
Comprehensive Analysis of Human Colorectal Cancers Harboring Polymerase Epsilon Mutations

 , ,
, ,
Abstract
1. Introduction
2. Results and Discussion
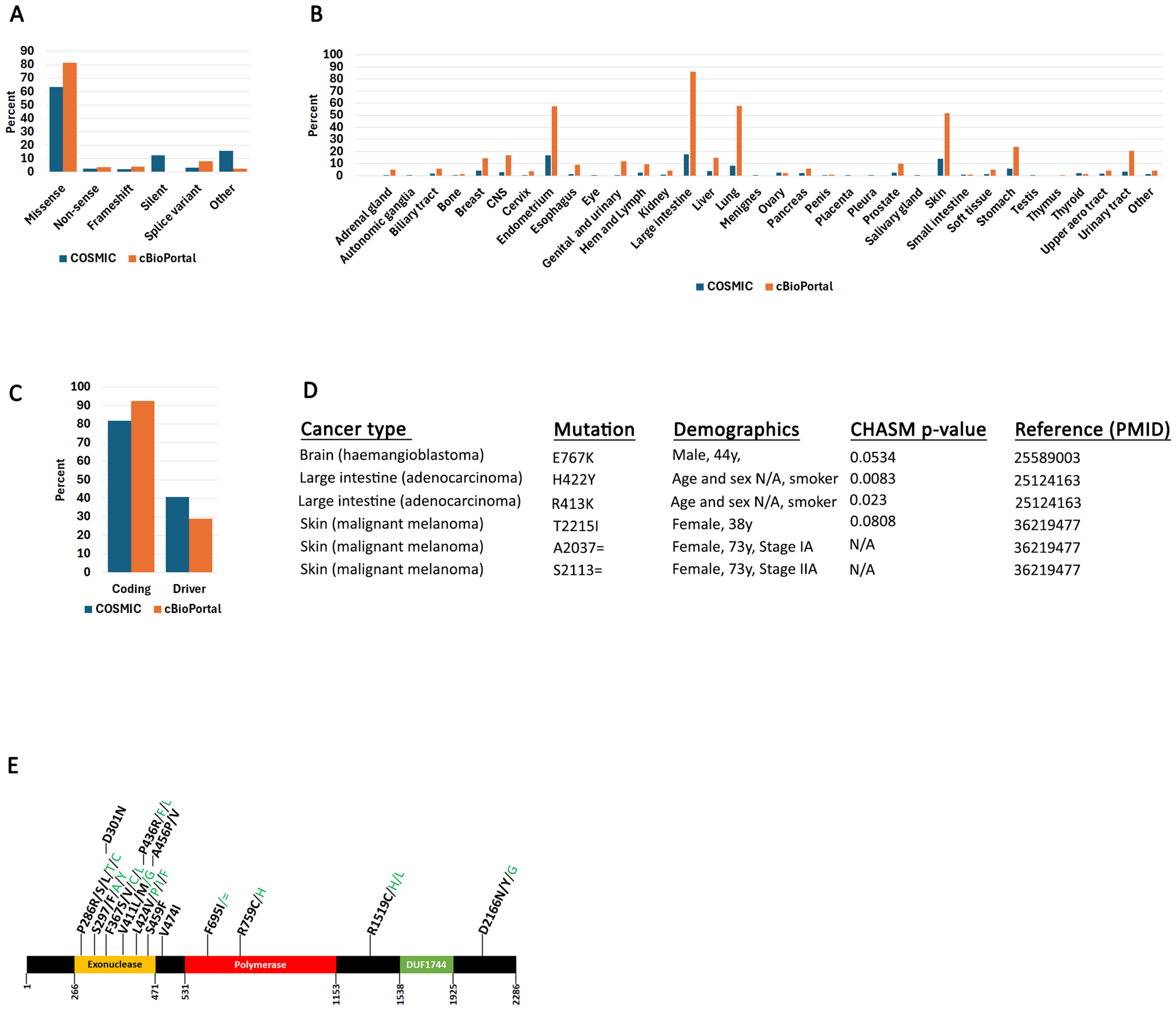
2.1. POLe Mutation Spectrum in Human Cancers
2.2. POLe-Associated Mutations in Colorectal Cancers
2.3. Colorectal Cancer Differential Gene Expression Patterns in POLe Mutant Samples
2.4. Evidence of POLe-Specific Mutation Signatures from In Vivo Mutagenic Screens and Experiments
3. Materials and Methods
3.1. Data Accession and Processing
3.2. Determination of Mutation Driver Potential
3.3. TCGA Expression
3.4. Pan-Cancer Analysis of POLe Mutations
3.5. Protein Structure Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.F.; Ibrahim, A.E.; Arends, M.J. Molecular pathological classification of colorectal cancer. Virchows Arch. 2016, 469, 125–134. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef]
- Pursell, Z.F.; Isoz, I.; Lundstrom, E.B.; Johansson, E.; Kunkel, T.A. Yeast DNA polymerase epsilon participates in leading-strand DNA replication. Science 2007, 317, 127–130. [Google Scholar] [CrossRef]
- Barbari, S.R.; Shcherbakova, P.V. Replicative DNA polymerase defects in human cancers: Consequences, mechanisms, and implications for therapy. DNA Repair 2017, 56, 16–25. [Google Scholar] [CrossRef]
- Palles, C.; Cazier, J.B.; Howarth, K.M.; Domingo, E.; Jones, A.M.; Broderick, P.; Kemp, Z.; Spain, S.L.; Guarino, E.; Salguero, I.; et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat. Genet. 2013, 45, 136–144. [Google Scholar] [CrossRef] [PubMed]
- ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef]
- Pon, J.R.; Marra, M.A. Driver and passenger mutations in cancer. Annu. Rev. Pathol. 2015, 10, 25–50. [Google Scholar] [CrossRef]
- McFarland, C.D.; Korolev, K.S.; Kryukov, G.V.; Sunyaev, S.R.; Mirny, L.A. Impact of deleterious passenger mutations on cancer progression. Proc. Natl. Acad. Sci. USA 2013, 110, 2910–2915. [Google Scholar] [CrossRef]
- McFarland, C.D.; Yaglom, J.A.; Wojtkowiak, J.W.; Scott, J.G.; Morse, D.L.; Sherman, M.Y.; Mirny, L.A. The Damaging Effect of Passenger Mutations on Cancer Progression. Cancer Res. 2017, 77, 4763–4772. [Google Scholar] [CrossRef]
- McFarland, C.D.; Mirny, L.A.; Korolev, K.S. Tug-of-war between driver and passenger mutations in cancer and other adaptive processes. Proc. Natl. Acad. Sci. USA 2014, 111, 15138–15143. [Google Scholar] [CrossRef]
- Bozic, I.; Gerold, J.M.; Nowak, M.A. Quantifying Clonal and Subclonal Passenger Mutations in Cancer Evolution. PLoS Comput. Biol. 2016, 12, e1004731. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Warrell, J.; Li, S.; McGillivray, P.D.; Meyerson, W.; Salichos, L.; Harmanci, A.; Martinez-Fundichely, A.; Chan, C.W.Y.; Nielsen, M.M.; et al. Passenger Mutations in More Than 2500 Cancer Genomes: Overall Molecular Functional Impact and Consequences. Cell 2020, 180, 915–927.E16. [Google Scholar] [CrossRef]
