
Metabolomics and Lipidomics Explore Phenotype-Specific Molecular Signatures for Phenylketonuria

Abstract
1. Introduction
2. Results
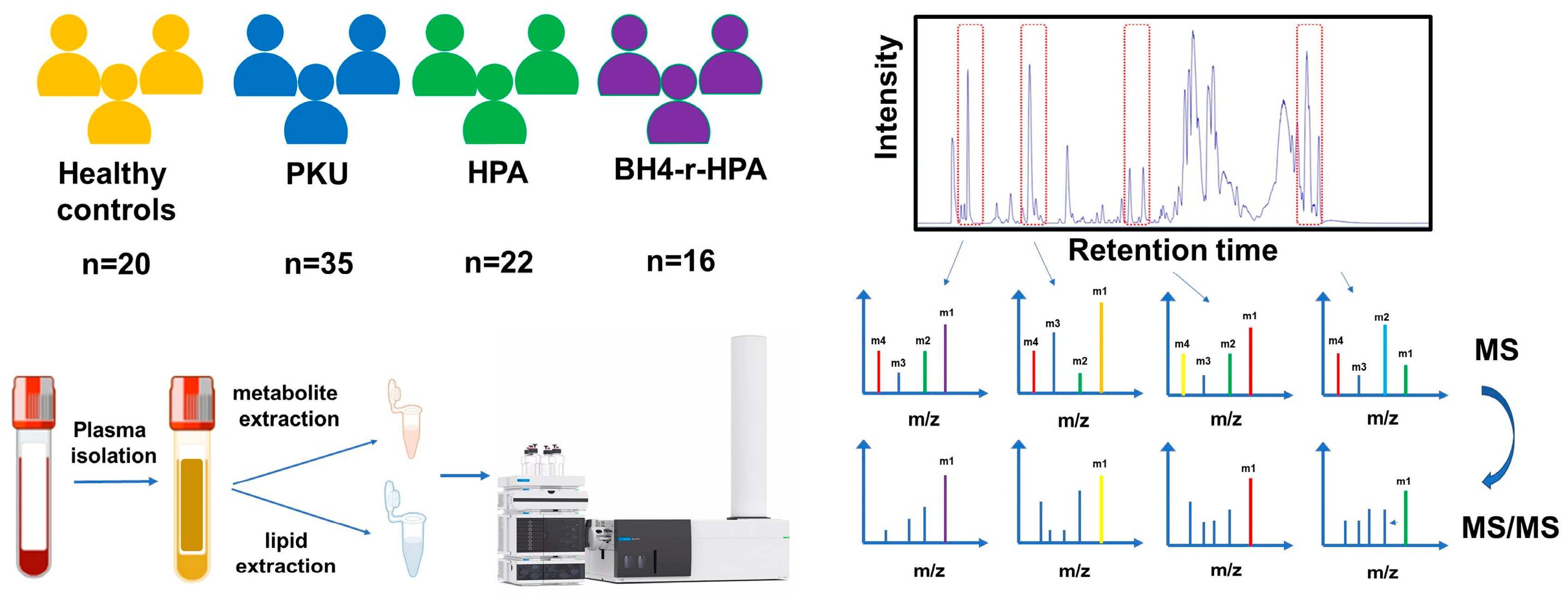
2.1. Clinical Evaluation and Patient Stratification
2.2. Quantitative Targeted Amino Acid Analysis Results
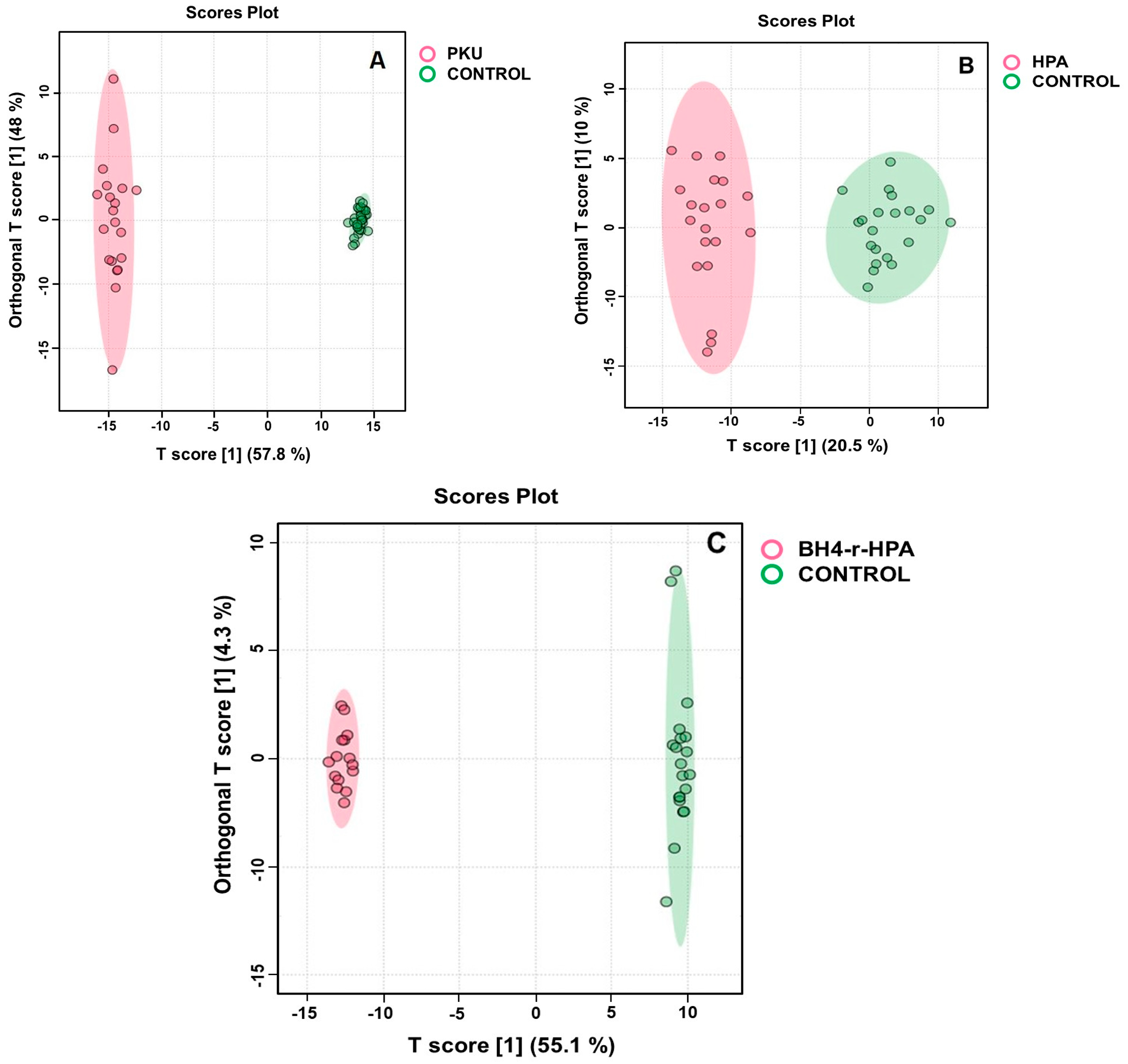
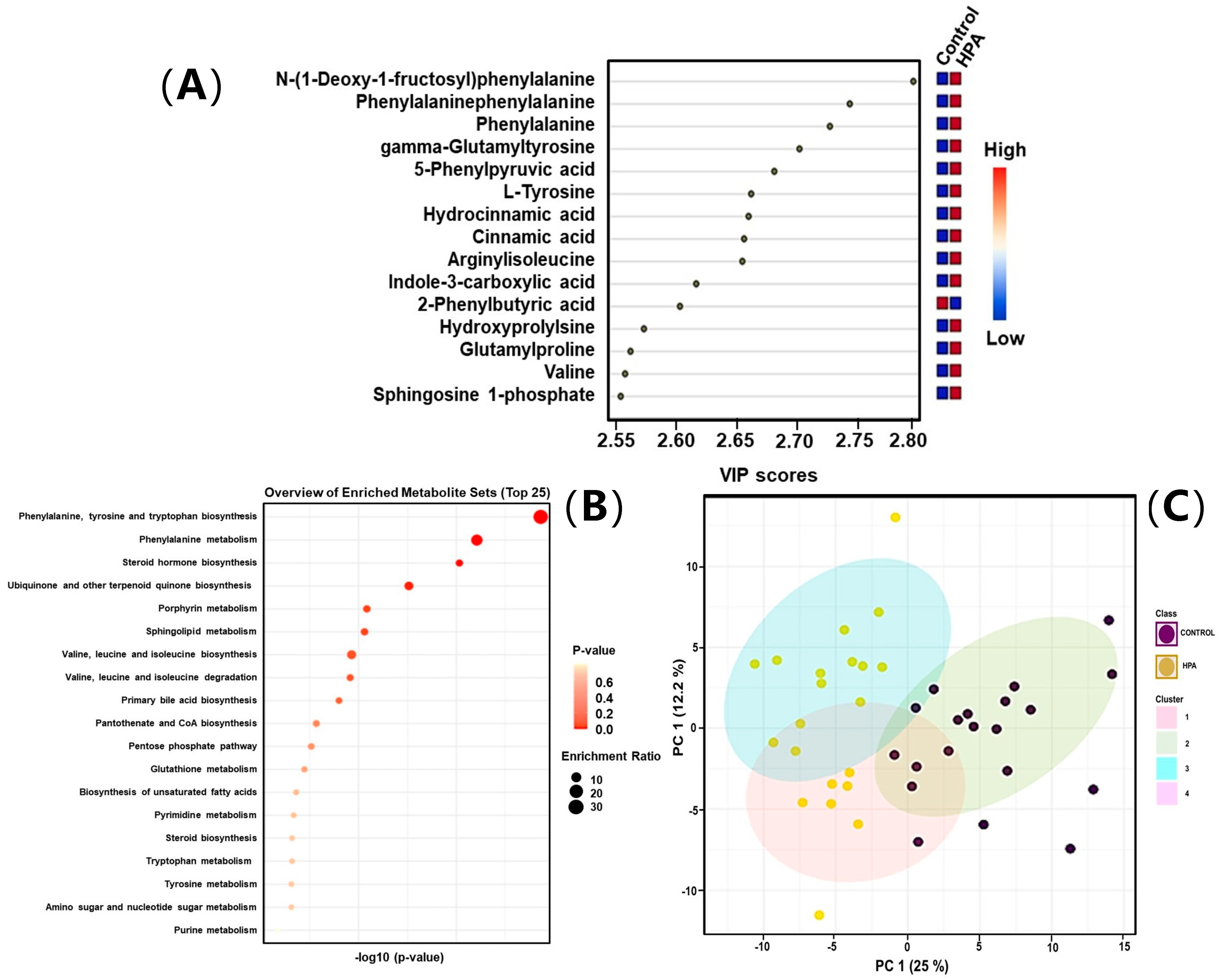
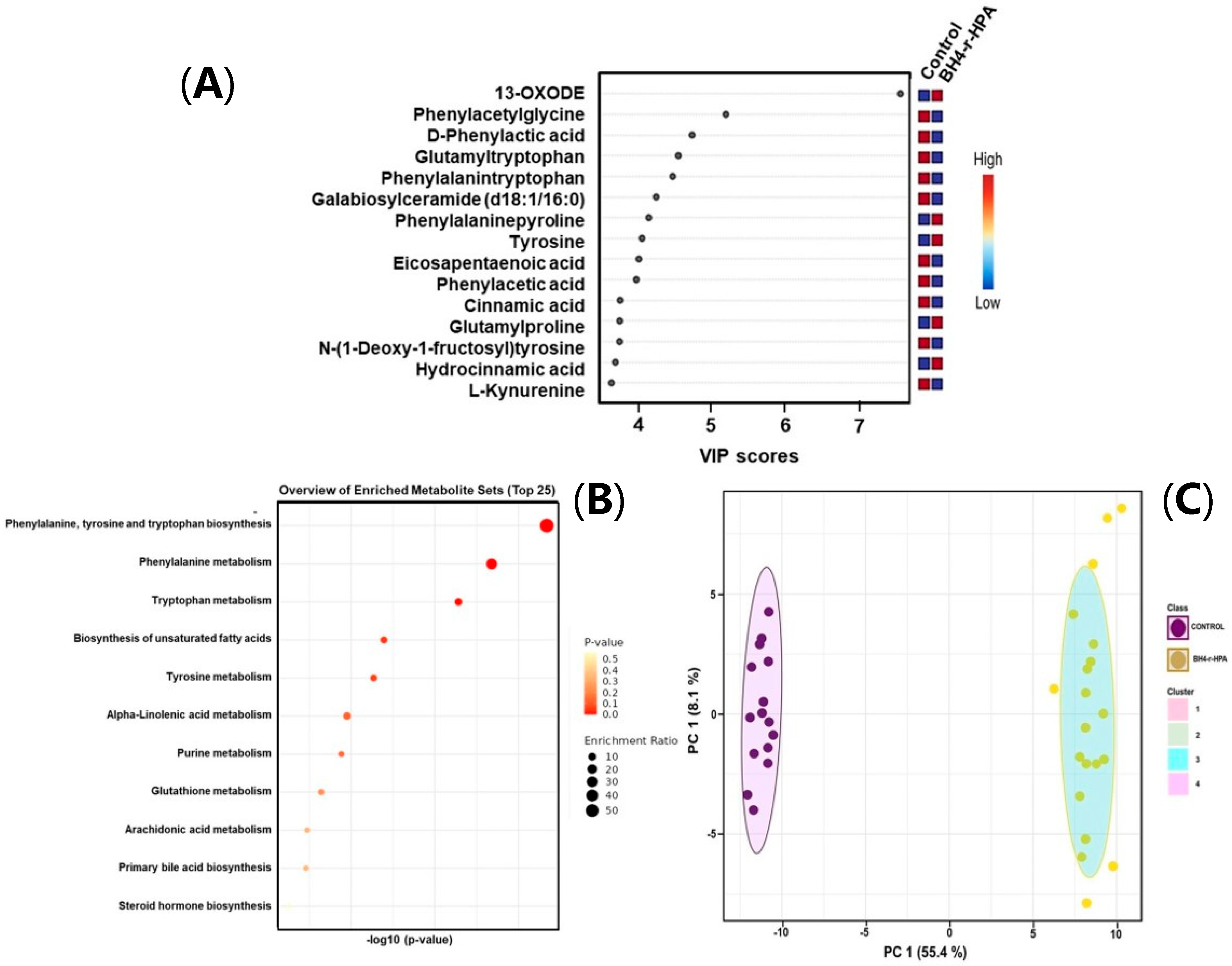
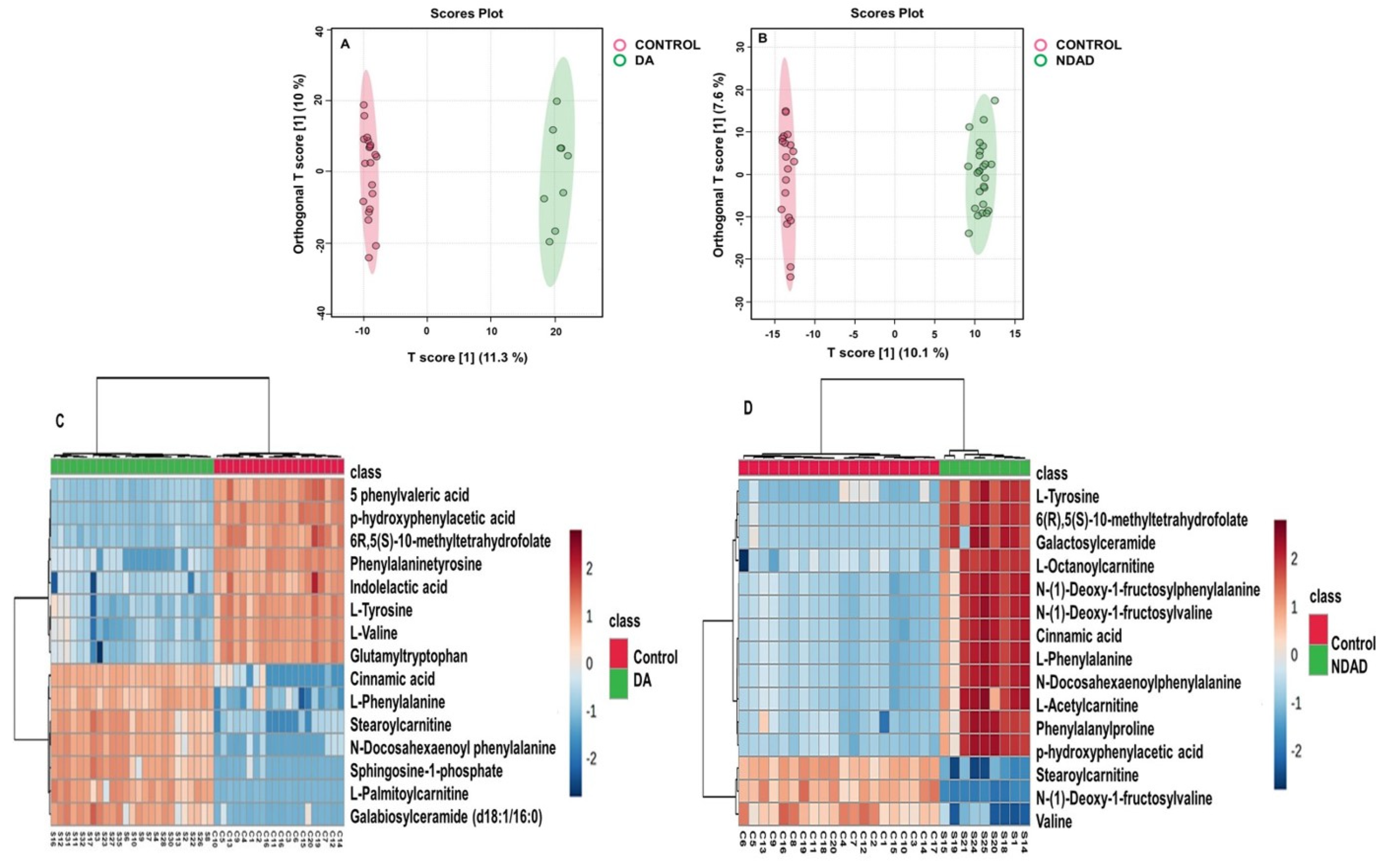
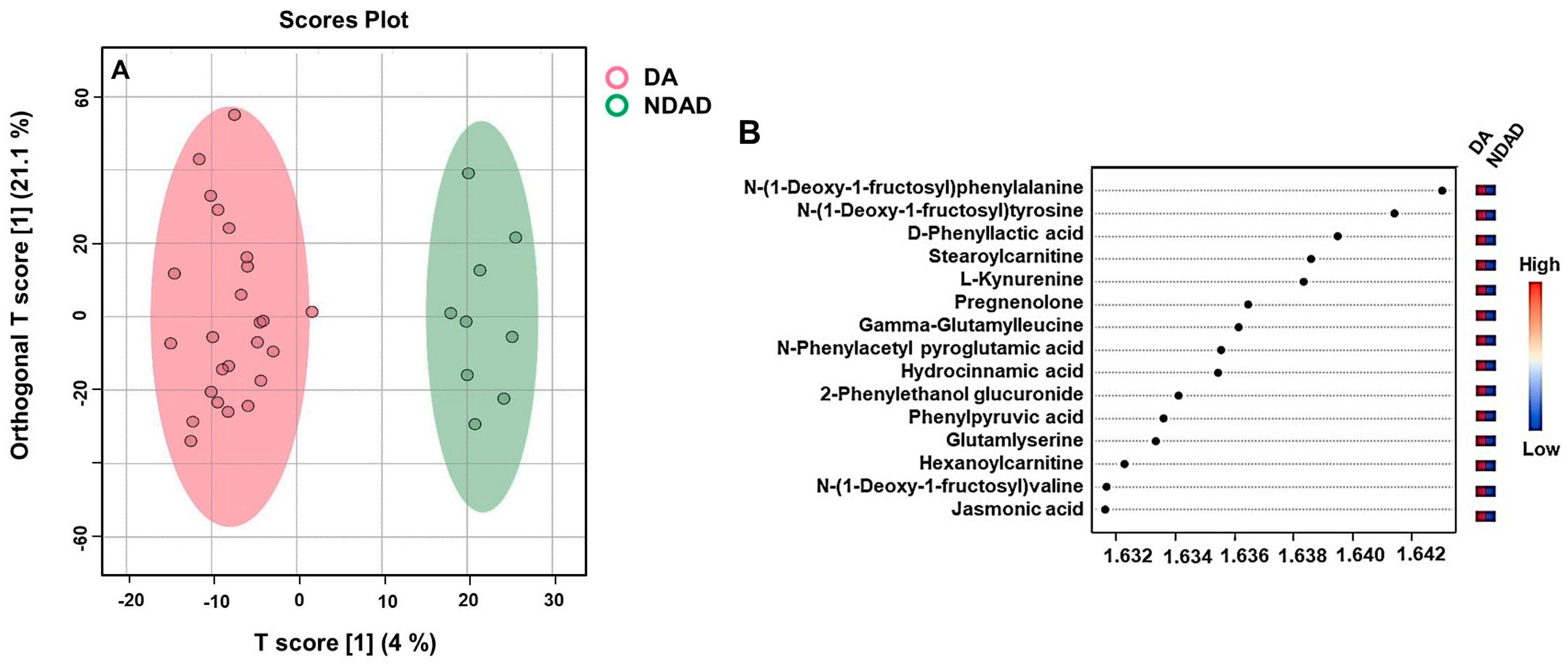
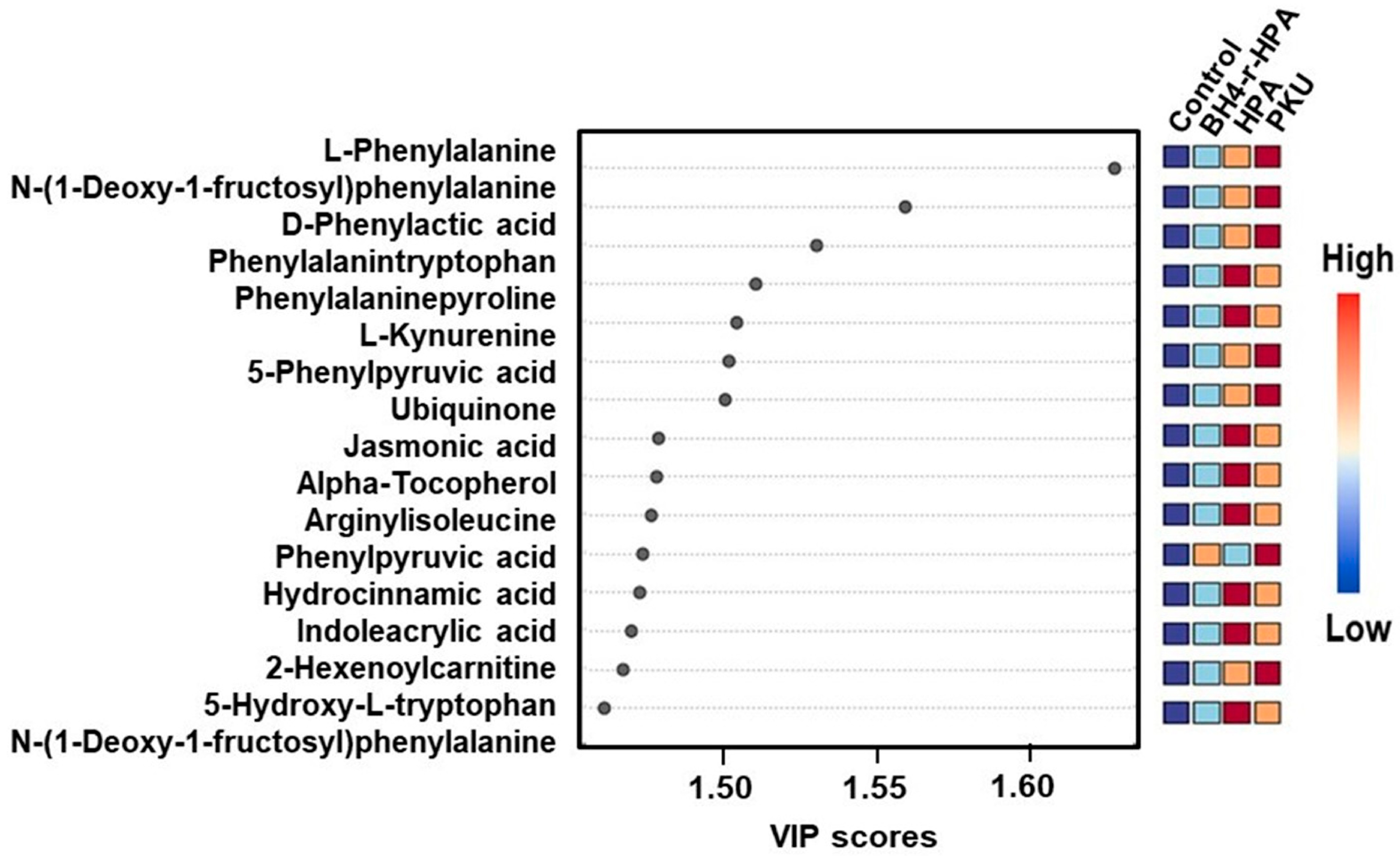
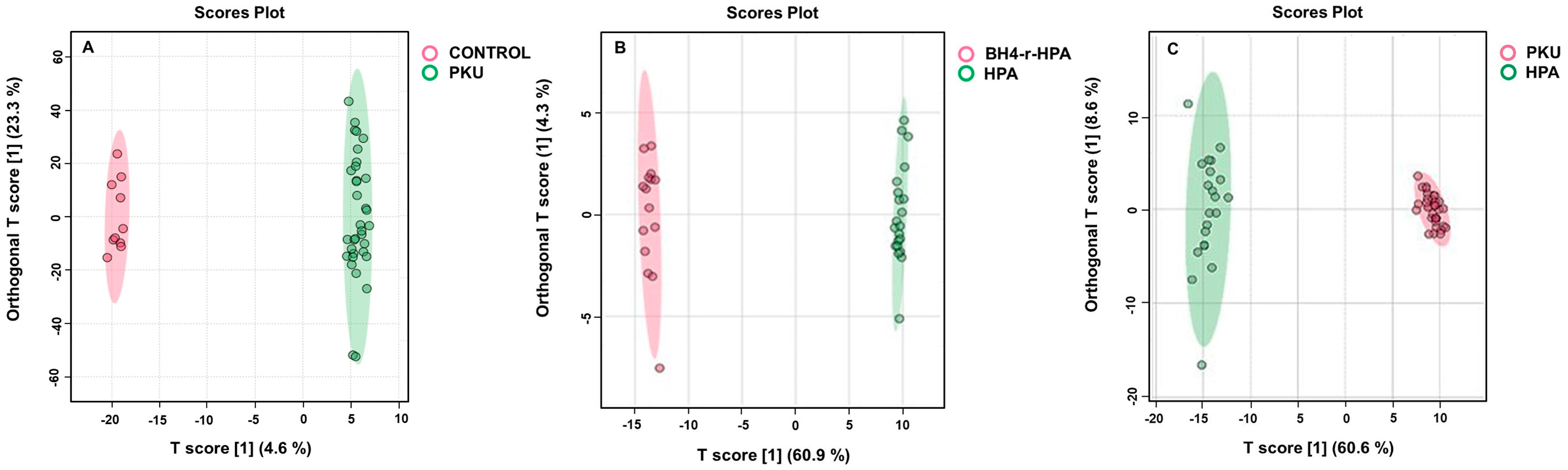
2.3. Untargeted Metabolomics Results
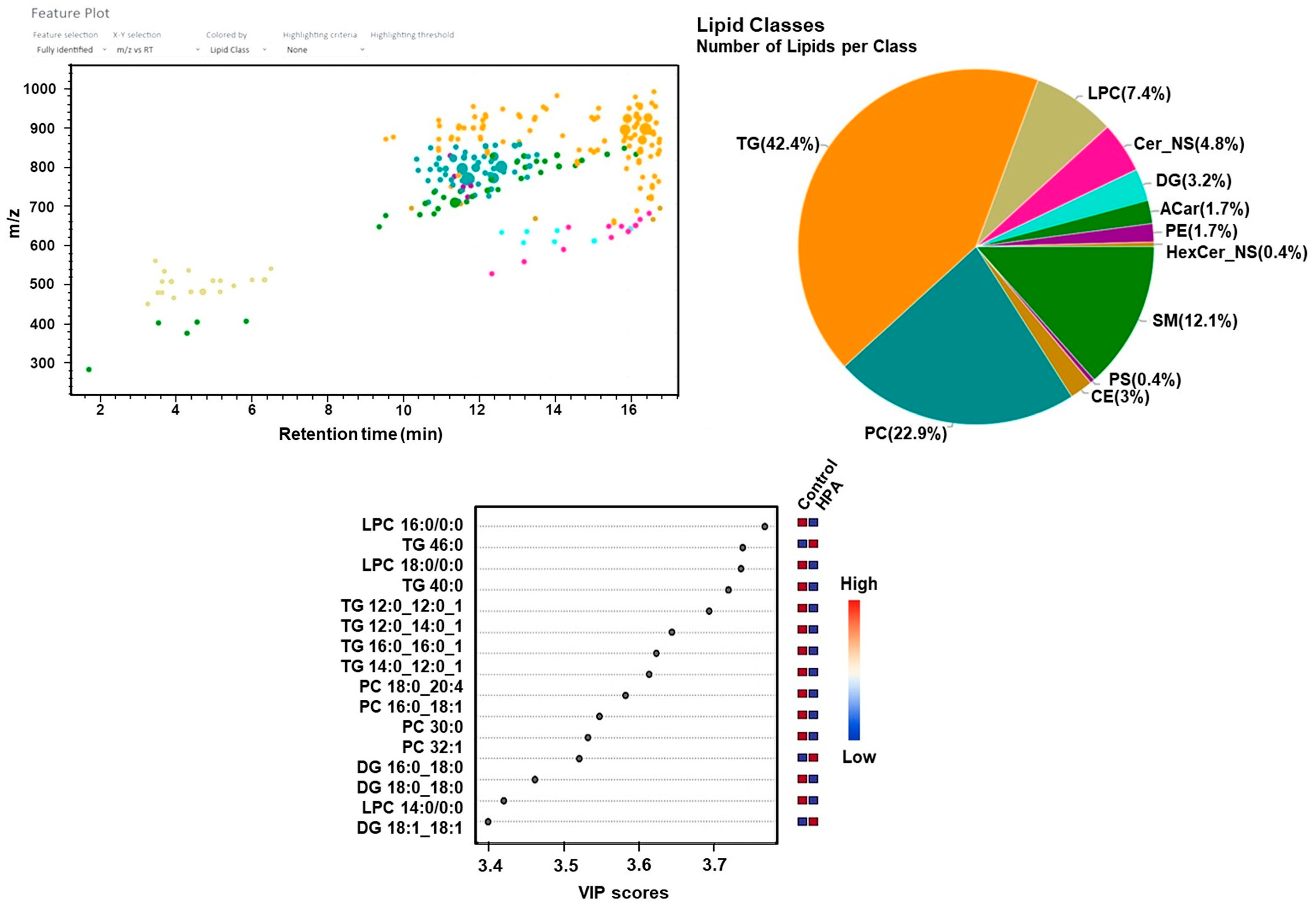
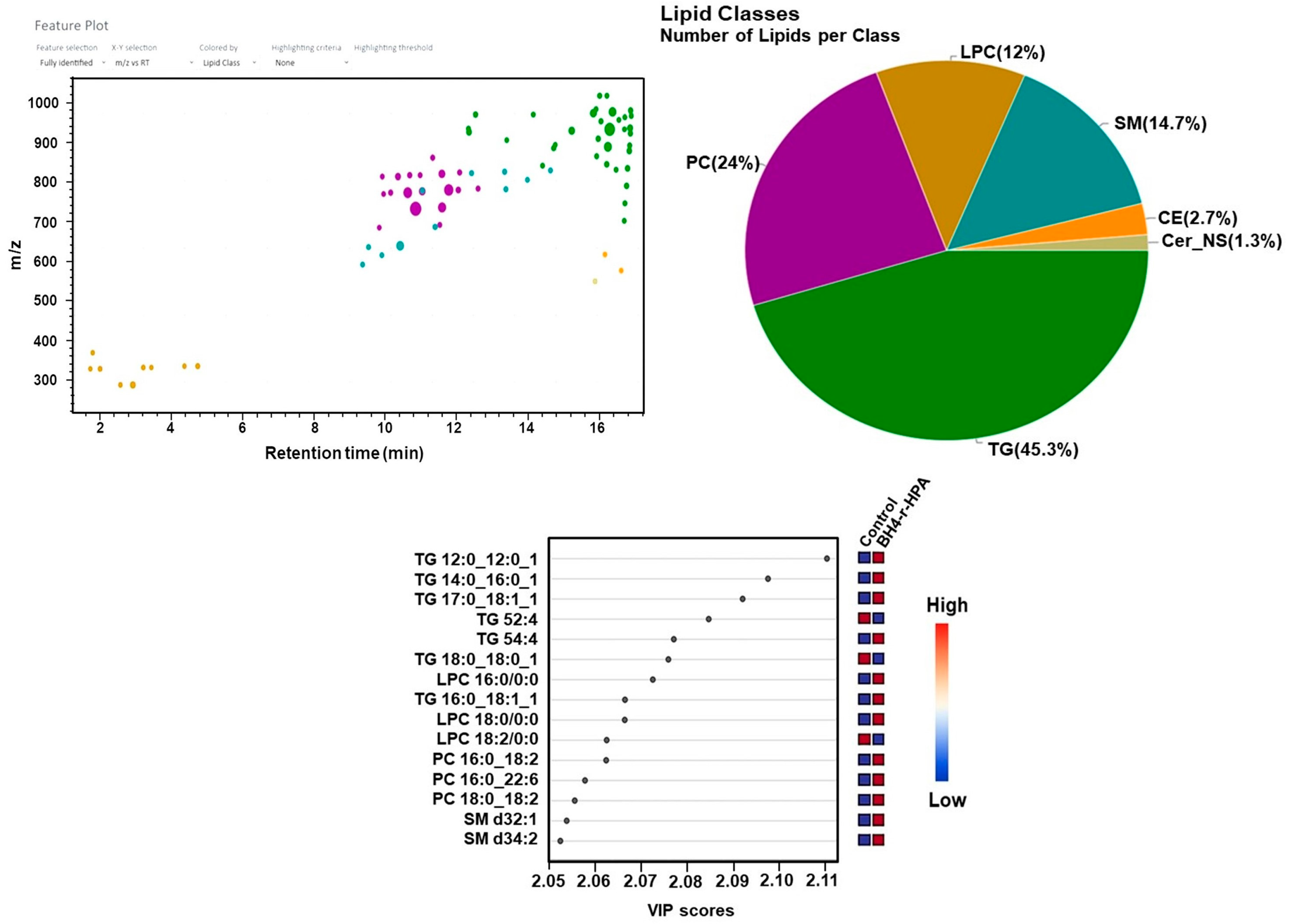
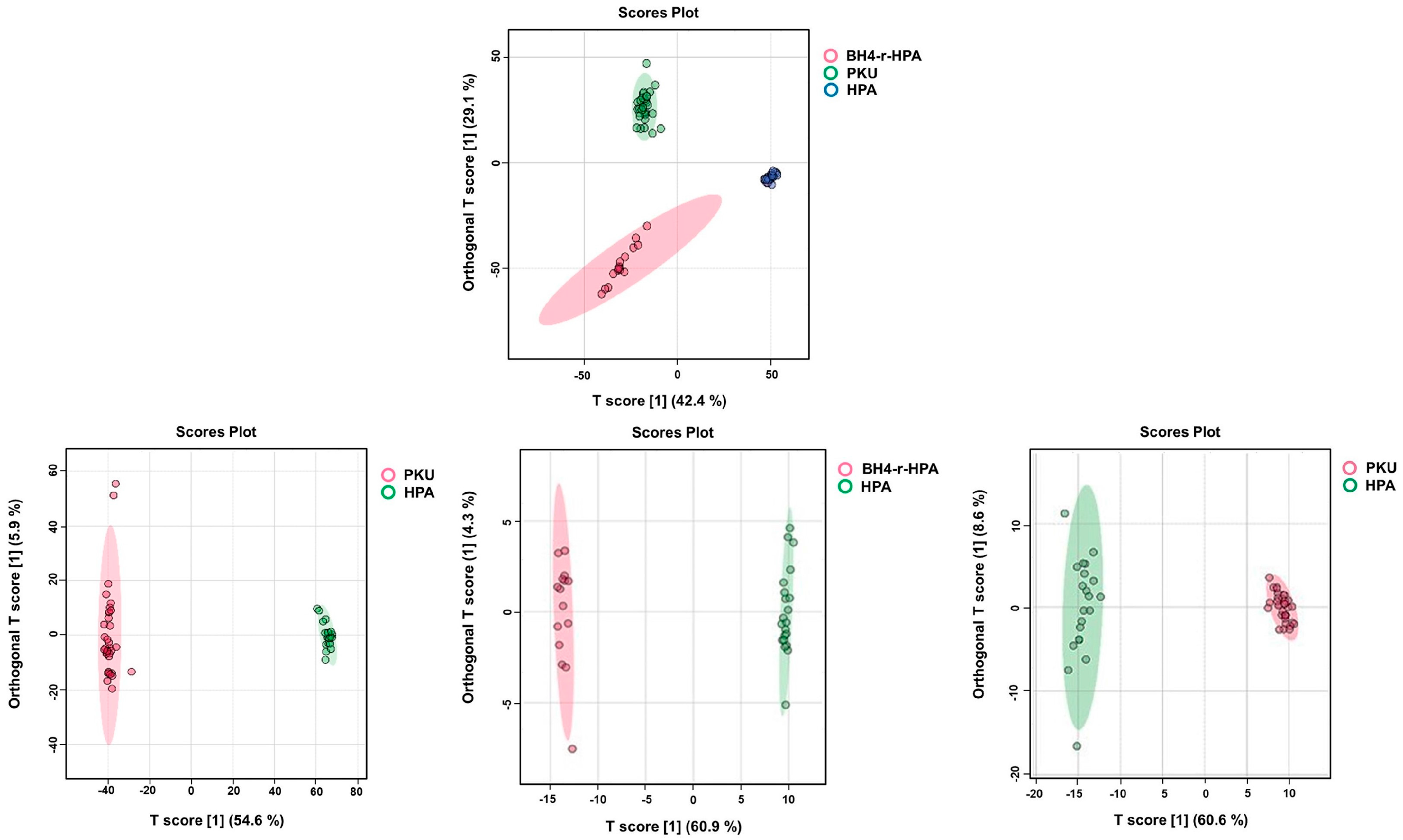
2.4. Untargeted Lipidomics Analysis Results
3. Discussion
4. Materials and Methods
4.1. Study Design Overview
4.2. Sample Collection and Plasma Extraction
4.3. Metabolite Extraction from Plasma Samples
4.4. Lipid Extraction from Plasma Samples
4.5. Characterization of Metabolite and Lipid Profiles by Liquid Chromatography–Hybrid Quadrupole Time-of-Flight Mass Spectrometry (LC–QTOF-MS)
4.6. Data Analysis
4.7. Quantitative Amino Acid Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| PKU | Phenylketonuria |
| HPA | Hyperphenylalaninemia |
| BH4-r-HPA | Tetrahydrobiopterin-Responsive Hyperphenylalaninemia |
| CPKU | Classical Phenylketonuria |
| Phe | Phenylalanine |
| Tyr | Tyrosine |
| Trp | Tryptophan |
| Phe/Tyr | Phenylalanine to Tyrosine ratio |
| BMI | Body Mass Index |
| BW | Body Weight |
| IQR | Interquartile Range |
| SD | Standard Deviation |
| LC-MS/MS | Liquid Chromatography–Mass Spectrometry |
| Q-TOF | Quadrupole Time-of-Flight |
| CE | Collision Energy |
| DDA | Data-Dependent Analysis |
| PCA | Principal Component Analysis |
| PLS-DA | Partial Least Squares Discriminant Analysis |
| OPLS-DA | Orthogonal Partial Least Squares Discriminant Analysis |
| VIP | Variable Importance in Projection |
| MS | Mass Spectrometry |
| NMR | Nuclear Magnetic Resonance |
| ESI | Electrospray Ionization |
| QC | Quality Control |
