1. Introduction
Pseudomonas is one of the most extensively studied microorganisms and represents the genus with the highest number of species among Gram-negative bacteria.
Pseudomonas spp. are strictly aerobic organisms commonly found in soil, water, and marine environments [
1]. These bacteria may constitute part of the normal human flora, but they are predominantly isolated in association with nosocomial opportunistic infections [
2]. The pathogenic mechanisms of
Pseudomonas-induced disease are complex and involve the production of extracellular proteases and other toxic proteins, as well as hemolysins, endotoxins, and exotoxin A [
3].
It is well known that infections caused by
Pseudomonas spp. are particularly challenging to treat due to their intrinsic resistance to many conventional antibiotics [
4]. Consequently, treatment of infections caused by these pathogens generally requires the administration of two or three antimicrobial agents or the use of antibiotics that are infrequently utilized in routine clinical practice, such as polymyxins, including colistin. Therefore, it is justified to take an interdisciplinary approach to combating infections caused by strains of the
Pseudomonas genus and to supplement traditional antibiotic therapy with antibacterial photodynamic inactivation (aPDI), which represents a promising method for treating infections caused by Gram-negative pathogens [
5].
Photodynamic inactivation utilizes non-toxic molecules known as a photosensitizer (PS) along with low-intensity visible light. When combined with oxygen, this interaction generates toxic reactive species [
6]. One of aPDI’s key benefits is its dual selectivity: the photosensitizer can be directed specifically to the target cells or tissues, and the light can be precisely focused on the affected area.
Photodynamic inactivation has been proven to be effective in the killing of bacteria from the genus
Pseudomonas, and several approaches to light-induced eradication of these pathogens have been described to date. For example, the effectiveness of aPDI utilizing methylene blue (MB) against planktonic
P. aeruginosa was evaluated at various MB concentrations and different light exposure parameters to identify the most effective combination for bacterial inactivation. The obtained results indicated the greatest reduction in bacterial viability—corresponding to a 3.48 and 4.32 log reduction—at light doses of 90 and 108 J cm
−2, respectively, with MB concentrations of 500 μg mL
−1 and 250 μg mL
−1 [
7]. In another study, the combined effect of curcumin as a photosensitizer with polymyxin B on
P. aeruginosa was evaluated, revealing that the synergistic action of curcumin and polymyxin B effectively killed these bacteria [
8]. An in vitro study assessed the efficacy of chlorin e6-PDI in combating suspensions of multiple bacterial species, including
P. aeruginosa, demonstrating a significant reduction in viable bacteria following treatment with chlorin e6 at a concentration of 10 μM and a light dose of 20 J cm
−2 [
9].
An alternative approach involves the combination of inorganic salts with photosensitizers. Michael Hamblin investigated the effects of selected inorganic salts—such as sodium azide, potassium iodide, potassium bromide, and thiocyanate—in conjunction with antimicrobial photodynamic inactivation [
10]. A significant enhancement was demonstrated in the antimicrobial efficacy of these compounds. However, the authors also noted certain limitations, including toxicity, reduced oxidation capacity, and moderate effectiveness. Ultimately, the findings indicated that among the tested salts, potassium iodide was the most effective and non-toxic to mammalian cells at concentrations up to 100 mM [
11]. Similar studies were conducted by Vecchio et al. [
12]. The researchers employed aPDI in combination with potassium iodide (KI), demonstrating a synergistic antimicrobial effect both in vitro and in vivo on bacterial cultures.
The identification that 5-aminolevulinic acid (5-ALA) can serve as a natural precursor to the photosensitizer has facilitated numerous applications wherein this amino acid or its ester derivatives are regarded as prodrugs requiring metabolic conversion into endogenous porphyrins-like compounds to attain their active photosensitizing properties [
13]. The Food and Drug Administration has approved 5-ALA, in conjunction with light irradiation, for the treatment of certain carcinomas [
14]. Achieving an adequate photosensitizer concentration for effective lesion destruction necessitates exogenous administration of 5-ALA, and, consequently, in this therapy, 5-ALA is applied topically in the form of a cream or gel approximately 3–5 h prior to illumination. Additionally, 5-ALA can be administered orally, via infusion, or through inhalation routes. This amino acid exhibits several advantages over other photosensitizers. Due to its natural origin, it is biocompatible and minimizes adverse reactions, which translates into reduced toxicity and a lower risk of side effects. It is considered that the price of this drug is also satisfactory.