- Martinez-Jimenez, F.; Muinos, F.; Sentis, I.; Deu-Pons, J.; Reyes-Salazar, I.; Arnedo-Pac, C.; Mularoni, L.; Pich, O.; Bonet, J.; Kranas, H.; et al. A compendium of mutational cancer driver genes. Nat. Rev. Cancer 2020, 20, 555–572. [Google Scholar] [CrossRef] [PubMed]
- Ostroverkhova, D.; Przytycka, T.M.; Panchenko, A.R. Cancer driver mutations: Predictions and reality. Trends Mol. Med. 2023, 29, 554–566. [Google Scholar] [CrossRef]
- Buljan, M.; Blattmann, P.; Aebersold, R.; Boutros, M. Systematic characterization of pan-cancer mutation clusters. Mol. Syst. Biol. 2018, 14, e7974. [Google Scholar] [CrossRef]
- Brown, A.L.; Li, M.; Goncearenco, A.; Panchenko, A.R. Finding driver mutations in cancer: Elucidating the role of background mutational processes. PLoS Comput. Biol. 2019, 15, e1006981. [Google Scholar] [CrossRef] [PubMed]
- Torkamani, A.; Schork, N.J. Prediction of cancer driver mutations in protein kinases. Cancer Res. 2008, 68, 1675–1682. [Google Scholar] [CrossRef]
- Gonzalez-Perez, A.; Perez-Llamas, C.; Deu-Pons, J.; Tamborero, D.; Schroeder, M.P.; Jene-Sanz, A.; Santos, A.; Lopez-Bigas, N. IntOGen-mutations identifies cancer drivers across tumor types. Nat. Methods 2013, 10, 1081–1082. [Google Scholar] [CrossRef]
- Roy, D.M.; Walsh, L.A.; Chan, T.A. Driver mutations of cancer epigenomes. Protein Cell 2014, 5, 265–296. [Google Scholar] [CrossRef]
- Iranzo, J.; Martincorena, I.; Koonin, E.V. Cancer-mutation network and the number and specificity of driver mutations. Proc. Natl. Acad. Sci. USA 2018, 115, E6010–E6019. [Google Scholar] [CrossRef]
- Wood, L.D.; Parsons, D.W.; Jones, S.; Lin, J.; Sjoblom, T.; Leary, R.J.; Shen, D.; Boca, S.M.; Barber, T.; Ptak, J.; et al. The genomic landscapes of human breast and colorectal cancers. Science 2007, 318, 1108–1113. [Google Scholar] [CrossRef]
- Tokheim, C.; Karchin, R. CHASMplus Reveals the Scope of Somatic Missense Mutations Driving Human Cancers. Cell Syst. 2019, 9, 9–23.e8. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N.; Kandoth, C.; Schultz, N.; Cherniack, A.D.; Akbani, R.; Liu, Y.; Shen, H.; Robertson, A.G.; Pashtan, I.; Shen, R.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [CrossRef]
- Briggs, S.; Tomlinson, I. Germline and somatic polymerase epsilon and delta mutations define a new class of hypermutated colorectal and endometrial cancers. J. Pathol. 2013, 230, 148–153. [Google Scholar] [CrossRef]
- Stenzinger, A.; Pfarr, N.; Endris, V.; Penzel, R.; Jansen, L.; Wolf, T.; Herpel, E.; Warth, A.; Klauschen, F.; Kloor, M.; et al. Mutations in POLE and survival of colorectal cancer patients--link to disease stage and treatment. Cancer Med. 2014, 3, 1527–1538. [Google Scholar] [CrossRef]