| TG | Triglyceride |
| DG | Diglyceride |
| PC | Phosphatidylcholine |
| LPC | Lysophosphatidylcholine |
| SM | Sphingomyelin |
| PUFA | Polyunsaturated Fatty Acids |
| AGE | Advanced Glycation End Products |
| ARPs | Amadori Rearrangement Products |
| HbA1c | Hemoglobin A1c (glycosylated hemoglobin) |
| IRIS | Infrared Ion Spectroscopy |
| MRM | Multiple Reaction Monitoring |
| UHPLC | Ultra-High-Performance Liquid Chromatography |
| v/v | Volume/Volume |
| m/z | Mass-to-Charge-Ratio |
References
- Elhawary, N.A.; AlJahdali, I.A.; Abumansour, I.S.; Elhawary, E.N.; Gaboon, N.; Dandini, M.; Madkhali, A.; Alosaimi, W.; Alzahrani, A.; Aljohani, F.; et al. Genetic etiology and clinical challenges of phenylketonuria. Hum. Genom. 2022, 16, 22. [Google Scholar] [CrossRef] [PubMed]
- Opladen, T.; López-Laso, E.; Cortès-Saladelafont, E.; Pearson, T.S.; Sivri, H.S.; Yildiz, Y.; Assmann, B.; Kurian, M.A.; Leuzzi, V.; Heales, S.; et al. Consensus guideline for the diagnosis and treatment of tetrahydrobiopterin (BH4) deficiencies. Orphanet J. Rare Dis. 2020, 15, 126. [Google Scholar] [CrossRef] [PubMed]
- Güttler, F.; Lou, H. Phenylketonuria and Hyperphenylalaninemia. In Inborn Metabolic Diseases: Diagnosis and Treatment; Fernandes, J., Saudubray, J.-M., Tada, K., Eds.; Springer: Berlin, Germany, 1990; pp. 161–174. [Google Scholar]
- Guldberg, P.; Rey, F.; Zschocke, J.; Romano, V.; François, B.; Michiels, L.; Ullrich, K.; Hoffmann, G.F.; Burgard, P.; Schmidt, H.; et al. A European multicenter study of phenylalanine hydroxylase deficiency: Classification of 105 mutations and a general system for genotype-based prediction of metabolic phenotype. Am. J. Hum. Genet. 1998, 63, 71–79. [Google Scholar] [CrossRef] [PubMed]
- van Wegberg, A.M.J.; MacDonald, A.; Ahring, K.; Bélanger-Quintana, A.; Beblo, S.; Blau, N.; Bosch, A.M.; Burlina, A.; Campistol, J.; Coşkun, T.; et al. European guidelines on diagnosis and treatment of phenylketonuria: First revision. Mol. Genet. Metab. 2025, 145, 109125. [Google Scholar] [CrossRef] [PubMed]
