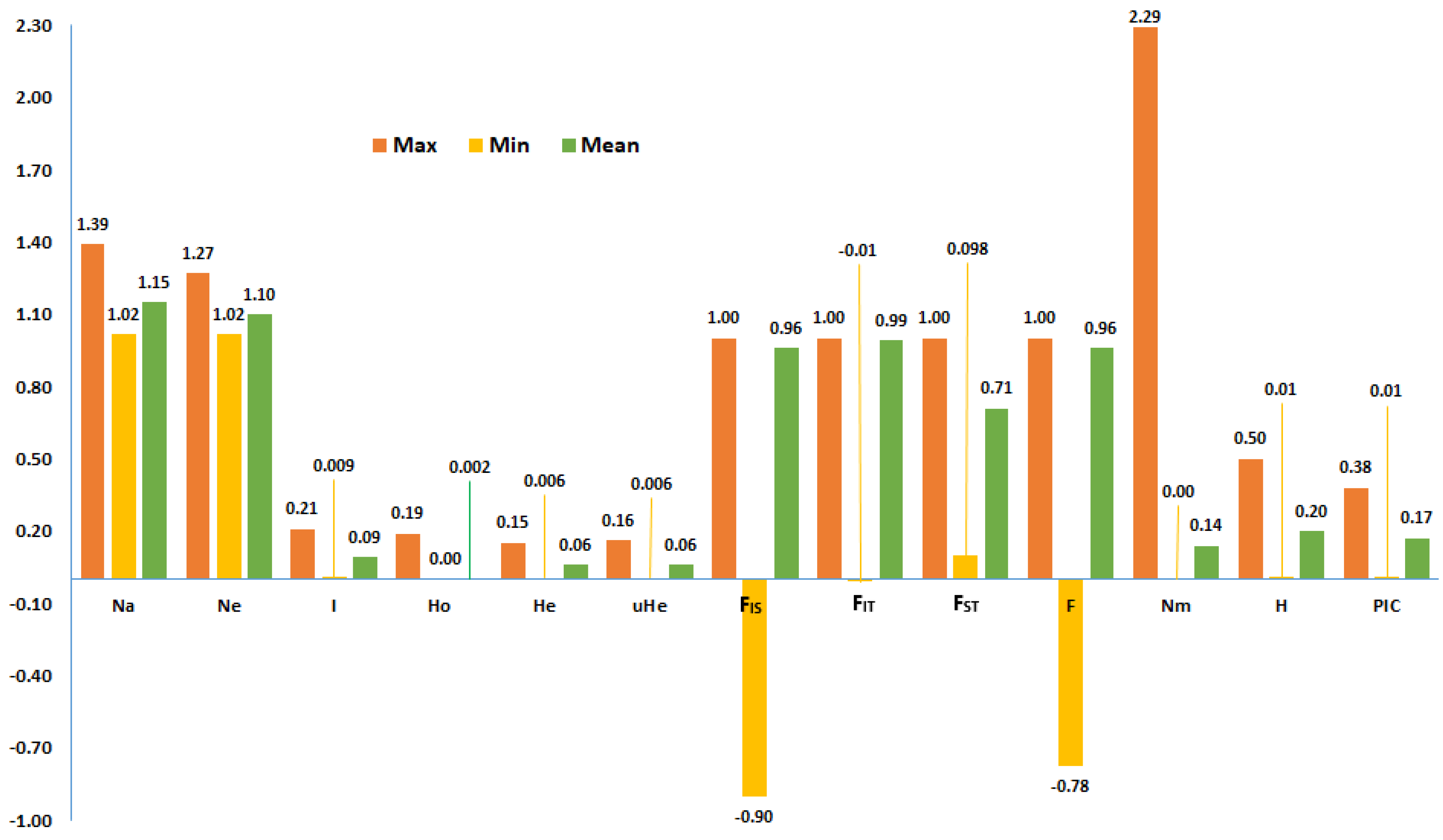
| Accession | Na | Ne | I | Ho | He | uHe | F | %PL | NLPA | %LPA | MFPA | ND | TD |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 31132 | 1.00 | 1.00 | 0.00 | 0.00 | 0.00 | 0.00 | −1.00 | 0.01 | 10 | 0.13 | 1.00 | 0.00 | 1.46 |
| 31137 | 1.00 | 1.00 | 0.00 | 0.00 | 0.00 | 0.00 | −0.75 | 0.04 | 0 | 0.00 | na | 0.00 | 1.38 |
| 31141 | 1.00 | 1.00 | 0.00 | 0.00 | 0.00 | 0.00 | −1.00 | 0.03 | 0 | 0.00 | na | 0.00 | 1.84 |
| 31146 | 1.11 | 1.10 | 0.07 | 0.00 | 0.05 | 0.06 | 1.00 | 10.65 | 1 | 0.01 | 0.40 | 0.06 | 2.55 ** |
| 31151 | 1.00 | 1.00 | 0.00 | 0.00 | 0.00 | 0.00 | −1.00 | 0.01 | 0 | 0.00 | na | 0.00 | 1.46 |
| 31158 | 1.00 | 1.00 | 0.00 | 0.00 | 0.00 | 0.00 | na | 0.00 | 0 | 0.00 | na | 0.00 | 0.00 |
| 31170 | 1.00 | 1.00 | 0.00 | 0.00 | 0.00 | 0.00 | −1.00 | 0.01 | 2 | 0.03 | 1.00 | 0.00 | 1.46 |
| 31176 | 1.17 | 1.12 | 0.10 | 0.00 | 0.07 | 0.07 | 1.00 | 16.74 | 0 | 0.00 | na | 0.07 | 1.24 |
| 31209 | 1.02 | 1.01 | 0.01 | 0.00 | 0.01 | 0.01 | 0.97 | 2.38 | 2 | 0.03 | 0.80 | 0.01 | −0.003 |
| 31215 | 1.17 | 1.12 | 0.10 | 0.00 | 0.07 | 0.07 | 0.99 | 16.79 | 11 | 0.14 | 0.38 | 0.07 | 1.24 |
| 31220 | 1.00 | 1.00 | 0.00 | 0.00 | 0.00 | 0.00 | na | 0.00 | 0 | 0.00 | na | 0.00 | 0.00 |
| 31230 | 1.00 | 1.00 | 0.00 | 0.00 | 0.00 | 0.00 | 1.00 | 0.01 | 0 | 0.00 | na | 0.00 | 1.30 |
| 31237 | 1.00 | 1.00 | 0.00 | 0.00 | 0.00 | 0.00 | −0.56 | 0.03 | 4 | 0.05 | 1.00 | 0.00 | 0.22 |
| 31239 | 1.02 | 1.01 | 0.01 | 0.00 | 0.01 | 0.01 | 0.98 | 1.63 | 1 | 0.01 | 0.20 | 0.01 | 0.03 |