- Elsayed, F.A.; Kets, C.M.; Ruano, D.; van den Akker, B.; Mensenkamp, A.R.; Schrumpf, M.; Nielsen, M.; Wijnen, J.T.; Tops, C.M.; Ligtenberg, M.J.; et al. Germline variants in POLE are associated with early onset mismatch repair deficient colorectal cancer. Eur. J. Hum. Genet. 2015, 23, 1080–1084. [Google Scholar] [CrossRef]
- Cornish, A.J.; Gruber, A.J.; Kinnersley, B.; Chubb, D.; Frangou, A.; Caravagna, G.; Noyvert, B.; Lakatos, E.; Wood, H.M.; Thorn, S.; et al. The genomic landscape of 2023 colorectal cancers. Nature 2024, 633, 127–136. [Google Scholar] [CrossRef]
- Church, D.N.; Briggs, S.E.; Palles, C.; Domingo, E.; Kearsey, S.J.; Grimes, J.M.; Gorman, M.; Martin, L.; Howarth, K.M.; Hodgson, S.V.; et al. DNA polymerase epsilon and delta exonuclease domain mutations in endometrial cancer. Hum. Mol. Genet. 2013, 22, 2820–2828. [Google Scholar] [CrossRef] [PubMed]
- Heitzer, E.; Tomlinson, I. Replicative DNA polymerase mutations in cancer. Curr. Opin. Genet. Dev. 2014, 24, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Freeman-Mills, L.; Rayner, E.; Glaire, M.; Briggs, S.; Vermeulen, L.; Fessler, E.; Medema, J.P.; Boot, A.; Morreau, H.; et al. Somatic POLE proofreading domain mutation, immune response, and prognosis in colorectal cancer: A retrospective, pooled biomarker study. Lancet Gastroenterol. Hepatol. 2016, 1, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Hino, H.; Shiomi, A.; Kusuhara, M.; Kagawa, H.; Yamakawa, Y.; Hatakeyama, K.; Kawabata, T.; Oishi, T.; Urakami, K.; Nagashima, T.; et al. Clinicopathological and mutational analyses of colorectal cancer with mutations in the POLE gene. Cancer Med. 2019, 8, 4587–4597. [Google Scholar] [CrossRef]
- Guerra, J.; Pinto, C.; Pinto, D.; Pinheiro, M.; Silva, R.; Peixoto, A.; Rocha, P.; Veiga, I.; Santos, C.; Santos, R.; et al. POLE somatic mutations in advanced colorectal cancer. Cancer Med. 2017, 6, 2966–2971. [Google Scholar] [CrossRef]
- Spier, I.; Holzapfel, S.; Altmuller, J.; Zhao, B.; Horpaopan, S.; Vogt, S.; Chen, S.; Morak, M.; Raeder, S.; Kayser, K.; et al. Frequency and phenotypic spectrum of germline mutations in POLE and seven other polymerase genes in 266 patients with colorectal adenomas and carcinomas. Int. J. Cancer 2015, 137, 320–331. [Google Scholar] [CrossRef]
- Hu, H.; Cai, W.; Wu, D.; Hu, W.; Dong Wang, L.; Mao, J.; Zheng, S.; Ge, W. Ultra-mutated colorectal cancer patients with POLE driver mutations exhibit distinct clinical patterns. Cancer Med. 2021, 10, 135–142. [Google Scholar] [CrossRef]
- Fang, H.; Barbour, J.A.; Poulos, R.C.; Katainen, R.; Aaltonen, L.A.; Wong, J.W.H. Mutational processes of distinct POLE exonuclease domain mutants drive an enrichment of a specific TP53 mutation in colorectal cancer. PLoS Genet. 2020, 16, e1008572. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Riaz, N.; Samstein, R.M.; Lee, M.; Makarov, V.; Valero, C.; Chowell, D.; Kuo, F.; Hoen, D.; Fitzgerald, C.W.R.; et al. Functional landscapes of POLE and POLD1 mutations in checkpoint blockade-dependent antitumor immunity. Nat. Genet. 2022, 54, 996–1012. [Google Scholar] [CrossRef]