- Evers, R.A.F.; van Vliet, D.; van Spronsen, F.J. Tetrahydrobiopterin treatment in phenylketonuria: A repurposing approach. J. Inherit. Metab. Dis. 2020, 43, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Moat, S.J.; George, R.S.; Carling, R.S. Use of Dried Blood Spot Specimens to Monitor Patients with Inherited Metabolic Disorders. Int. J. Neonatal Screen. 2020, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- McWhorter, N.; Ndugga-Kabuye, M.K.; Puurunen, M.; Ernst, S.L. Complications of the Low Phenylalanine Diet for Patients with Phenylketonuria and the Benefits of Increased Natural Protein. Nutrients 2022, 14, 4960. [Google Scholar] [CrossRef] [PubMed]
- van Spronsen, F.J.; Blau, N.; Harding, C.; Burlina, A.; Longo, N.; Bosch, A.M. Phenylketonuria. Nat. Rev. Dis. Primers 2021, 7, 36. [Google Scholar] [CrossRef] [PubMed]
- Rovelli, V.; Longo, N. Phenylketonuria and the brain. Mol. Genet. Metab. 2023, 139, 107583. [Google Scholar] [CrossRef] [PubMed]
- Burlina, A.P.; Cazzorla, C.; Massa, P.; Polo, G.; Loro, C.; Gueraldi, D.; Burlina, A.B. Large Neutral Amino Acid Therapy Increases Tyrosine Levels in Adult Patients with Phenylketonuria: A Long-Term Study. Nutrients 2019, 11, 2541. [Google Scholar] [CrossRef] [PubMed]
- van Vliet, D.; van der Goot, E.; van Ginkel, W.G.; van Faassen, H.J.R.; de Blaauw, P.; Kema, I.P.; Heiner-Fokkema, M.R.; van der Zee, E.A.; van Spronsen, F.J. The increasing importance of LNAA supplementation in phenylketonuria at higher plasma phenylalanine concentrations. Mol. Genet. Metab. 2022, 135, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Thau-Zuchman, O.; Pallier, P.N.; Savelkoul, P.J.M.; Kuipers, A.A.M.; Verkuyl, J.M.; Michael-Titus, A.T. High phenylalanine concentrations induce demyelination and microglial activation in mouse cerebellar organotypic slices. Front. Neurosci. 2022, 16, 926023. [Google Scholar] [CrossRef] [PubMed]
- Rondelli, V.; Koutsioubas, A.; Di Cola, E.; Fragneto, G.; Grillo, I.; Del Favero, E.; Colombo, L.; Cantù, L.; Brocca, P.; Salmona, M. Dysmyelination and glycolipid interference caused by phenylalanine in phenylketonuria. Int. J. Biol. Macromol. 2022, 221, 784–795. [Google Scholar] [CrossRef] [PubMed]