| 31246 | 1.10 | 1.05 | 0.05 | 0.00 | 0.03 | 0.04 | 0.99 | 10.18 | 1 | 0.01 | 0.80 | 0.04 | 0.04 |
| 31248 | 1.49 | 1.42 | 0.32 | 0.03 | 0.22 | 0.25 | 0.83 | 49.07 | 1685 | 21.49 | 0.40 | 0.25 | 2.21 * |
| 31250 | 1.00 | 1.00 | 0.00 | 0.00 | 0.00 | 0.00 | −1.00 | 0.01 | 4 | 0.05 | 1.00 | 0.00 | 1.46 |
| 31252 | 1.09 | 1.09 | 0.06 | 0.00 | 0.04 | 0.05 | 1.00 | 9.23 | 0 | 0.00 | na | 0.05 | 2.55 ** |
| 31260 | 1.13 | 1.06 | 0.06 | 0.00 | 0.04 | 0.05 | 1.00 | 12.90 | 0 | 0.00 | na | 0.05 | 0.03 |
| 31266 | 1.00 | 1.00 | 0.00 | 0.00 | 0.00 | 0.00 | −1.00 | 0.03 | 1 | 0.01 | 1.00 | 0.00 | 1.84 |
| 31269 | 1.26 | 1.12 | 0.13 | 0.00 | 0.08 | 0.09 | 1.00 | 26.43 | 17 | 0.22 | 0.20 | 0.09 | 0.03 |
| 31292 | 1.01 | 1.01 | 0.00 | 0.00 | 0.00 | 0.00 | 0.84 | 0.78 | 0 | 0.00 | na | 0.00 | 1.07 |
| 31299 | 1.00 | 1.00 | 0.00 | 0.00 | 0.00 | 0.00 | −0.70 | 0.04 | 3 | 0.04 | 1.00 | 0.00 | 0.84 |
| 31326 | 1.12 | 1.06 | 0.06 | 0.00 | 0.04 | 0.04 | 0.99 | 11.91 | 9 | 0.11 | 0.80 | 0.04 | 0.06 |
| 31334 | 1.07 | 1.03 | 0.04 | 0.00 | 0.02 | 0.03 | 0.99 | 7.24 | 0 | 0.00 | na | 0.03 | 0.04 |
| 31356 | 1.00 | 1.00 | 0.00 | 0.00 | 0.00 | 0.00 | na | 0.00 | 0 | 0.00 | na | 0.00 | 0.00 |
| 31358 | 1.16 | 1.14 | 0.11 | 0.00 | 0.08 | 0.08 | 1.00 | 16.48 | 6 | 0.08 | 0.37 | 0.08 | 2.27 ** |
| 31361 | 1.00 | 1.00 | 0.00 | 0.00 | 0.00 | 0.00 | na | 0.00 | 0 | 0.00 | na | 0.00 | 0.00 |
| 31368 | 1.10 | 1.05 | 0.05 | 0.00 | 0.03 | 0.03 | 0.98 | 9.81 | 0 | 0.00 | na | 0.03 | 0.02 |