One significant area of research pertains to the role of glucose in photodynamic therapy using 5-ALA as a precursor of the intracellular photosensitizer. Given that most studies utilizing this amino acid have focused on the effective photoinactivation of tumors, the majority of existing research findings relate to this group of pathogenic cells. It is well established that tumor cells exhibit higher glucose consumption compared to normal cells and, consequently, this preferential uptake has been exploited in light-induced therapy through the use of photosensitizer conjugates with glucose, facilitating targeted delivery to tumor-affected sites. For example, the synergistic effect of administering glucose in conjunction with 5-ALA in tumor inactivation was investigated by Nelson et al. [
15], and the results obtained demonstrated that glucose, when combined with PDT, led to a higher proportion of cured animals compared to the use of PDT alone. Furthermore, the findings indicated that this approach required a lower light dose and could potentially allow for a reduction in the photosensitizer dosage, thereby achieving optimal therapeutic response with minimal side effects in cancer treatment.
Ambiguous data are available regarding the antibacterial efficacy of photodynamic inactivation utilizing 5-aminolevulinic acid (5-ALA) against
P. aeruginosa [
16,
17,
18,
19], and to the best of our knowledge, there are no such data pertaining to
P. putida. Furthermore, the role of glucose in the light-induced inactivation of prokaryotic cells mediated by this amino acid as a prodrug has not been previously demonstrated.
This study evaluated the application of 5-ALA in antimicrobial photodynamic inactivation targeting two clinically significant pathogens: P. aeruginosa and P. putida. An effective strategy to enhance the light-mediated inactivation of these bacteria was proposed, involving the exogenous administration of glucose in conjunction with this amino acid. The impact of multiple photodynamic inactivation treatments on the reduction in certain virulence factors and the emergence of tolerance to aPDI in these pathogens was also examined.
