
Gut–Brain Interactions in Neuronal Ceroid Lipofuscinoses: A Systematic Review Beyond the Brain in Paediatric Dementias

 , , and
, , and
Abstract
1. Introduction
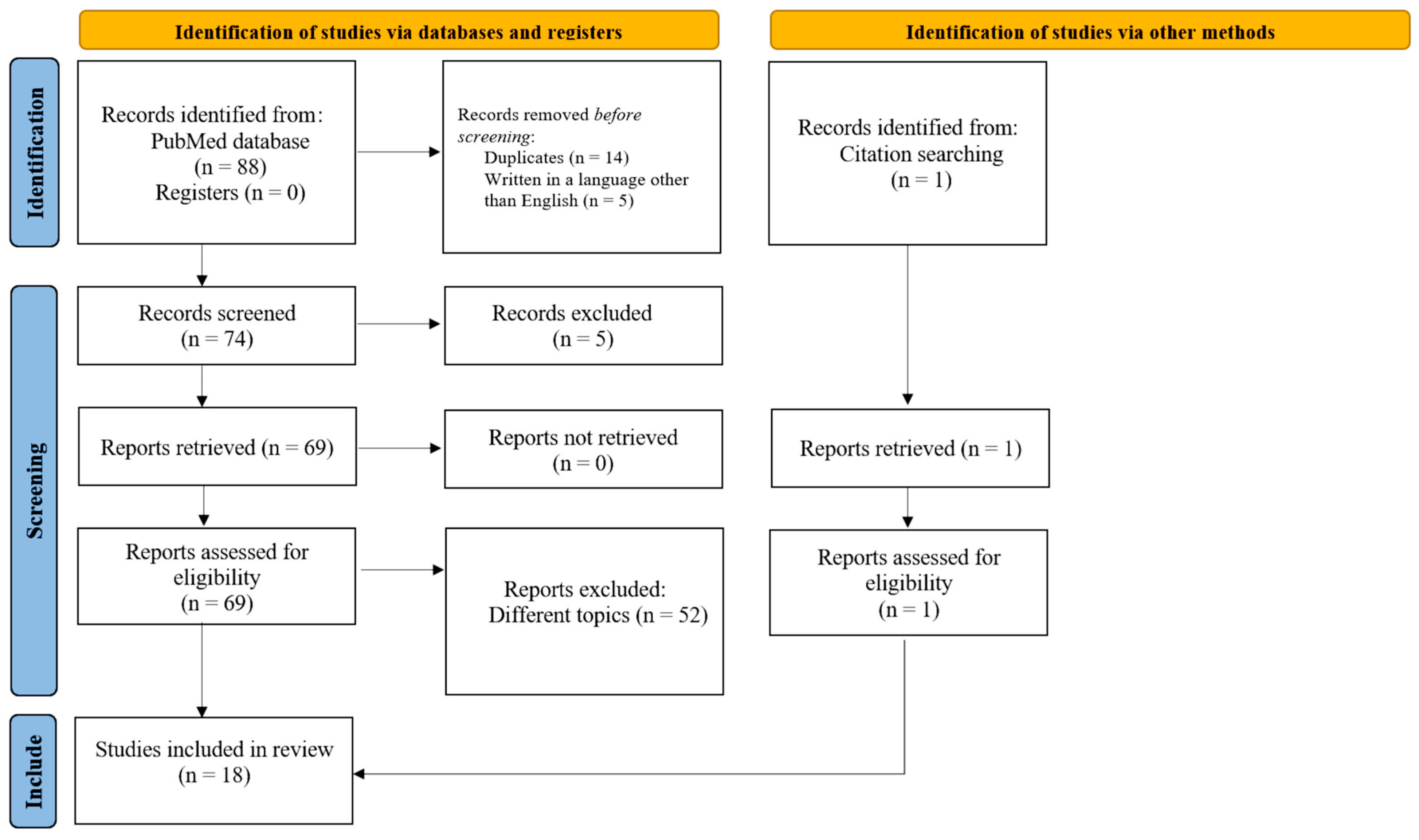
2. Methods
3. Results
3.1. Human Evidence of ENS and GI Pathology in NCLs
3.2. ENS Findings in NCL Animal Models
3.3. Gut Microbiota Alterations in NCL Animal Models
3.4. Gene Therapy Efficacy on GI Features in NCL Mice Models
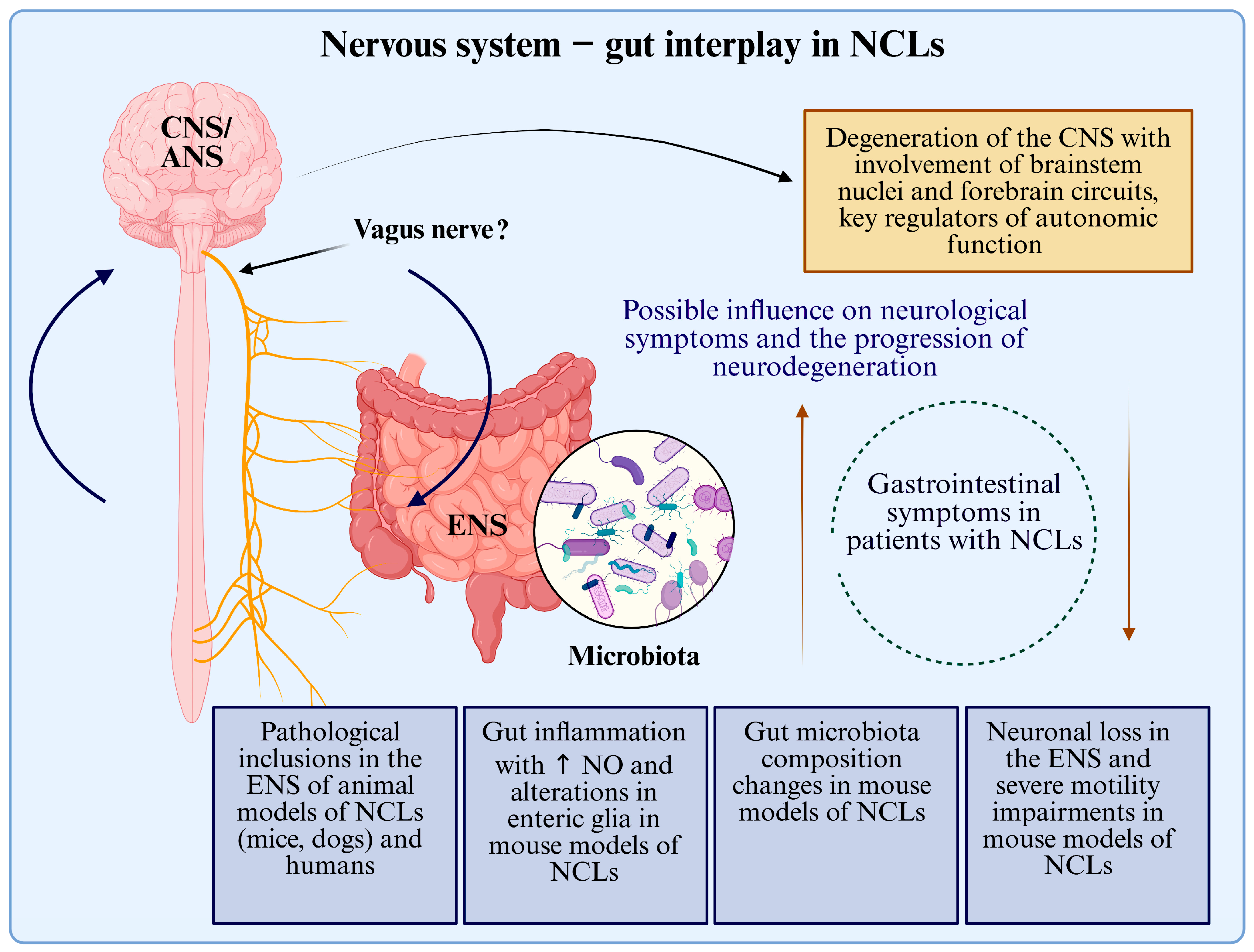
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mole, S.E.; Williams, R.E.; Goebel, H.H. The Neuronal Ceroid Lipofuscinoses (Batten Disease), 2nd ed.; Oxford University Press: Oxford, UK, 2011; pp. 361–365. [Google Scholar]
- Seehafer, S.S.; APearce, D. You say lipofuscin, we say ceroid: Defining autofluorescent storage material. Neurobiol. Aging 2006, 27, 576–588. [Google Scholar] [CrossRef] [PubMed]
- Palmer, D.N.; Fearnley, I.M.; Walker, J.E.; Hall, N.A.; Lake, B.D.; Wolfe, L.S.; Haltia, M.; Martinus, R.D.; Jolly, R.D. Mitochondrial ATP synthase subunit c storage in the ceroid-lipofuscinoses (Batten disease). Am. J. Med Genet. 1992, 42, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Tyynelä, J.; Palmer, D.N.; Baumann, M.; Haltia, M. Storage of saposins A and D in infantile neuronal ceroid-lipofuscinosis. FEBS Lett. 1993, 330, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.E.; Mole, S.E. New nomenclature and classification scheme for the neuronal ceroid lipofuscinoses. Neurology 2012, 79, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Specchio, N.; Pietrafusa, N.; Trivisano, M. Changing Times for CLN2 Disease: The Era of Enzyme Replacement Therapy. Ther. Clin. Risk Manag. 2020, 16, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Simonati, A.; Williams, R.E. Neuronal Ceroid Lipofuscinosis: The Multifaceted Approach to the Clinical Issues, an Overview. Front. Neurol. 2022, 13, 811686. [Google Scholar] [CrossRef] [PubMed]
- On behalf of the Pediatric Quality of Life Inventory™ Gastrointestinal Symptoms Module Testing Study Consortium; Varni, J.W.; Shulman, R.J.; Self, M.M.; Nurko, S.; Saps, M.; Saeed, S.A.; Patel, A.S.; Dark, C.V.; Bendo, C.B.; et al. Gastrointestinal symptoms predictors of health-related quality of life in pediatric patients with functional gastrointestinal disorders. Qual. Life Res. 2016, 26, 1015–1025. [Google Scholar] [CrossRef] [PubMed]
- Febo-Rodriguez, L.; Chumpitazi, B.P.; Musaad, S.; Sher, A.C.; Varni, J.W.; Shulman, R.J. Gastrointestinal Symptoms Profile in Pediatric Patients With Gastroparesis Compared to Healthy Controls. J. Pediatr. Gastroenterol. Nutr. 2022, 75, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Han, M.N.; Finkelstein, D.I.; McQuade, R.M.; Diwakarla, S. Gastrointestinal Dysfunction in Parkinson’s Disease: Current and Potential Therapeutics. J. Pers. Med. 2022, 12, 144. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Huh, J.R.; Shah, K. Microbiota and the gut-brain-axis: Implications for new therapeutic design in the CNS. EBioMedicine 2022, 77, 103908. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Bonfili, L.; Wei, T.; Eleuteri, A.M. Understanding the Gut–Brain Axis and Its Therapeutic Implications for Neurodegenerative Disorders. Nutrients 2023, 15, 4631. [Google Scholar] [CrossRef] [PubMed]