- van Spronsen, F.J.; Hoeksma, M.; Reijngoud, D.-J. Brain dysfunction in phenylketonuria: Is phenylalanine toxicity the only possible cause? J. Inherit. Metab. Dis. 2009, 32, 46. [Google Scholar] [CrossRef] [PubMed]
- Dobrowolski, S.F.; Phua, Y.L.; Vockley, J.; Goetzman, E.; Blair, H.C. Phenylketonuria oxidative stress and energy dysregulation: Emerging pathophysiological elements provide interventional opportunity. Mol. Genet. Metab. 2022, 136, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Moat, S.J.; Schulenburg-Brand, D.; Lemonde, H.; Bonham, J.R.; Weykamp, C.W.; Mei, J.V.; Shortland, G.S.; Carling, R.S. Performance of laboratory tests used to measure blood phenylalanine for the monitoring of patients with phenylketonuria. J. Inherit. Metab. Dis. 2020, 43, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, R.; Schallert, M.; Nguyen, T.; Och, U.; Rutsch, F.; Weglage, J. Children and adolescents with phenylketonuria display fluctuations in their blood phenylalanine levels. Acta Paediatr. 2019, 108, 541–543. [Google Scholar] [CrossRef] [PubMed]
- Wada, Y.; Totsune, E.; Mikami-Saito, Y.; Kikuchi, A.; Miyata, T.; Kure, S. A method for phenylalanine self-monitoring using phenylalanine ammonia-lyase and a pre-existing portable ammonia detection system. Mol. Genet. Metab. Rep. 2023, 35, 100970. [Google Scholar] [CrossRef] [PubMed]
- Messina, M.A.; Maugeri, L.; Spoto, G.; Puccio, R.; Ruggieri, M.; Petralia, S. Fully Integrated Point-of-Care Platform for the Self-Monitoring of Phenylalanine in Finger-Prick Blood. ACS Sens. 2023, 8, 4152–4160. [Google Scholar] [CrossRef] [PubMed]
- Wild, J.; Shanmuganathan, M.; Hayashi, M.; Potter, M.; Britz-McKibbin, P. Metabolomics for improved treatment monitoring of phenylketonuria: Urinary biomarkers for non-invasive assessment of dietary adherence and nutritional deficiencies. Analyst 2019, 144, 6595–6608. [Google Scholar] [CrossRef] [PubMed]