| 31372 | 1.13 | 1.06 | 0.07 | 0.00 | 0.04 | 0.05 | 1.00 | 13.45 | 0 | 0.00 | na | 0.05 | 0.02 |
| 31609 | 1.11 | 1.06 | 0.06 | 0.00 | 0.04 | 0.04 | 1.00 | 10.74 | 1 | 0.01 | 0.20 | 0.04 | 0.43 |
| 31696 | 1.00 | 1.00 | 0.00 | 0.00 | 0.00 | 0.00 | −1.00 | 0.05 | 0 | 0.00 | na | 0.00 | 2.19 * |
| 31979 | 1.20 | 1.13 | 0.11 | 0.00 | 0.08 | 0.09 | 1.00 | 19.89 | 4 | 0.05 | 0.33 | 0.09 | 1.05 |
| 33235 | 1.00 | 1.00 | 0.00 | 0.00 | 0.00 | 0.00 | −0.11 | 0.01 | 0 | 0.00 | na | 0.00 | −1.11 |
| 33239 | 1.00 | 1.00 | 0.00 | 0.00 | 0.00 | 0.00 | −0.33 | 0.05 | 7 | 0.09 | 1.00 | 0.00 | −0.7 |
| 33244 | 1.20 | 1.12 | 0.11 | 0.00 | 0.07 | 0.08 | 0.99 | 20.42 | 48 | 0.61 | 0.44 | 0.08 | 0.68 |
| 33283 | 1.19 | 1.13 | 0.11 | 0.00 | 0.08 | 0.08 | 0.90 | 19.15 | 8 | 0.10 | 0.35 | 0.08 | 1.24 |
| 33286 | 1.16 | 1.15 | 0.11 | 0.00 | 0.08 | 0.09 | 1.00 | 15.95 | 1 | 0.01 | 0.40 | 0.09 | 2.55 ** |
| 33296 | 1.00 | 1.00 | 0.00 | 0.00 | 0.00 | 0.00 | na | 0.00 | 35 | 0.45 | 1.00 | 0.00 | 0.00 |
| 33496 | 1.10 | 1.08 | 0.06 | 0.00 | 0.04 | 0.05 | 1.00 | 9.68 | 1 | 0.01 | 0.40 | 0.05 | 2.19 * |
| 33511 | 1.15 | 1.07 | 0.07 | 0.00 | 0.05 | 0.05 | 0.99 | 14.61 | 4 | 0.05 | 0.80 | 0.05 | 0.02 |
| 33517 | 1.00 | 1.00 | 0.00 | 0.00 | 0.00 | 0.00 | −1.00 | 0.08 | 17 | 0.22 | 1.00 | 0.00 | 2.36 ** |
| 33523 | 1.04 | 1.01 | 0.01 | 0.01 | 0.01 | 0.01 | −0.11 | 4.13 | 1 | 0.01 | 1.00 | 0.01 | −2.12 *** |
| 33555 | 1.13 | 1.09 | 0.08 | 0.00 | 0.05 | 0.06 | 0.88 | 13.06 | 4 | 0.05 | 0.35 | 0.06 | 1.40 |