- Fung, T.C.; Olson, C.A.; Hsiao, E.Y. Interactions between the microbiota, immune and nervous systems in health and disease. Nat. Neurosci. 2017, 20, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Cheon, J.H. Microbial Modulation in Inflammatory Bowel Diseases. Immune Netw. 2022, 22, e44. [Google Scholar] [CrossRef] [PubMed]
- Rothhammer, V.; Mascanfroni, I.D.; Bunse, L.; Takenaka, M.C.; Kenison, J.E.; Mayo, L.; Chao, C.-C.; Patel, B.; Yan, R.; Blain, M.; et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat. Med. 2016, 22, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Yano, J.M.; Yu, K.; Donaldson, G.P.; Shastri, G.G.; Ann, P.; Ma, L.; Nagler, C.R.; Ismagilov, R.F.; Mazmanian, S.K.; Hsiao, E.Y. Indigenous Bacteria from the Gut Microbiota Regulate Host Serotonin Biosynthesis. Cell 2015, 161, 264–276, Erratum in Cell 2015, 163, 258. [Google Scholar] [CrossRef] [PubMed]
- Bravo, J.A.; Forsythe, P.; Chew, M.V.; Escaravage, E.; Savignac, H.M.; Dinan, T.G.; Bienenstock, J.; Cryan, J.F. Ingestion of Lactobacillus strain regulates emotional behavior and central GABA receptor expression in a mouse via the vagus nerve. Proc. Natl. Acad. Sci. USA 2011, 108, 16050–16055. [Google Scholar] [CrossRef] [PubMed]
- Asano, Y.; Hiramoto, T.; Nishino, R.; Aiba, Y.; Kimura, T.; Yoshihara, K.; Koga, Y.; Sudo, N. Critical role of gut microbiota in the production of biologically active, free catecholamines in the gut lumen of mice. Am. J. Physiol. Liver Physiol. 2012, 303, G1288–G1295. [Google Scholar] [CrossRef] [PubMed]
- Hou, K.; Wu, Z.-X.; Chen, X.-Y.; Wang, J.-Q.; Zhang, D.; Xiao, C.; Zhu, D.; Koya, J.B.; Wei, L.; Li, J.; et al. Microbiota in health and diseases. Signal Transduct. Target. Ther. 2022, 7, 135. [Google Scholar] [CrossRef] [PubMed]
- Cryan, J.F.; O’Riordan, K.J.; Sandhu, K.; Peterson, V.; Dinan, T.G. The gut microbiome in neurological disorders. Lancet Neurol. 2020, 19, 179–194. [Google Scholar] [CrossRef] [PubMed]
- Vogt, N.M.; Kerby, R.L.; Dill-McFarland, K.A.; Harding, S.J.; Merluzzi, A.P.; Johnson, S.C.; Carlsson, C.M.; Asthana, S.; Zetterberg, H.; Blennow, K.; et al. Gut microbiome alterations in Alzheimer’s disease. Sci. Rep. 2017, 7, 13537. [Google Scholar] [CrossRef] [PubMed]
- Kwon, D.; Zhang, K.; Paul, K.C.; Folle, A.D.; Del Rosario, I.; Jacobs, J.P.; Keener, A.M.; Bronstein, J.M.; Ritz, B. Diet and the gut microbiome in patients with Parkinson’s disease. npj Park. Dis. 2024, 10, 89. [Google Scholar] [CrossRef] [PubMed]
- Mole, S.E.; Cotman, S.L. Genetics of the neuronal ceroid lipofuscinoses (Batten disease). Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2015, 1852, 2237–2241. [Google Scholar] [CrossRef] [PubMed]