3. Discussion
In these studies, two Gram-negative bacterial species were used:
Pseudomonas aeruginosa and
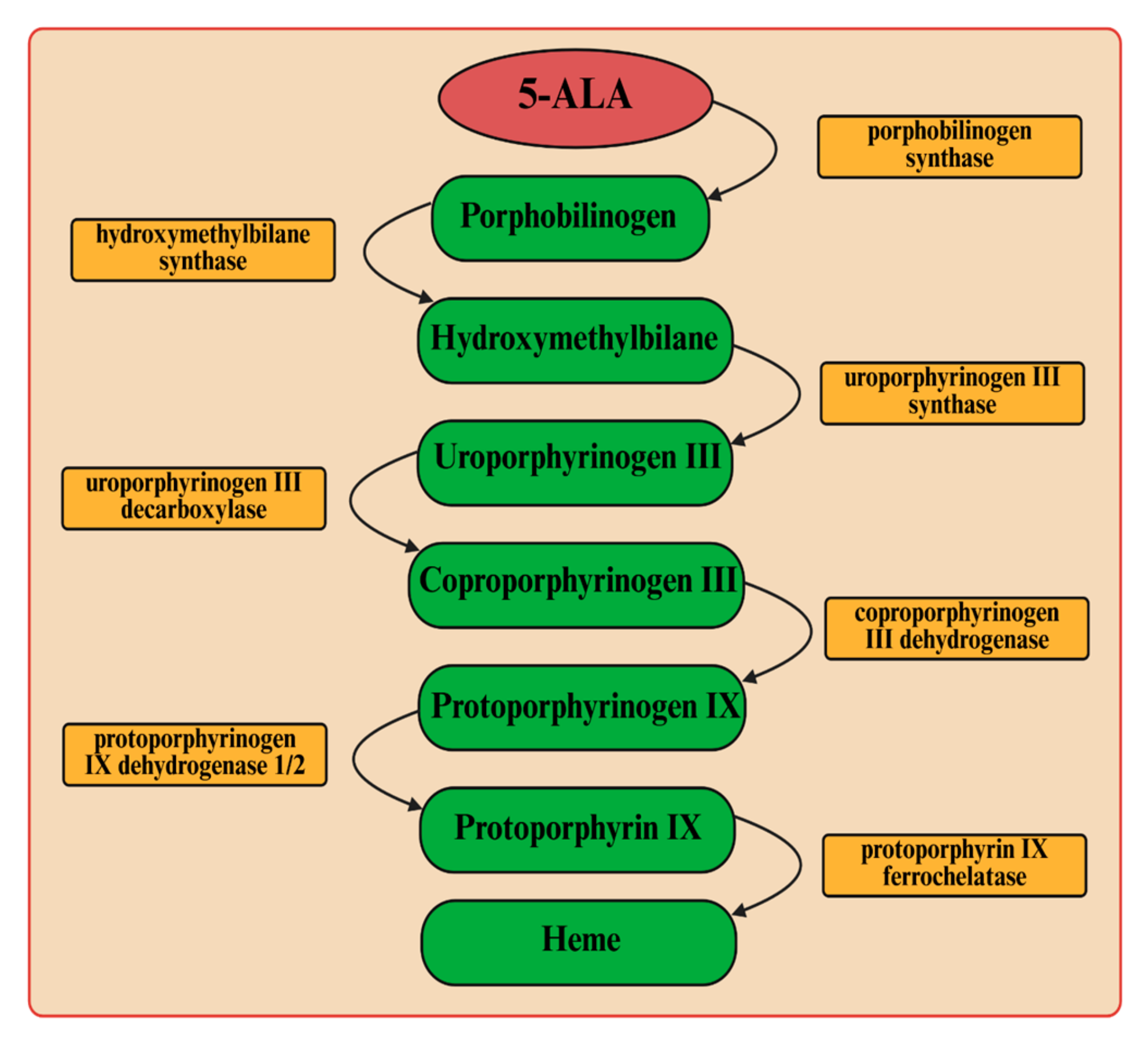
Pseudomonas putida. To combat these pathogenic bacteria, we employed light-induced inactivation (aPDI), a technique with a longstanding history that has garnered renewed interest in recent years owing to the significant increase in bacterial antibiotic resistance. The endogenous production of porphyrin-like compounds from 5-ALA was used as a tool to enable bacterial control (
Figure 6). These molecules act as photosensitizers and, upon excitation, interact with molecular oxygen (in its triplet state (
3O
2)), which is the predominant form of oxygen in biological systems. The energy transfer from the excited triplet state of photosensitizer to molecular oxygen results in the formation of singlet oxygen (
1O
2), a highly reactive molecule [
14,
20]. Singlet oxygen reacts with various biological substrates, leading to oxidative damage of cellular biomolecules, including lipids, proteins, and nucleic acids [
21]. It is important to note that singlet oxygen has a very limited diffusion radius—approximately 0.02 μm—and a short lifetime within biological environments. These characteristics enable its use for localized therapeutic effects, minimizing damage to surrounding healthy cells and tissues [
22].
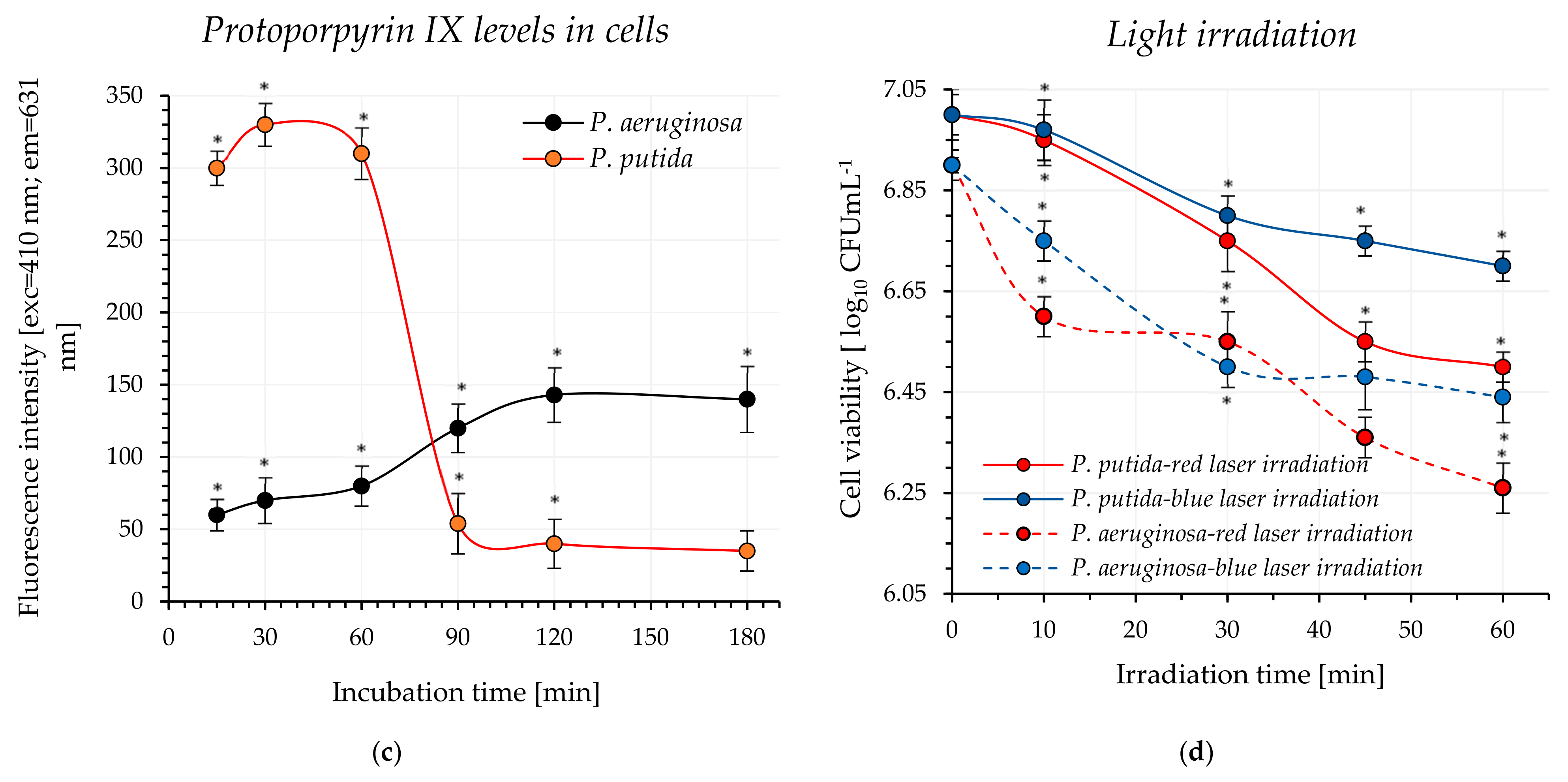
In our experiments, light at wavelengths of 405 nm and 635 nm was used for photodynamic destruction of
P. aeruginosa and
P. putida. It is assumed that, in the case of Gram-negative bacteria, intracellular protoporphyrin IX is responsible for cell photosensitization using visible light [