| 33681 | 1.16 | 1.11 | 0.09 | 0.00 | 0.06 | 0.07 | 1.00 | 15.67 | 7 | 0.09 | 0.23 | 0.07 | 1.28 |
| 33761 | 1.15 | 1.11 | 0.09 | 0.00 | 0.06 | 0.07 | 1.00 | 15.44 | 7 | 0.09 | 0.20 | 0.07 | 1.21 |
| 33840 | 1.02 | 1.01 | 0.01 | 0.00 | 0.01 | 0.01 | 0.99 | 1.99 | 0 | 0.00 | na | 0.01 | 0.34 |
| Bakalcha | 1.24 | 1.16 | 0.14 | 0.00 | 0.09 | 0.10 | 0.96 | 24.09 | 45 | 0.57 | 0.43 | 0.10 | 1.12 |
| Denbi | 1.05 | 1.04 | 0.03 | 0.00 | 0.02 | 0.02 | 0.97 | 4.60 | 0 | 0.00 | na | 0.02 | 1.95 |
| Ginchi | 1.14 | 1.06 | 0.07 | 0.00 | 0.04 | 0.05 | 0.99 | 13.50 | 1 | 0.01 | 0.20 | 0.05 | 0.03 |
| Leliso | 1.15 | 1.07 | 0.08 | 0.00 | 0.05 | 0.05 | 0.99 | 15.00 | 2 | 0.03 | 0.40 | 0.05 | 0.08 |
| PON19CD_162 | 1.10 | 1.05 | 0.05 | 0.00 | 0.03 | 0.04 | 0.98 | 10.46 | 7 | 0.09 | 0.57 | 0.04 | 0.05 |
| PON19CD_251 | 1.00 | 1.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.14 | 0.09 | 6 | 0.08 | 0.87 | 0.00 | 1.05 |
| PON19CD_262 | 1.01 | 1.01 | 0.01 | 0.00 | 0.00 | 0.00 | 0.83 | 1.15 | 38 | 0.48 | 0.99 | 0.00 | 0.68 |
| PON19CD_270 | 1.01 | 1.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.79 | 0.51 | 3 | 0.04 | 1.00 | 0.00 | 1.03 |
| PON19CD_276 | 1.01 | 1.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.63 | 0.69 | 25 | 0.32 | 1.00 | 0.00 | 0.11 |
| PON19CD_311 | 1.01 | 1.01 | 0.01 | 0.00 | 0.01 | 0.01 | 0.92 | 1.36 | 22 | 0.28 | 0.97 | 0.01 | 1.22 |
| Mean | 1.08 | 1.05 | 0.04 | 0.00 | 0.03 | 0.03 | 0.48 | 7.86 | 35.98 | 0.46 | 0.64 | 0.03 | 0.82 |