- Nelvagal, H.R.; Lange, J.; Takahashi, K.; Tarczyluk-Wells, M.A.; Cooper, J.D. Pathomechanisms in the neuronal ceroid lipofuscinoses. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2020, 1866, 165570. [Google Scholar] [CrossRef] [PubMed]
- Fung, C.; Van den Berghe, P. Functional circuits and signal processing in the enteric nervous system. Cell. Mol. Life Sci. 2020, 77, 4505–4522. [Google Scholar] [CrossRef] [PubMed]
- Goyal, D.; Ali, S.A.; Singh, R.K. Emerging role of gut microbiota in modulation of neuroinflammation and neurodegeneration with emphasis on Alzheimer’s disease. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2021, 106, 110112. [Google Scholar] [CrossRef] [PubMed]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. PLoS Med. 2021, 18, e1003583. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.; Dickson, J.A.S.; Lake, B.D. Rectal biopsy in the diagnosis of neurological disease in childhood. Br. J. Surg. 1976, 63, 313–316. [Google Scholar] [CrossRef] [PubMed]
- Lake, B.; Brett, E.; Boyd, S. A Form of Juvenile Batten Disease with Granular Osmiophilic Deposits. Neuropediatrics 1996, 27, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Åberg, L.; Järvelä, I.; Rapola, J.; Autti, T.; Kirveskari, E.; Lappi, M.; Sipilä, L.; Santavuori, P. Atypical juvenile neuronal ceroid liposfuscinosis with granular osmiophilic deposit-like inclusions in the autonomic nerve cells of the gut wall. Acta Neuropathol. 1998, 95, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Rowan, S.A.; Lake, B.D. Tissue and cellular distribution of subunit c of ATP synthase in Batten disease (neuronal ceroid-lipofuscinosis). Am. J. Med. Genet. 1995, 57, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Simonati, A.; Rizzuto, N. Neuronal ceroid lipofuscinoses: Pathological features of bioptic specimens from 28 patients. Neurol. Sci. 2000, 21, S63–S70. [Google Scholar] [CrossRef] [PubMed]
- Pasquinelli, G.; Cenacchi, G.; Piane, E.L.; Russo, C.; Aguglia, U. The problematic issue of Kufs disease diagnosis as performed on rectal biopsies: A case report. Ultrastruct Pathol. 2004, 28, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Ferlazzo, E.; Gasparini, S.; Pasquinelli, G.; Labate, A.; Gambardella, A.; Sofia, V.; Cianci, V.; Branca, D.; Quattrone, A.; Aguglia, U. Usefulness of rectal biopsy for the diagnosis of Kufs disease: A controlled study and review of the literature. Eur. J. Neurol. 2012, 19, 1331–1336. [Google Scholar] [CrossRef] [PubMed]
- Moro, F.; Gismondi, F.; Pezzini, F.; Santorelli, F.M.; Simonati, A. Clinical, ultrastructural, and molecular studies in a patient with Kufs disease. Neurol. Sci. 2014, 35, 605–607. [Google Scholar] [CrossRef] [PubMed]
- Ziółkowska, E.A.; Jansen, M.J.; Williams, L.L.; Wang, S.H.; Eultgen, E.M.; Takahashi, K.; Le, S.Q.; Nelvagal, H.R.; Sharma, J.; Sardiello, M.; et al. Gene therapy ameliorates bowel dysmotility and enteric neuron degeneration and extends survival in lysosomal storage disorder mouse models. Sci. Transl. Med. 2025, 17, eadj1445. [Google Scholar] [CrossRef] [PubMed]