- Moritz, L.; Klotz, K.; Grünert, S.C.; Hannibal, L.; Spiekerkoetter, U. Metabolic phenotyping in phenylketonuria reveals disease clustering independently of metabolic control. Mol. Genet. Metab. 2023, 138, 107509. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Sheng, X.; Liu, D.; Zeng, T.; Peng, Y.; Wang, Y. A GC/MS-based metabolomic approach for reliable diagnosis of phenylketonuria. Anal. Bioanal. Chem. 2015, 407, 8825–8833. [Google Scholar] [CrossRef] [PubMed]
- Cannet, C.; Bayat, A.; Frauendienst-Egger, G.; Freisinger, P.; Spraul, M.; Himmelreich, N.; Kockaya, M.; Ahring, K.; Godejohann, M.; MacDonald, A.; et al. Phenylketonuria (PKU) Urinary Metabolomic Phenotype Is Defined by Genotype and Metabolite Imbalance: Results in 51 Early Treated Patients Using Ex Vivo 1H-NMR Analysis. Molecules 2023, 28, 4916. [Google Scholar] [CrossRef] [PubMed]
- van Outersterp, R.E.; Moons, S.J.; Engelke, U.F.H.; Bentlage, H.; Peters, T.M.A.; van Rooij, A.; Huigen, M.C.D.G.; de Boer, S.; van der Heeft, E.; Kluijtmans, L.A.J.; et al. Amadori rearrangement products as potential biomarkers for inborn errors of amino-acid metabolism. Commun. Biol. 2021, 4, 367. [Google Scholar] [CrossRef] [PubMed]
- Weerd, J.C.V.; Wegberg, A.; Boer, T.S.; Engelke, U.F.H.; Coene, K.L.M.; Wevers, R.A.; Bakker, S.J.L.; Blaauw, P.; Groen, J.; Spronsen, F.J.V.; et al. Impact of Phenylketonuria on the Serum Metabolome and Plasma Lipidome: A Study in Early-Treated Patients. Metabolites 2024, 14, 479. [Google Scholar] [CrossRef] [PubMed]
- van Wegberg, A.M.J.; van der Weerd, J.C.; Engelke, U.F.H.; Coene, K.L.M.; Jahja, R.; Bakker, S.J.L.; Huijbregts, S.C.J.; Wevers, R.A.; Heiner-Fokkema, M.R.; van Spronsen, F.J. The clinical relevance of novel biomarkers as outcome parameter in adults with phenylketonuria. J. Inherit. Metab. Dis. 2024, 47, 624–635. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, Y.; Emond, P.; Schwartz, I.V.D.; Lefèvre, A.; Dupuy, C.; Chicheri, G.; Blasco, H.; Maillot, F. Multimodal Metabolomic Analysis Reveals Novel Metabolic Disturbances in Adults with Early Treated Phenylketonuria. JIMD Rep. 2025, 66, e70010. [Google Scholar] [CrossRef] [PubMed]