| Accession Type | Na | Ne | I | Ho | He | uHe | F | %PL | NLPA | %LPA | MFPA | ND | TD |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Amhara | 1.08 | 1.05 | 0.05 | 0.000 | 0.030 | 0.034 | 0.60 | 7.9 | 2 | 0.02 | 0.62 | 0.03 | 1.06 |
| Oromia | 1.08 | 1.05 | 0.05 | 0.000 | 0.031 | 0.035 | 0.45 | 8.0 | 7 | 0.09 | 0.56 | 0.03 | 1.09 |
| Tigray | 1.07 | 1.05 | 0.05 | 0.000 | 0.030 | 0.033 | 0.33 | 7.5 | 2 | 0.02 | 0.37 | 0.03 | 0.94 |
| Below 2000 masl | 1.06 | 1.04 | 0.04 | 0.000 | 0.022 | 0.026 | 0.97 | 6 | 3 | 0.03 | 0.58 | 0.03 | 0.56 |
| 2000–2500 masl | 1.09 | 1.07 | 0.06 | 0.000 | 0.040 | 0.044 | 0.65 | 10 | 12 | 0.15 | 0.50 | 0.04 | 1.27 |
| Above 2500 masl | 1.08 | 1.05 | 0.04 | 0.000 | 0.030 | 0.033 | 0.32 | 7 | 4 | 0.05 | 0.56 | 0.03 | 1.14 |
| Breeding population | 1.02 | 1.01 | 0.01 | 0.000 | 0.007 | 0.008 | 0.72 | 2.4 | 17 | 0.22 | 0.90 | 0.01 | 0.69 |
| Improved cultivar | 1.15 | 1.08 | 0.08 | 0.000 | 0.050 | 0.055 | 0.98 | 14.3 | 12 | 0.15 | 0.34 | 0.06 | 0.80 |
| Landrace | 1.08 | 1.05 | 0.05 | 0.001 | 0.031 | 0.035 | 0.40 | 8.0 | 40 | 0.52 | 0.62 | 0.03 | 0.84 |
| Dense | 1.07 | 1.04 | 0.04 | 0.001 | 0.024 | 0.027 | 0.14 | 6.5 | 3 | 0.03 | 0.57 | 0.03 | 0.88 |
| lax | 1.07 | 1.05 | 0.04 | 0.000 | 0.028 | 0.031 | 0.55 | 7.1 | 3 | 0.04 | 0.66 | 0.03 | 0.52 |
| very Dense | 1.09 | 1.06 | 0.05 | 0.001 | 0.034 | 0.038 | 0.67 | 9.0 | 66 | 0.84 | 0.67 | 0.04 | 0.64 |
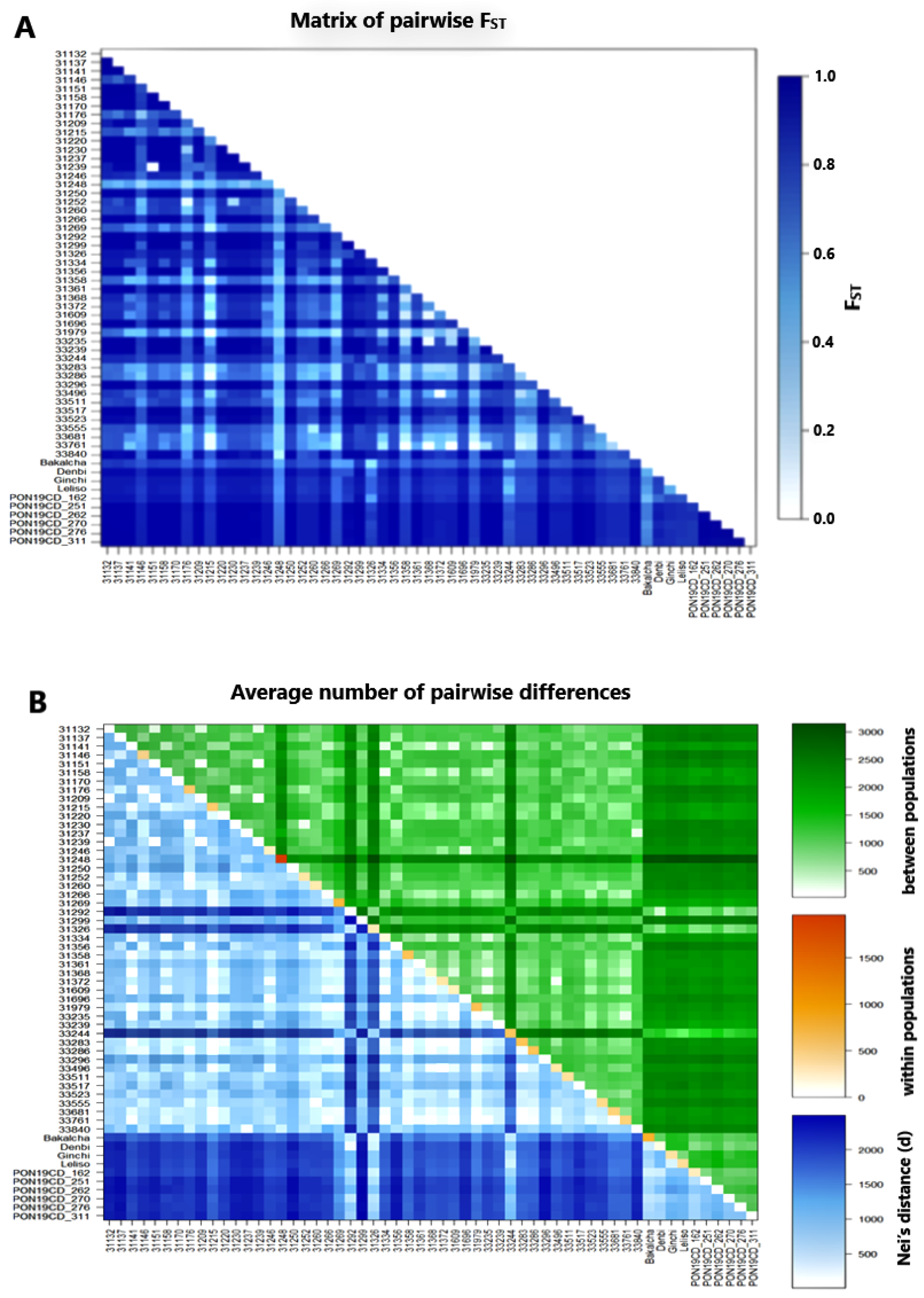
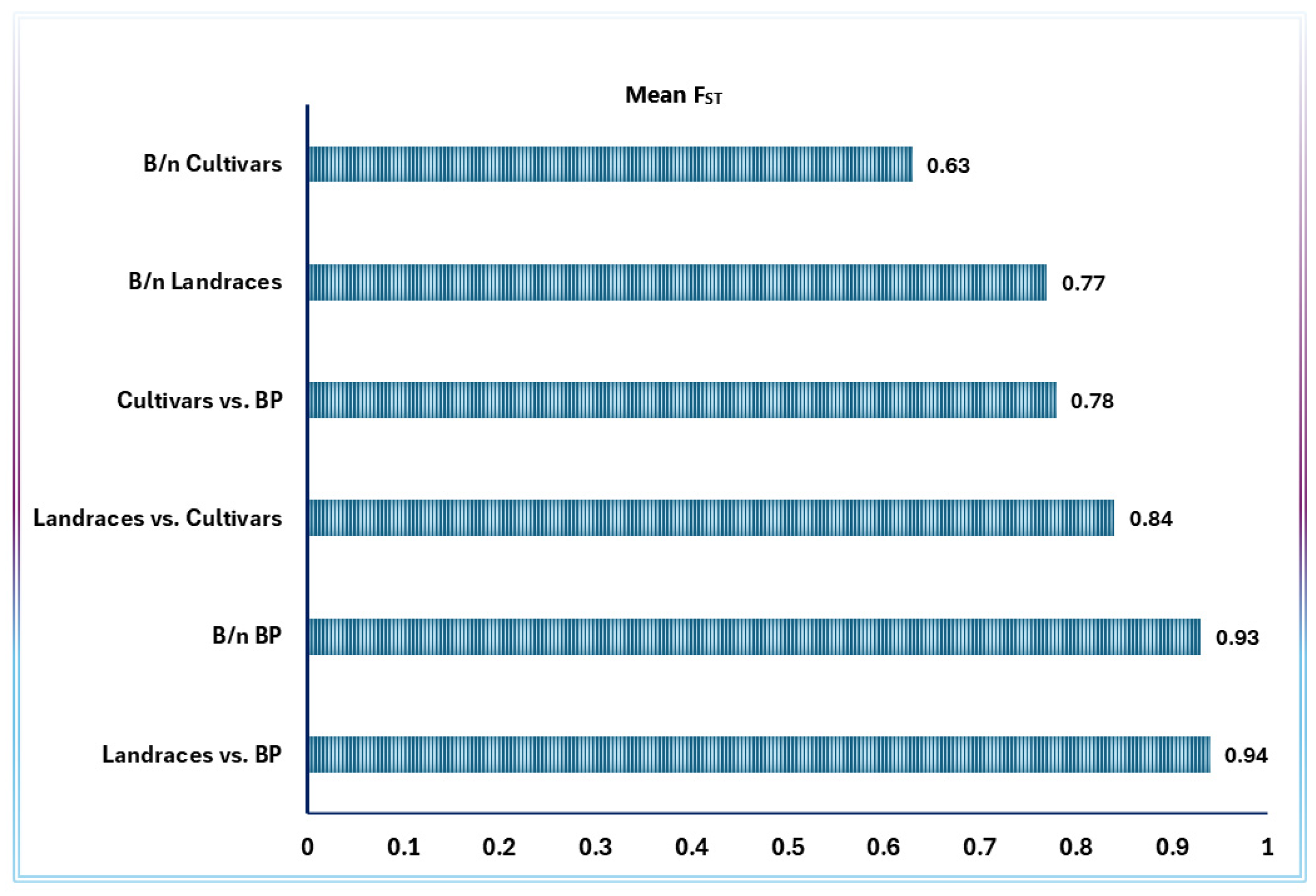