- Ziółkowska, E.A.; Jablonka-Shariff, A.; Williams, L.L.; Jansen, M.J.; Wang, S.H.; Eultgen, E.M.; Wood, M.D.; Hunter, D.A.; Sharma, J.; Sardiello, M.; et al. Identifying and treating CLN3 disease outside the central nervous system. bioRxiv 2025, 2025, 635518. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.H.; Williams, E.M.; Takahash, K.; Nelvagal, H.R. Enteric Nervous System defects underlie bowel dysfunction in Cln1, Cln2 and Cln3 disease mice: A new therapeutic target? In Proceedings of the 17th International Conference on Neuronal Ceroid Lipofuscinoses (Batten Disease), St. Louis, MO, USA, 6–10 October 2021. [Google Scholar]
- Hirz, M.; Drögemüller, M.; Schänzer, A.; Jagannathan, V.; Dietschi, E.; Goebel, H.; Hecht, W.; Laubner, S.; Schmidt, M.; Steffen, F.; et al. Neuronal ceroid lipofuscinosis (NCL) is caused by the entire deletion of CLN8 in the Alpenländische Dachsbracke dog. Mol. Genet. Metab. 2017, 120, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Jolly, R.; Hartley, W.; Jones, B.; Johnstone, A.; Palmer, A.; Blakemore, W. Generalised ceroid-lipofuscinosis and brown bowel syndrome in Cocker spaniel dogs. N. Z. Vet. J. 1994, 42, 236–239. [Google Scholar] [CrossRef] [PubMed]
- Minatel, L.; Underwood, S.C.; Carfagnini, J.C. Ceroid-Lipofuscinosis in a Cocker Spaniel Dog. Vet. Pathol. 2000, 37, 488–490. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, H.; Zhang, J.; Koike, M.; Nishioku, T.; Okamoto, Y.; Kominami, E.; von Figura, K.; Peters, C.; Yamamoto, K.; Saftig, P.; et al. Involvement of Nitric Oxide Released from Microglia–Macrophages in Pathological Changes of Cathepsin D-Deficient Mice. J. Neurosci. 2001, 21, 7526–7533. [Google Scholar] [CrossRef] [PubMed]
- Patani, R.; Hardingham, G.E.; Liddelow, S.A. Functional roles of reactive astrocytes in neuroinflammation and neurodegeneration. Nat. Rev. Neurol. 2023, 19, 395–409. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.B.; Langin, L.M.; Zhao, J.; Weimer, J.M.; Pearce, D.A.; Kovács, A.D. Changes in motor behavior, neuropathology, and gut microbiota of a Batten disease mouse model following administration of acidified drinking water. Sci. Rep. 2019, 9, 14962. [Google Scholar] [CrossRef] [PubMed]
- Parker, C.; Zhao, J.; Pearce, D.A.; Kovács, A.D. Comparative analysis of the gut microbiota composition in the Cln1R151X and Cln2R207X mouse models of Batten disease and in three wild-type mouse strains. Arch. Microbiol. 2021, 203, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chia, N.; Kalari, K.R.; Yao, J.Z.; Novotna, M.; Paz Soldan, M.M.; Luckey, D.H.; Marietta, E.V.; Jeraldo, P.R.; Chen, X.; et al. Multiple sclerosis patients have a distinct gut microbiota compared to healthy controls. Sci. Rep. 2016, 6, 28484. [Google Scholar] [CrossRef] [PubMed]