13]. Protoporphyrin IX is an endogenous fluorescent heterocyclic organic molecule that occurs in all living cells. This molecule is an essential precursor with specific physiological functions in bacteria, such as oxygen transport and storage via heme (iron/zinc-PPIX). It is also a ubiquitous prosthetic group of proteins such as cytochrome, catalases, peroxidases, and nitrate reductase. It was previously shown that the UV–visible absorption spectra of protoporphyrin IX are characterized by a prominent Soret band and four weaker Q bands (QI, QII, QIII, and QIV) [
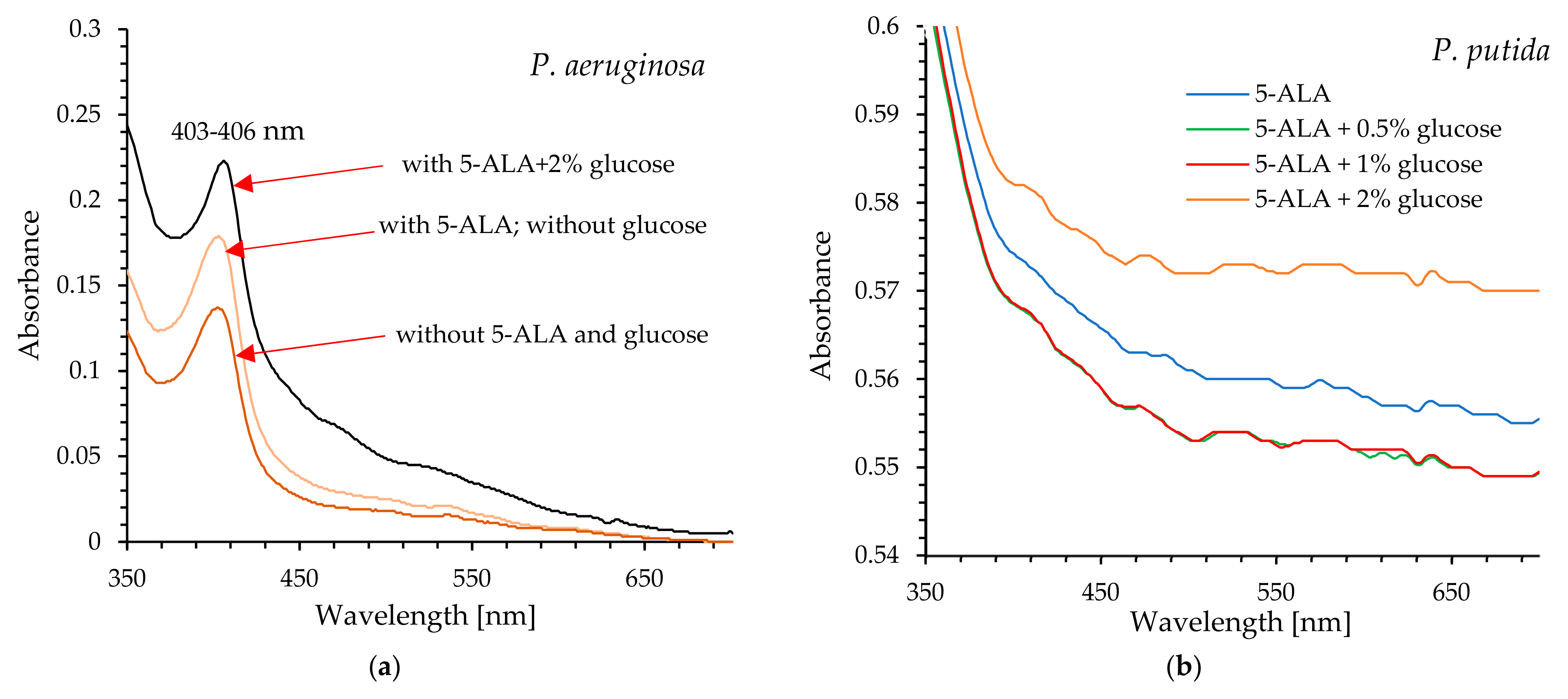
23]. The Soret band is centered around 406 nm, while the Q bands are observed in the range of approximately 532–641 nm (their exact positions depend on the solvent used).
The results obtained showed that red light (without the addition of an exogenous photosensitizer) induced bacterial cell death, with the efficacy depending on the microorganism and the light dose. The bacterial mortality rate ranged from approximately 55% to 77%, with P. aeruginosa exhibiting greater sensitivity to red light.
A blue light was also effective in destroying the studied pathogens, which was not an unexpected finding. It is well known that blue light (aBL) within the wavelength range of 400–470 nm caused inactivation of bacteria, and this antimicrobial approach, particularly in the context of skin infections, is being increasingly explored [
24]. Numerous studies have confirmed the efficacy of aBL in bacterial eradication but the precise mechanism of BL action remains unclear. It is widely accepted that aBL stimulates endogenous porphyrin-like compounds and activates the production cascade of reactive oxygen species (ROS). These toxic ROS interact with various bacterial cellular components, ultimately leading to cell lysis.
Our study primarily focused on evaluating the efficacy of pathogen eradication using laser blue and red light, employing 5-ALA as an exogenous precursor of the active “biocide”(protoporphyrin IX). It was determined that achieving the targeted mortality rate of