| Source of Variation | DF | Sum of Squares | Variance Component | %Age of Variation | Fixation Index | p-Value |
|---|---|---|---|---|---|---|
| Among accessions | 56.0 | 348,300.4 | 592.96Va | 80.12 | FST = 0.80 | Va and FST < 0.001 |
| AIWA | 228.0 | 66,133.2 | 142.95Vb | 19.32 | FIS = 0.97 | Vb and FIS < 0.001 |
| Within individuals | 285.0 | 1,182.5 | 4.15 Vc | 0.56 | FIT = 0.99 | Vc and FIT < 0.001 |
| Total | 569.0 | 415,616.1 | 740.063 | |||
| Among groups a | 2.0 | 6,691.9 | −30.83 Va | −4.86 | FST = 0.79 | Vc and FST < 0.001 |
| AAWG | 19 | 103,781.4 | 533.04 Vb | 84.09 | FSC = 0.80 | Vb and FSC < 0.001 |
| within accessions | 198 | 26,080.9 | 131.72 Vc | 20.78 | FCT = −0.05 | Va and FCT = 0.930 |
| Total | 219 | 136,554.2 | 633.93 | |||
| Among groups b | 2.0 | 6,621.78 | −21.8 Va | −3.80 | FST = 0.77 | Vc and FST < 0.001 |
| AAWG | 23 | 109,557.7 | 462.93 Vb | 80.49 | FSC = 0.78 | Vb and FSC < 0.001 |
| within accessions | 234 | 31,364.8 | 134.04 Vc | 23.31 | FCT = −0.038 | Va and FCT = 0.930 |
| Total | 259.0 | 147,544.3 | 575.14 | |||
| Among groups c | 2.0 | 90,639.9 | 467.87 Va | 44.0 | FST = 0.88 | Vc and FST < 0.001 |
| AAWG | 54 | 257,660.4 | 464.03 Vb | 43.65 | FSC = 0.78 | Vb and FSC < 0.001 |
| within accessions | 513 | 67,315.7 | 131.22 Vc | 12.34 | FCT = 0.44 | Va and FCT < 0.001 |
| Total | 569.0 | 415,616,07 | 1063.11 | |||
| Among groups d | 2.0 | 15,599.9 | 9.71 Va | 1.31 | FST = 0.82 | Vc and FST < 0.001 |
| AAWG | 54 | 332,700.51 | 602.99 Vb | 81.06 | FSC = 0.82 | Vb and FSC < 0.001 |