- Schaubeck, M.; Clavel, T.; Calasan, J.; Lagkouvardos, I.; Haange, S.B.; Jehmlich, N.; Basic, M.; Dupont, A.; Hornef, M.; von Bergen, M.; et al. Dysbiotic gut microbiota causes transmissible Crohn’s disease-like ileitis independent of failure in antimicrobial defence. Gut 2016, 65, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Ling, Z.; Zhang, Y.; Mao, H.; Ma, Z.; Yin, Y.; Wang, W.; Tang, W.; Tan, Z.; Shi, J.; et al. Altered fecal microbiota composition in patients with major depressive disorder. Brain Behav. Immun. 2015, 48, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Finegold, S.M.; Dowd, S.E.; Gontcharova, V.; Liu, C.; Henley, K.E.; Wolcott, R.D.; Youn, E.; Summanen, P.H.; Granpeesheh, D.; Dixon, D.; et al. Pyrosequencing study of fecal microflora of autistic and control children. Anaerobe 2010, 16, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Kovács, A.D.; Langin, L.M.; Hernandez, J.L.G.; Pearce, D.A. Acidified drinking water attenuates motor deficits and brain pathology in a mouse model of a childhood neurodegenerative disorder. Sci. Rep. 2022, 12, 9025. [Google Scholar] [CrossRef] [PubMed]
- Kovács, A.D.; Hernandez, J.L.G.; Pearce, D.A. Acidified drinking water improves motor function, prevents tremors and changes disease trajectory in Cln2R207X mice, a model of late infantile Batten disease. Sci. Rep. 2023, 13, 19229. [Google Scholar] [CrossRef] [PubMed]
- Politei, J.; Thurberg, B.; Wallace, E.; Warnock, D.; Serebrinsky, G.; Durand, C.; Schenone, A. Gastrointestinal involvement in Fabry disease. So important, yet often neglected. Clin. Genet. 2016, 89, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Valstar, M.J.; Ruijter, G.J.G.; van Diggelen, O.P.; Poorthuis, B.J.; Wijburg, F.A. Sanfilippo syndrome: A mini-review. J. Inherit. Metab. Dis. 2008, 31, 240–252. [Google Scholar] [CrossRef] [PubMed]
- Delgadillo, V.; del O’cAllaghan, M.; Gort, L.; Coll, M.J.; Pineda, M. Natural history of Sanfilippo syndrome in Spain. Orphanet J. Rare Dis. 2013, 8, 189. [Google Scholar] [CrossRef] [PubMed]
- Malcolm, C.; Hain, R.; Gibson, F.; Adams, S.; Anderson, G.; Forbat, L. Challenging symptoms in children with rare life-limiting conditions: Findings from a prospective diary and interview study with families. Acta Paediatr. 2012, 101, 985–992. [Google Scholar] [CrossRef] [PubMed]
- Dinari, G.; Rosenbach, Y.; Grunebaum, M.; Zahavi, I.; Alpert, G.; Nitzan, M. Gastrointestinal Manifestations of Niemann-Pick Disease. Enzyme 1980, 25, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Camilleri, M. Gastrointestinal motility disorders in neurologic disease. J. Clin. Investig. 2021, 131, e143771. [Google Scholar] [CrossRef] [PubMed]
- Ostergaard, J.R.; Nelvagal, H.R.; Cooper, J.D. Top-down and bottom-up propagation of disease in the neuronal ceroid lipofuscinoses. Front. Neurol. 2022, 13, 1061363. [Google Scholar] [CrossRef] [PubMed]
- Baekmann, C.; Handrup, M.M.; Molgaard, H.; Ejerskov, C.; Jensen, H.K.; Ostergaard, J.R. Insight of autonomic dysfunction in CLN3 disease: A study on episodes resembling paroxysmal sympathetic hyperactivity (PSH). Orphanet J. Rare Dis. 2024, 19, 374. [Google Scholar] [CrossRef] [PubMed]