P. aeruginosa (99.999%) required a minimum of 30 min of exposure to blue light, corresponding to a light dose of 81 J cm
−2. In the case of red light, even 45 min of irradiation, delivering a dose of 143 J cm
−2, did not result in the desired eradication effect. One possible reason for the limited inactivation effectiveness is the weak absorption of PpIX in the red spectral region compared to its absorption near the Soret band. This assumption was previously confirmed in
Propionibacterium acnes (
Cutibacterium acnes), which accumulates high levels of PpIX [
25], but inactivation of this pathogen with red light required high light doses to achieve a significant decrease in viability [
26]. Similar results were also observed with another Gram-negative bacterium,
E. coli [
27]. Hsieh et al. [
18] observed a significant decrease in bacterial viability for
P. aeruginosa, where 5.0 mM 5-ALA was photosensitized by accumulating a red light dose of 162 J cm
−2.
Analysis of the photoinactivation efficiency of
P. aeruginosa demonstrated by other researchers has shown that aPDI based on 5-ALA is an effective method for combating these pathogens. However, it requires relatively high concentrations of this amino acid as a precursor to the active photosensitizer and substantial light doses. For example, Lee et al. [
16] reported that pre-incubation of
P. aeruginosa with 5-ALA at a concentration of 20 mM combined with red light irradiation at 630 nm (light dose of 240 J cm
−2) resulted in lethal photoinactivation. Similarly, using 7.5 mM 5-ALA with a light dose of 360 J cm
−2 also achieved effective inactivation.
In a different study [
19], the authors demonstrated that effective inactivation of
P.
aeruginosa via visible light-induced photoinactivation (using a halogen-tungsten lamp) required a 5-aminolevulinic acid (5-ALA) concentration of 40 mM and a light dose of 142 J cm
−2.