- Stroup, B.M.; Nair, N.; Murali, S.G.; Broniowska, K.; Rohr, F.; Levy, H.L.; Ney, D.M. Metabolomic Markers of Essential Fatty Acids, Carnitine, and Cholesterol Metabolism in Adults and Adolescents with Phenylketonuria. J. Nutr. 2018, 148, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Mütze, U.; Beblo, S.; Kortz, L.; Matthies, C.; Koletzko, B.; Bruegel, M.; Rohde, C.; Thiery, J.; Kiess, W.; Ceglarek, U. Metabolomics of Dietary Fatty Acid Restriction in Patients with Phenylketonuria. PLoS ONE 2012, 7, e43021. [Google Scholar] [CrossRef] [PubMed]
- Guerra, I.M.S.; Diogo, L.; Pinho, M.; Melo, T.; Domingues, P.; Domingues, M.R.; Moreira, A.S.P. Plasma Phospholipidomic Profile Differs between Children with Phenylketonuria and Healthy Children. J. Proteome Res. 2021, 20, 2651–2661. [Google Scholar] [CrossRef] [PubMed]
- Guerra, I.M.S.; Ferreira, H.B.; Neves, B.; Melo, T.; Diogo, L.M.; Domingues, M.R.; Moreira, A.S.P. Lipids and phenylketonuria: Current evidences pointed the need for lipidomics studies. Arch. Biochem. Biophys. 2020, 688, 108431. [Google Scholar] [CrossRef] [PubMed]
- Sanjurjo, P.; Aldamiz, L.; Georgi, G.; Jelinek, J.; Ruiz, J.I.; Boehm, G. Dietary threonine reduces plasma phenylalanine levels in patients with hyperphenylalaninemia. J. Pediatr. Gastroenterol. Nutr. 2003, 36, 23–26. [Google Scholar] [CrossRef] [PubMed]
- Beyer, B.A.; Fang, M.; Sadrian, B.; Montenegro-Burke, J.R.; Plaisted, W.C.; Kok, B.P.C.; Saez, E.; Kondo, T.; Siuzdak, G.; Lairson, L.L. Metabolomics-based discovery of a metabolite that enhances oligodendrocyte maturation. Nat. Chem. Biol. 2018, 14, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Matuszewska, E.; Matysiak, J.; Kałużny, Ł.; Walkowiak, D.; Plewa, S.; Duś-Żuchowska, M.; Rzetecka, N.; Jamka, M.; Klupczyńska-Gabryszak, A.; Piorunek, M.; et al. Amino Acid Profile Alterations in Phenylketonuria: Implications for Clinical Practice. Metabolites 2024, 14, 397. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro de Oliveira, F.; Mendes, R.H.; Dobbler, P.T.; Mai, V.; Pylro, V.S.; Waugh, S.G.; Vairo, F.; Refosco, L.F.; Roesch, L.F.W.; Schwartz, I.V.D. Phenylketonuria and Gut Microbiota: A Controlled Study Based on Next-Generation Sequencing. PLoS ONE 2016, 11, e0157513. [Google Scholar] [CrossRef] [PubMed]