| within accessions | 513 | 67,315.7 | 131.22 Vc | 17.64 | FCT = 0.01 | Va and FCT = 0.192 |
| Total | 569.0 | 415,616,07 | 743.92 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ararsa, L.; Mulugeta, B.; Bekele, E.; Geleta, N.; Abreha, K.B.; Geleta, M. A 25K Wheat SNP Array Revealed the Genetic Diversity and Population Structure of Durum Wheat (Triticum turgidum subsp. durum) Landraces and Cultivars. Int. J. Mol. Sci. 2025, 26, 7220. https://doi.org/10.3390/ijms26157220
Ararsa L, Mulugeta B, Bekele E, Geleta N, Abreha KB, Geleta M. A 25K Wheat SNP Array Revealed the Genetic Diversity and Population Structure of Durum Wheat (Triticum turgidum subsp. durum) Landraces and Cultivars. International Journal of Molecular Sciences. 2025; 26(15):7220. https://doi.org/10.3390/ijms26157220
Chicago/Turabian StyleArarsa, Lalise, Behailu Mulugeta, Endashaw Bekele, Negash Geleta, Kibrom B. Abreha, and Mulatu Geleta. 2025. "A 25K Wheat SNP Array Revealed the Genetic Diversity and Population Structure of Durum Wheat (Triticum turgidum subsp. durum) Landraces and Cultivars" International Journal of Molecular Sciences 26, no. 15: 7220. https://doi.org/10.3390/ijms26157220
APA StyleArarsa, L., Mulugeta, B., Bekele, E., Geleta, N., Abreha, K. B., & Geleta, M. (2025). A 25K Wheat SNP Array Revealed the Genetic Diversity and Population Structure of Durum Wheat (Triticum turgidum subsp. durum) Landraces and Cultivars. International Journal of Molecular Sciences, 26(15), 7220. https://doi.org/10.3390/ijms26157220