- Rietdorf, K.; Coode, E.E.; Schulz, A.; Wibbeler, E.; Bootman, M.D.; Ostergaard, J.R. Cardiac pathology in neuronal ceroid lipofuscinoses (NCL): More than a mere co-morbidity. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2020, 1866, 165643. [Google Scholar] [CrossRef] [PubMed]
- Miao, N.; Levin, S.W.; Baker, E.H.; Caruso, R.C.; Zhang, Z.; Gropman, A.; Koziol, D.; Wesley, R.; Mukherjee, A.B.; Quezado, Z.M.N. Children with Infantile Neuronal Ceroid Lipofuscinosis Have an Increased Risk of Hypothermia and Bradycardia During Anesthesia. Anesthesia Analg. 2009, 109, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Karemaker, J.M. An introduction into autonomic nervous function. Physiol. Meas. 2017, 38, R89–R118. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Ortega, J.F.; Baguley, I.J.; Gates, T.A.; Garcia-Caballero, M.; Quesada-Garcia, J.G.; Prieto-Palomino, M.A. Catecholamines and Paroxysmal Sympathetic Hyperactivity after Traumatic Brain Injury. J. Neurotrauma 2017, 34, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Ostergaard, J.R. Paroxysmal sympathetic hyperactivity in Juvenile neuronal ceroid lipofuscinosis (Batten disease). Auton. Neurosci. 2018, 214, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Ostergaard, J.R. Treatment of non-epileptic episodes of anxious, fearful behavior in adolescent juvenile neuronal ceroid lipofuscinosis (CLN3 disease). Front. Neurol. 2023, 14, 1216861. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Miao, Z.; Liu, Y.; Chen, X.; Wang, H.; Su, J.; Chen, J. The Brain–Gut–Bone Axis in Neurodegenerative Diseases: Insights, Challenges, and Future Prospects. Adv. Sci. 2024, 11, e2307971. [Google Scholar] [CrossRef] [PubMed]
- Keshavarzian, A.; Green, S.J.; Engen, P.A.; Voigt, R.M.; Naqib, A.; Forsyth, C.B.; Mutlu, E.; Shannon, K.M. Colonic bacterial composition in Parkinson’s disease. Mov. Disord. 2015, 30, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Sachdeva, K.; Sundaramurthy, V. The Interplay of Host Lysosomes and Intracellular Pathogens. Front. Cell. Infect. Microbiol. 2020, 10, 595502. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Hara, T. Intracellular Quality Control by Autophagy: How Does Autophagy Prevent Neurodegeneration? Autophagy 2006, 2, 302–304. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.T.T.; Lapaquette, P.; Bringer, M.-A.; Darfeuille-Michaud, A. Autophagy and Crohn’s Disease. J. Innate Immun. 2013, 5, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Larabi, A.; Barnich, N.; Nguyen, H.T.T. New insights into the interplay between autophagy, gut microbiota and inflammatory responses in IBD. Autophagy 2020, 16, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Pezzini, F.; Fiorini, M.; Doccini, S.; Santorelli, F.M.; Zanusso, G.; Simonati, A. Enhanced expression of the autophagosomal marker LC3-II in detergent-resistant protein lysates from a CLN3 patient’s post-mortem brain. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2023, 1869, 166756. [Google Scholar] [CrossRef] [PubMed]