When P. putida was used in our experiments, it was observed that, regardless of exposure duration up to 45 min to either blue or red light (light dose of 121 J cm−2 and 143 J cm−2, respectively), the mortality rate did not reach the 99.999% threshold. To the best of our knowledge, no research results have been published to date regarding the use of 5-ALA in the photodynamic eradication of P. putida; therefore, the data obtained cannot be compared with the results presented by other authors. This is likely due to the fact that this microorganism has not been identified as a medical concern.
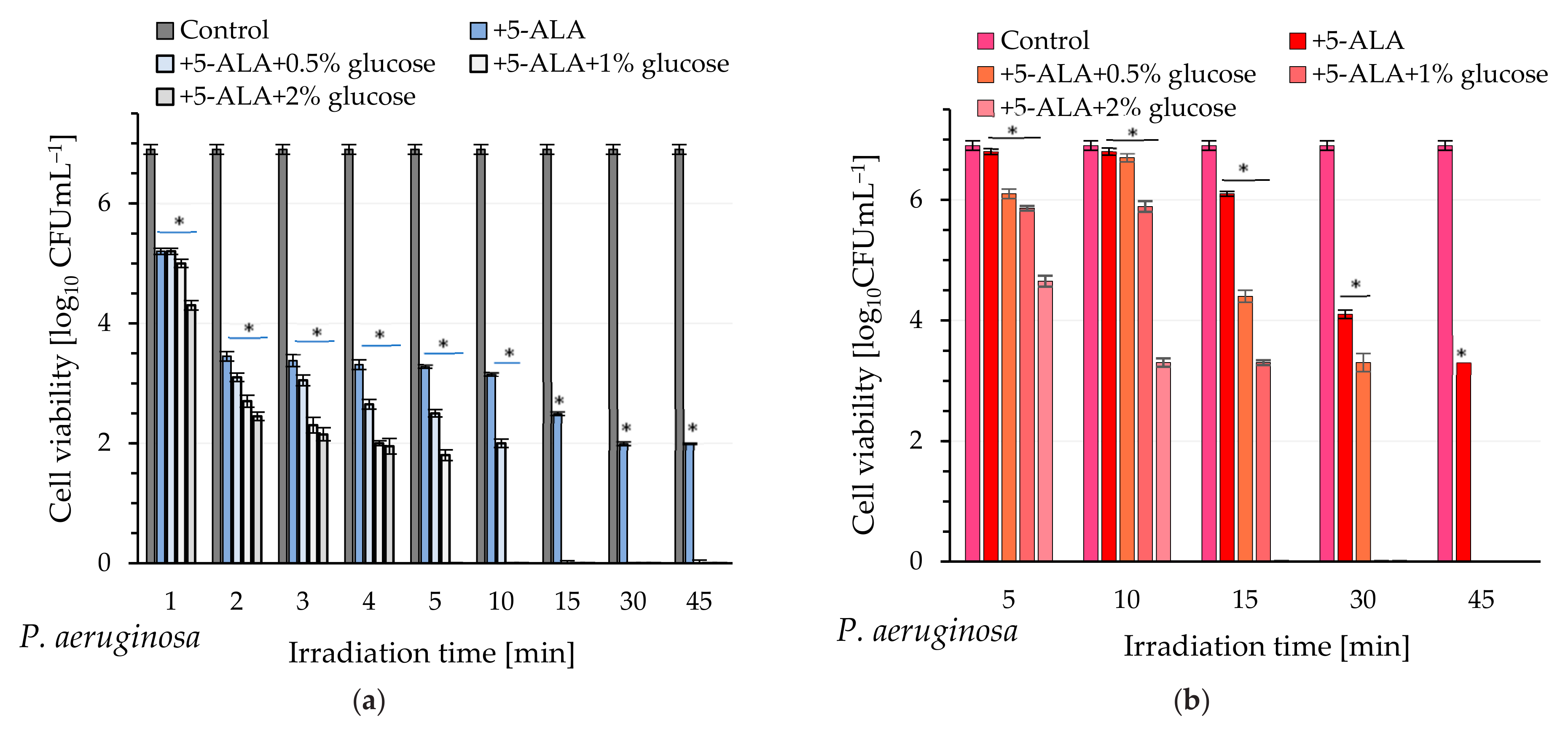
Inspired by our previous discoveries [
28], this study proposed a simple method to enhance the efficiency of aPDI, involving the exogenous delivery of not only 5-ALA but also glucose to the cells. The light-induced mortality of the examined pathogens, which depended on the glucose concentration and the duration of light exposure, was studied. It was demonstrated that incubation of
P. aeruginosa cells with 5-ALA in the presence of 2% glucose and exposing these bacteria to blue light resulted in a lethal effect (visible cell count below the detection level) after just 5 min (light dose of 13.5 J cm
−2). Lower glucose concentrations (1% and 0.5%) also induced a lethal effect, but the required light dose was in the range of 27–40 J cm
−2. A similar phenomenon was observed with red light, but achieving a lethal effect required prolonged exposure times of 15 and 30 min for glucose concentrations of 1% and 0.5%, respectively (light dose of 48–95 J cm
−2).