- Rayner, E.; van Gool, I.C.; Palles, C.; Kearsey, S.E.; Bosse, T.; Tomlinson, I.; Church, D.N. A panoply of errors: Polymerase proofreading domain mutations in cancer. Nat. Rev. Cancer 2016, 16, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Temko, D.; Van Gool, I.C.; Rayner, E.; Glaire, M.; Makino, S.; Brown, M.; Chegwidden, L.; Palles, C.; Depreeuw, J.; Beggs, A.; et al. Somatic POLE exonuclease domain mutations are early events in sporadic endometrial and colorectal carcinogenesis, determining driver mutational landscape, clonal neoantigen burden and immune response. J. Pathol. 2018, 245, 283–296. [Google Scholar] [CrossRef]
- Robinson, P.S.; Coorens, T.H.H.; Palles, C.; Mitchell, E.; Abascal, F.; Olafsson, S.; Lee, B.C.H.; Lawson, A.R.J.; Lee-Six, H.; Moore, L.; et al. Increased somatic mutation burdens in normal human cells due to defective DNA polymerases. Nat. Genet. 2021, 53, 1434–1442. [Google Scholar] [CrossRef]
- Sondka, Z.; Dhir, N.B.; Carvalho-Silva, D.; Jupe, S.; Madhumita; McLaren, K.; Starkey, M.; Ward, S.; Wilding, J.; Ahmed, M.; et al. COSMIC: A curated database of somatic variants and clinical data for cancer. Nucleic Acids Res. 2024, 52, D1210–D1217. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Sharma, Y.; Miladi, M.; Dukare, S.; Boulay, K.; Caudron-Herger, M.; Gross, M.; Backofen, R.; Diederichs, S. A pan-cancer analysis of synonymous mutations. Nat. Commun. 2019, 10, 2569. [Google Scholar] [CrossRef]
- Shankar, G.M.; Taylor-Weiner, A.; Lelic, N.; Jones, R.T.; Kim, J.C.; Francis, J.M.; Abedalthagafi, M.; Borges, L.F.; Coumans, J.V.; Curry, W.T.; et al. Sporadic hemangioblastomas are characterized by cryptic VHL inactivation. Acta Neuropathol. Commun. 2014, 2, 167. [Google Scholar] [CrossRef] [PubMed]
- Birkealv, S.; Harland, M.; Matsuyama, L.; Rashid, M.; Mehta, I.; Laye, J.P.; Haase, K.; Mell, T.; Iyer, V.; Robles-Espinoza, C.D.; et al. Mutually exclusive genetic interactions and gene essentiality shape the genomic landscape of primary melanoma. J. Pathol. 2023, 259, 56–68. [Google Scholar] [CrossRef]
- Juul, R.I.; Nielsen, M.M.; Juul, M.; Feuerbach, L.; Pedersen, J.S. The landscape and driver potential of site-specific hotspots across cancer genomes. NPJ Genom. Med. 2021, 6, 33. [Google Scholar] [CrossRef]
- Gao, J.; Chang, M.T.; Johnsen, H.C.; Gao, S.P.; Sylvester, B.E.; Sumer, S.O.; Zhang, H.; Solit, D.B.; Taylor, B.S.; Schultz, N.; et al. 3D clusters of somatic mutations in cancer reveal numerous rare mutations as functional targets. Genome Med. 2017, 9, 4. [Google Scholar] [CrossRef]