- Mancilla, V.J.; Mann, A.E.; Zhang, Y.; Allen, M.S. The Adult Phenylketonuria (PKU) Gut Microbiome. Microorganisms 2021, 9, 530. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.S.; Jacob-Dolan, J.W.; Pham, V.T.T.; Sjoblom, N.M.; Scheck, R.A. The chemical language of protein glycation. Nat. Chem. Biol. 2024, 21, 324–336. [Google Scholar] [CrossRef] [PubMed]
- Santos, H.O.; Penha-Silva, N. Translating the advanced glycation end products (AGEs) knowledge into real-world nutrition strategies. Eur. J. Clin. Nutr. 2022, 76, 922–928. [Google Scholar] [CrossRef] [PubMed]
- Wyse, A.T.S.; dos Santos, T.M.; Seminotti, B.; Leipnitz, G. Insights from Animal Models on the Pathophysiology of Hyperphenylalaninemia: Role of Mitochondrial Dysfunction, Oxidative Stress and Inflammation. Mol. Neurobiol. 2021, 58, 2897–2909. [Google Scholar] [CrossRef] [PubMed]
- Yılmaz, B.K.; Baykan, A.; Kardaş, F.; Kendirci, M. Evaluation of the effect of obesity, dietary glycemic index and metabolic profiles on the cardiovascular risk in children with classical phenylketonuria. Mol. Genet. Metab. 2023, 140, 107677. [Google Scholar] [CrossRef] [PubMed]
- Cannet, C.; Pilotto, A.; Rocha, J.C.; Schäfer, H.; Spraul, M.; Berg, D.; Nawroth, P.; Kasperk, C.; Gramer, G.; Haas, D.; et al. Lower plasma cholesterol, LDL-cholesterol and LDL-lipoprotein subclasses in adult phenylketonuria (PKU) patients compared to healthy controls: Results of NMR metabolomics investigation. Orphanet J. Rare Dis. 2020, 15, 61. [Google Scholar] [CrossRef] [PubMed]
- Gürses Cila, H.E.; Dursun, A.; Vardar Acar, N.; Geçici, N.N.; Ayhan, S.; Oskay Halaçlı, S.; Özgül, R.K. Endoplasmic reticulum stress pathways and cellular death mechanisms in patients with phenylketonuria. Mol. Biol. Rep. 2025, 52, 597. [Google Scholar] [CrossRef] [PubMed]