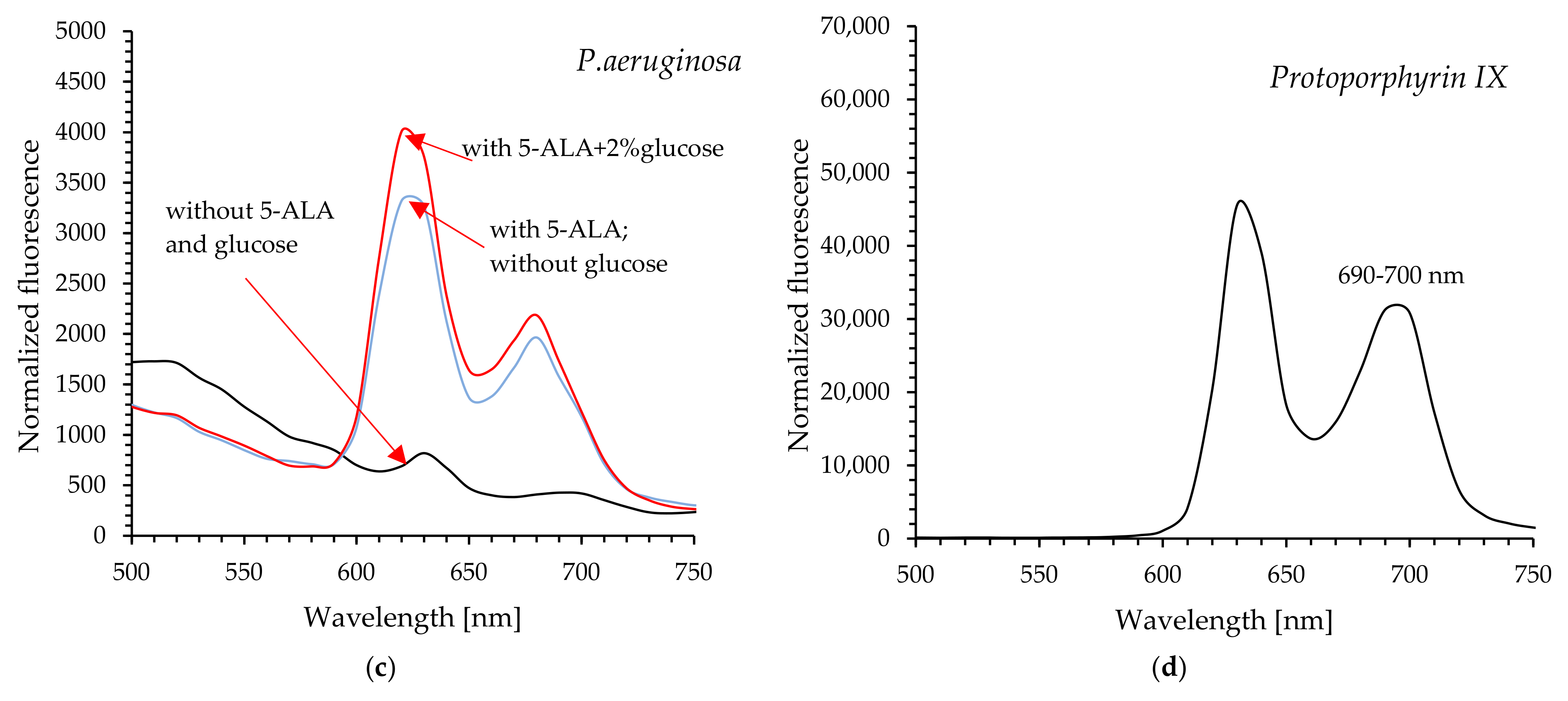
The acquisition of cell-free lysates and the analysis of their UV-Vis and fluorescence spectra clearly showed that glucose increased the level of protoporphyrin IX in P. aeruginosa cells, and consequently, enhanced photodynamic inactivation of this pathogen was possible after exposure to light. It seems obvious that excitation of protoporphyrin IX at the Soret band maximum results in high efficacy of bacterial killing by blue light (compared to red light).