- Garcia-Nieto, P.E.; Morrison, A.J.; Fraser, H.B. The somatic mutation landscape of the human body. Genome Biol. 2019, 20, 298. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.T.; Asthana, S.; Gao, S.P.; Lee, B.H.; Chapman, J.S.; Kandoth, C.; Gao, J.; Socci, N.D.; Solit, D.B.; Olshen, A.B.; et al. Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat. Biotechnol. 2016, 34, 155–163. [Google Scholar] [CrossRef]
- Pursell, Z.F.; Kunkel, T.A. DNA polymerase epsilon: A polymerase of unusual size (and complexity). Prog. Nucleic Acid. Res. Mol. Biol. 2008, 82, 101–145. [Google Scholar] [CrossRef]
- Logan, C.V.; Murray, J.E.; Parry, D.A.; Robertson, A.; Bellelli, R.; Tarnauskaite, Z.; Challis, R.; Cleal, L.; Borel, V.; Fluteau, A.; et al. DNA Polymerase Epsilon Deficiency Causes IMAGe Syndrome with Variable Immunodeficiency. Am. J. Hum. Genet. 2018, 103, 1038–1044. [Google Scholar] [CrossRef] [PubMed]
- Parkash, V.; Kulkarni, Y.; Ter Beek, J.; Shcherbakova, P.V.; Kamerlin, S.C.L.; Johansson, E. Structural consequence of the most frequently recurring cancer-associated substitution in DNA polymerase epsilon. Nat. Commun. 2019, 10, 373. [Google Scholar] [CrossRef] [PubMed]
- Jenkyn-Bedford, M.; Jones, M.L.; Baris, Y.; Labib, K.P.M.; Cannone, G.; Yeeles, J.T.P.; Deegan, T.D. A conserved mechanism for regulating replisome disassembly in eukaryotes. Nature 2021, 600, 743–747. [Google Scholar] [CrossRef] [PubMed]
- Hogg, M.; Osterman, P.; Bylund, G.O.; Ganai, R.A.; Lundstrom, E.B.; Sauer-Eriksson, A.E.; Johansson, E. Structural basis for processive DNA synthesis by yeast DNA polymerase varepsilon. Nat. Struct. Mol. Biol. 2014, 21, 49–55. [Google Scholar] [CrossRef]
- Yang, W. Nucleases: Diversity of structure, function and mechanism. Q. Rev. Biophys. 2011, 44, 1–93. [Google Scholar] [CrossRef]
- Hansen, M.F.; Johansen, J.; Bjornevoll, I.; Sylvander, A.E.; Steinsbekk, K.S.; Saetrom, P.; Sandvik, A.K.; Drablos, F.; Sjursen, W. A novel POLE mutation associated with cancers of colon, pancreas, ovaries and small intestine. Fam. Cancer 2015, 14, 437–448. [Google Scholar] [CrossRef]
- Inamura, K. Colorectal Cancers: An Update on Their Molecular Pathology. Cancers 2018, 10, 26. [Google Scholar] [CrossRef]
- Zarkavelis, G.; Boussios, S.; Papadaki, A.; Katsanos, K.H.; Christodoulou, D.K.; Pentheroudakis, G. Current and future biomarkers in colorectal cancer. Ann. Gastroenterol. 2017, 30, 613–621. [Google Scholar] [CrossRef]
- Parthiban, V.; Gromiha, M.M.; Schomburg, D. CUPSAT: Prediction of protein stability upon point mutations. Nucleic Acids Res. 2006, 34, W239–W242. [Google Scholar] [CrossRef]
- Xing, X.; Jin, N.; Wang, J. Polymerase Epsilon-Associated Ultramutagenesis in Cancer. Cancers 2022, 14, 1467. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Lundwall, A. A locus on chromosome 20 encompassing genes that are highly expressed in the epididymis. Asian J. Androl. 2007, 9, 540–544. [Google Scholar] [CrossRef] [PubMed]