- Loh, J.S.; Mak, W.Q.; Tan, L.K.S.; Ng, C.X.; Chan, H.H.; Yeow, S.H.; Foo, J.B.; Ong, Y.S.; How, C.W.; Khaw, K.Y. Microbiota–gut–brain axis and its therapeutic applications in neurodegenerative diseases. Signal Transduct. Target. Ther. 2024, 9, 37. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Reference | Genetic or Clinical Form | Main Findings |
|---|---|---|
| Aberg L, et al., 1998 [30] | Atypical JNCL | Cytoplasmic granular osmiophilic inclusions were found in ENS cells |
| Rowan SA, 1995 [31] | NCLs | Accumulation of subunit c of mitochondrial ATP synthase in enteric neurons |
| Ferlazzo E, et al., 2012 [34] | Kufs disease | Validation of rectal biopsy in the diagnosis of Kufs disease, highlighting alterations in the ENS |
| Simonati A, Rizzuto N, 2000 [32] | 15 LINCL, 10 JNCL e 3 ANCL | Presence of different types of inclusions in the enteric nervous system |
| Pasquinelli G, et al., 2004 [33] | Kufs disease | Rectal biopsies can reveal abnormalities in the ENS |
| Moro F, et al., 2014 [35] | Kufs disease | Rectal biopsies, highlighting abnormalities in the ENS |
| Lake BD, et al., 1996 [29] | JNCL | Granular osmiocentric deposits (GRODs) were observed in the ganglion neurons of the rectum |
| Smith P, et al., 1976 [28] | Different NCL types | Accumulations in ENS ganglia |
| Ziółkowska EA, et al., 2025 [36] Ziółkowska EA, et al., 2025 [37] | NCL1, NCL3 | The anatomopathological analysis of a colon autopsy from a CLN1 case revealed loss of ENS ganglia, nerve fibres, and enteric glia, resembling mouse models. Similar pathology was found in the small intestine and colon of a CLN3 case. |
| Reference | Genetic or Clinical Form | Main Findings |
|---|---|---|
| Hirz M, et al., 2017 [39] | Dog with mutations in CLN8 | Ceroid-lipofuscin deposits were observed in the ENS |
| Jolly RD, et al., 1994 [40] | Cocker spaniel dogs with NCL | Significant intestinal involvement has been described in cocker spaniel dogs with NCL, manifested by a clinical picture known as “brown bowel syndrome”, probably related to pathological accumulations |
| Minatel L, et al., 2000 [41] | Cocker spaniel dogs with NCL | The brown discolouration of the intestine, described in cocker spaniel dogs with NCL is not always present |
| Nakanishi H, et al., 2001 [42] | Ctsd−/− mouse | Inflammation and increased NO are features present not only in the CNS but also in the intestines of these mice. Therapy with NO inhibitors improves intestinal manifestations and increases the survival of these mice |
| Ziółkowska EA, et al., 2025 [36] | Ppt1−/− and Tpp1R207X/R207X mice | Both mouse models exhibited a progressive decline in bowel transit with age, despite normal early development of the ENS. In adult mice, a significant loss of myenteric plexus neurons was observed, along with alterations in enteric glial cells. Neonatal administration of gene therapy prevented bowel transit defects, mitigated neuronal loss in the ENS, and extended survival. |