- Matyash, V.; Liebisch, G.; Kurzchalia, T.V.; Shevchenko, A.; Schwudke, D. Lipid extraction by methyl-tert-butyl ether for high-throughput lipidomics. J. Lipid Res. 2008, 49, 1137–1146. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cohort Characteristics | PKU | HPA | BH4-Responsive HPA |
|---|---|---|---|
| n | 35 (29 classical PKU, 3 moderate PKU, 3 mild PKU) | 22 mild HPA | 16 |
| Age | 10.29 ± 5.84 (IQR: 4.45–16.12) | 7.64 ± 4.08 (IQR: 3.55–11.72) | 8.5 ± 3.87 (IQR: 4.63–12.37) |
| BMI | 20.40 ± 4.98 (IQR: 15.42–25.37) | 17.48 ± 3.51 (IQR: 13.97–20.99) | 20.09 ± 5.15 (IQR: 14.94–25.24) |
| Gender | 19 Males, 16 Females | 10 Males, 12 Females | 8 Males, 8 Females |
| Metabolic Control status | 26 (Good) 9 (Poor) | 18 (Good) 4 (Poor) | 15 (Good) 1 (Poor) |
| Phe levels at diagnosis (µM) | 1574.5 ± 686.4 (IQR: 888.67–2261.0) | 254.8 ± 93.8 (IQR: 161.0–348.7) | 406.80 ± 151.3 (IQR: 256.1–558.14) |
| BW | 41.98 ± 20.23 (IQR: 21.75–62.21) | 27.31 ± 14.15 (13.17–41.46) | 35.52 ± 19.90 (IQR: 15.62–55.42) |
| Height | 138.44 ± 24.31 (IQR: 114.13–162.75) | 120.95 ± 25.71 (IQR: 95.25–146.66) | 127.91 ± 26.56 (IQR: 101.35–154.47) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Şahiner, B.Y.; Dursun, A.; Gülbakan, B. Metabolomics and Lipidomics Explore Phenotype-Specific Molecular Signatures for Phenylketonuria. Int. J. Mol. Sci. 2025, 26, 7171. https://doi.org/10.3390/ijms26157171
Şahiner BY, Dursun A, Gülbakan B. Metabolomics and Lipidomics Explore Phenotype-Specific Molecular Signatures for Phenylketonuria. International Journal of Molecular Sciences. 2025; 26(15):7171. https://doi.org/10.3390/ijms26157171
Chicago/Turabian StyleŞahiner, Buket Yurteri, Ali Dursun, and Basri Gülbakan. 2025. "Metabolomics and Lipidomics Explore Phenotype-Specific Molecular Signatures for Phenylketonuria" International Journal of Molecular Sciences 26, no. 15: 7171. https://doi.org/10.3390/ijms26157171
APA StyleŞahiner, B. Y., Dursun, A., & Gülbakan, B. (2025). Metabolomics and Lipidomics Explore Phenotype-Specific Molecular Signatures for Phenylketonuria. International Journal of Molecular Sciences, 26(15), 7171. https://doi.org/10.3390/ijms26157171