- Marra, G. An “expressionistic” look at serrated precancerous colorectal lesions. Diagn. Pathol. 2021, 16, 4. [Google Scholar] [CrossRef]
- Stanley, F.K.; Moore, S.; Goodarzi, A.A. CHD chromatin remodelling enzymes and the DNA damage response. Mutat. Res. 2013, 750, 31–44. [Google Scholar] [CrossRef]
- Zhang, B.; Liu, Q.; Wen, W.; Gao, H.; Wei, W.; Tang, A.; Qin, B.; Lyu, H.; Meng, X.; Li, K.; et al. The chromatin remodeler CHD6 promotes colorectal cancer development by regulating TMEM65-mediated mitochondrial dynamics via EGF and Wnt signaling. Cell Discov. 2022, 8, 130. [Google Scholar] [CrossRef]
- Burgos, M.; Jimenez, R.; Sanchez, A.; Diaz de la Guardia, R. Replication of the heterogeneous heterochromatin of the sex chromosomes of Microtus cabrerae. Experientia 1992, 48, 1151–1153. [Google Scholar] [CrossRef]
- Xu, Y.; Zhou, X.; Gao, L.; Yan, S.; Li, Z.; Zhang, D.; Pu, J.; Zou, S.; Mao, Z. Identification of HAGHL as a novel metabolic oncogene regulating human colorectal cancer progression. Clin. Transl. Oncol. 2023, 25, 1033–1042. [Google Scholar] [CrossRef]
- Ivanenkov, V.V.; Murphy-Piedmonte, D.M.; Kirley, T.L. Bacterial expression, characterization, and disulfide bond determination of soluble human NTPDase6 (CD39L2) nucleotidase: Implications for structure and function. Biochemistry 2003, 42, 11726–11735. [Google Scholar] [CrossRef]
- Hodel, K.P.; Sun, M.J.S.; Ungerleider, N.; Park, V.S.; Williams, L.G.; Bauer, D.L.; Immethun, V.E.; Wang, J.; Suo, Z.; Lu, H.; et al. POLE Mutation Spectra Are Shaped by the Mutant Allele Identity, Its Abundance, and Mismatch Repair Status. Mol. Cell 2020, 78, 1166–1177.e6. [Google Scholar] [CrossRef]
- Shah, S.M.; Demidova, E.V.; Ringenbach, S.; Faezov, B.; Andrake, M.; Gandhi, A.; Mur, P.; Viana-Errasti, J.; Xiu, J.; Swensen, J.; et al. Exploring Co-occurring POLE Exonuclease and Non-exonuclease Domain Mutations and Their Impact on Tumor Mutagenicity. Cancer Res. Commun. 2024, 4, 213–225. [Google Scholar] [CrossRef]
- Castellsague, E.; Li, R.; Aligue, R.; Gonzalez, S.; Sanz, J.; Martin, E.; Velasco, A.; Capella, G.; Stewart, C.J.R.; Vidal, A.; et al. Novel POLE pathogenic germline variant in a family with multiple primary tumors results in distinct mutational signatures. Hum. Mutat. 2019, 40, 36–41. [Google Scholar] [CrossRef]
- de Bruijn, I.; Kundra, R.; Mastrogiacomo, B.; Tran, T.N.; Sikina, L.; Mazor, T.; Li, X.; Ochoa, A.; Zhao, G.; Lai, B.; et al. Analysis and Visualization of Longitudinal Genomic and Clinical Data from the AACR Project GENIE Biopharma Collaborative in cBioPortal. Cancer Res. 2023, 83, 3861–3867. [Google Scholar] [CrossRef]
- Cheadle, C.; Vawter, M.P.; Freed, W.J.; Becker, K.G. Analysis of microarray data using Z score transformation. J. Mol. Diagn. 2003, 5, 73–81. [Google Scholar] [CrossRef]