| Ziółkowska EA, et al., 2025 [37]. | Cln3Δex7/8 mice | The mice exhibited intestinal smooth muscle atrophy, delayed bowel transit, and significant loss of enteric neurons and glial cells. Neonatal intravenous gene therapy improved bowel transit and largely preserved enteric neurons and glia. |
| Reference | Genetic or Clinical Form | Main Findings |
|---|---|---|
| Johnson TB, et al., 2019 [44] | Cln3−/− mice | The gut microbiota of Cln3−/− mice is markedly different compared to controls. Acidified water temporarily attenuated motor deficits, with benefits for some behavioural parameters and prevention of microglial activation in the brain. |
| Parker C, et al., 2021 [45] | Cln1R151X and Cln2R207X mice | Gut microbiota changes in Cln1R151X and Cln2R207X mice are model-specific and could affect neurological and neuropathological features |
| Kovács AD, et al., 2022 [50] | Cln1R151X mice | Acidified water had beneficial effects in Cln1R151X mice, reducing the accumulation of lysosomal material, astrocytosis, and microglial activation in specific brain areas, and improving motor capacity in behavioural tests. Acidified water also modified the composition of the gut microbiota, increasing beneficial bacteria such as Bifidobacterium and short-chain fatty acid producers, with potential neuroprotective effects. |
| Kovács AD, et al., 2023 [51] | Cln2R207X mice | Acidified water improved motor function and altered disease progression in mice. The overall composition of the gut microbiota was significantly altered by the acidified water and these changes may have contributed to the improved neurological function and delayed death, although a causal relationship could not be stated with certainty. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Della Vecchia, S.; Marchese, M.; Simonati, A.; Santorelli, F.M. Gut–Brain Interactions in Neuronal Ceroid Lipofuscinoses: A Systematic Review Beyond the Brain in Paediatric Dementias. Int. J. Mol. Sci. 2025, 26, 7192. https://doi.org/10.3390/ijms26157192
Della Vecchia S, Marchese M, Simonati A, Santorelli FM. Gut–Brain Interactions in Neuronal Ceroid Lipofuscinoses: A Systematic Review Beyond the Brain in Paediatric Dementias. International Journal of Molecular Sciences. 2025; 26(15):7192. https://doi.org/10.3390/ijms26157192
Chicago/Turabian StyleDella Vecchia, Stefania, Maria Marchese, Alessandro Simonati, and Filippo Maria Santorelli. 2025. "Gut–Brain Interactions in Neuronal Ceroid Lipofuscinoses: A Systematic Review Beyond the Brain in Paediatric Dementias" International Journal of Molecular Sciences 26, no. 15: 7192. https://doi.org/10.3390/ijms26157192
APA StyleDella Vecchia, S., Marchese, M., Simonati, A., & Santorelli, F. M. (2025). Gut–Brain Interactions in Neuronal Ceroid Lipofuscinoses: A Systematic Review Beyond the Brain in Paediatric Dementias. International Journal of Molecular Sciences, 26(15), 7192. https://doi.org/10.3390/ijms26157192