- Guo, Y.; Sheng, Q.; Li, J.; Ye, F.; Samuels, D.C.; Shyr, Y. Large scale comparison of gene expression levels by microarrays and RNAseq using TCGA data. PLoS ONE 2013, 8, e71462. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Jones, M.L.; Baris, Y.; Taylor, M.R.G.; Yeeles, J.T.P. Structure of a human replisome shows the organisation and interactions of a DNA replication machine. EMBO J. 2021, 40, e108819. [Google Scholar] [CrossRef]
- Huang, C.H.; Mandelker, D.; Schmidt-Kittler, O.; Samuels, Y.; Velculescu, V.E.; Kinzler, K.W.; Vogelstein, B.; Gabelli, S.B.; Amzel, L.M. The structure of a human p110alpha/p85alpha complex elucidates the effects of oncogenic PI3Kalpha mutations. Science 2007, 318, 1744–1748. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Protein | Mutation | Tertiary Structure Interaction Changes | Electrostatic Surface Potential Changes | ΔΔG Value (kcal/mol) |
|---|---|---|---|---|
| PIK3CA | R88Q | Yes, decreased | Neutral to negative | −4.53 |
| E545K | Yes, decreased | Negative to neutral | +0.08 | |
| H1074R | No | Negative to partial negative | −0.56 | |
| CNOT | R559Q | Yes, increased | No change | −0.08 |
| FBXW7 | R465Q | No | No change | −1.83 |
| R465H | No | No change | −1.40 | |
| R465C | No | No change | +7.88 | |
| CTNNB1 | S45F | No | No change | −1.87 |
| SF3B1 | R196Q | No | Positive to negative | −0.23 |
| R315Q | No | Positive to neutral | +0.54 | |
| CHD4 | R1338I | No | No change | −2.31 |
| CHD8 | R546C | No | Positive to partial positive | −0.37 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gibson, L.M.; Konda, P.; Bliss, H.J.; Nelakurti, D.D.; Mirzaei, G.; Bouley, R.A.; Wang, J.J.; Petreaca, R.C. Comprehensive Analysis of Human Colorectal Cancers Harboring Polymerase Epsilon Mutations. Int. J. Mol. Sci. 2025, 26, 7208. https://doi.org/10.3390/ijms26157208
Gibson LM, Konda P, Bliss HJ, Nelakurti DD, Mirzaei G, Bouley RA, Wang JJ, Petreaca RC. Comprehensive Analysis of Human Colorectal Cancers Harboring Polymerase Epsilon Mutations. International Journal of Molecular Sciences. 2025; 26(15):7208. https://doi.org/10.3390/ijms26157208
Chicago/Turabian StyleGibson, Louis M., Phanithan Konda, Hunter J. Bliss, Devi D. Nelakurti, Golrokh Mirzaei, Renee A. Bouley, Jing J. Wang, and Ruben C. Petreaca. 2025. "Comprehensive Analysis of Human Colorectal Cancers Harboring Polymerase Epsilon Mutations" International Journal of Molecular Sciences 26, no. 15: 7208. https://doi.org/10.3390/ijms26157208
APA StyleGibson, L. M., Konda, P., Bliss, H. J., Nelakurti, D. D., Mirzaei, G., Bouley, R. A., Wang, J. J., & Petreaca, R. C. (2025). Comprehensive Analysis of Human Colorectal Cancers Harboring Polymerase Epsilon Mutations. International Journal of Molecular Sciences, 26(15), 7208. https://doi.org/10.3390/ijms26157208