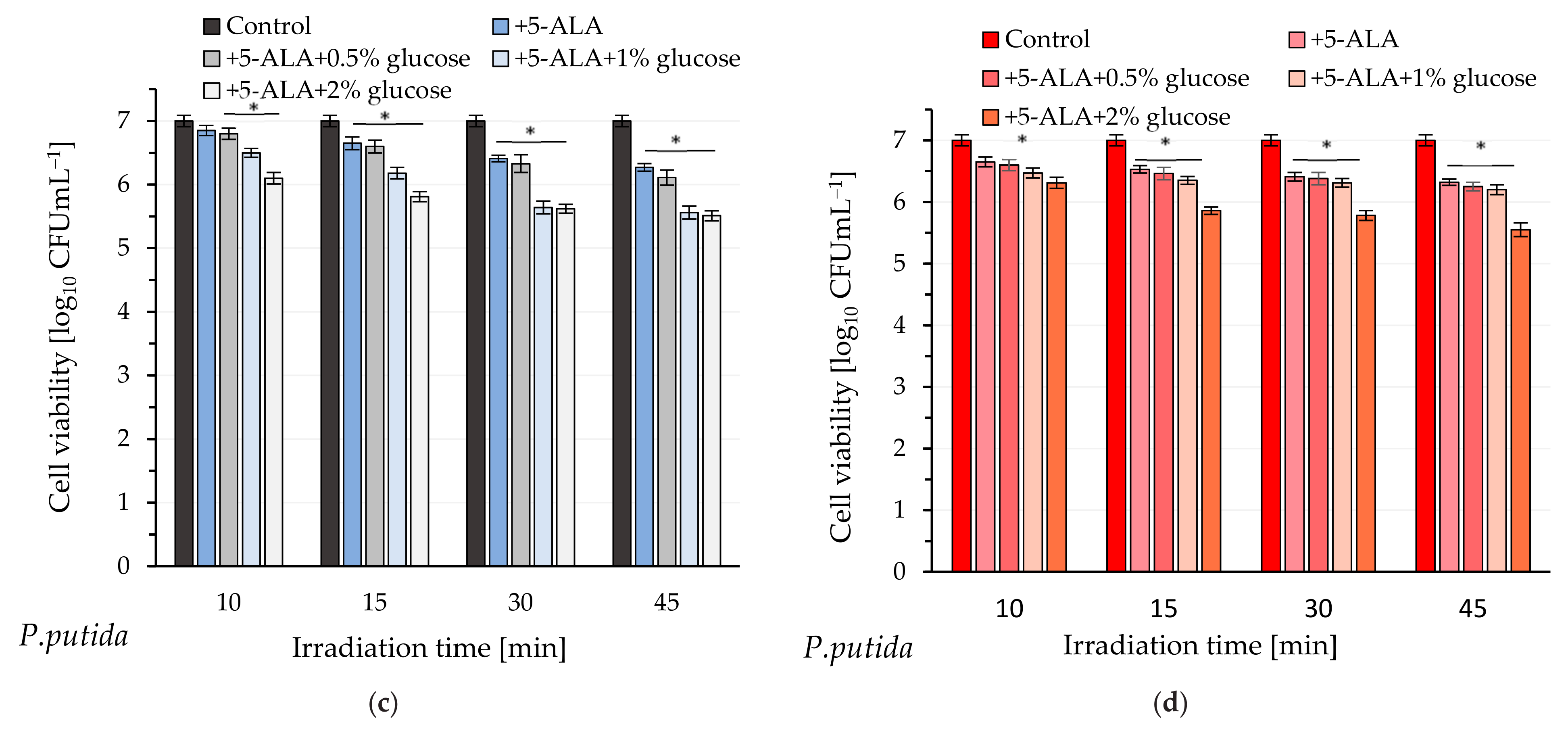
P. putida cells exhibited reduced sensitivity to light even in the presence of glucose. Although the efficiency of photo-destruction was higher in the presence of this monosaccharide, a lethal effect was not observed within 45 min of exposure to blue or red light. Exogenous administration of 5-ALA and glucose at a concentration of 2% resulted in approximately 96.8% bacterial mortality; however, this outcome remains unsatisfactory. The UV-Vis spectral analysis of cell-free lysates obtained from this pathogen does not definitively confirm the presence of absorption maxima characteristic of protoporphyrin IX. It appears that minor absorption maxima are observable near 410 nm and 630 nm, particularly in the spectrum of bacterial lysate obtained from cells supplemented with 5-ALA and 2% glucose (these findings are not definitive). Additionally, the lysates did not exhibit emission spectra characteristic of protoporphyrin IX (
Figure S3; Supplementary Information). It is believed that the formation of photodynamically inactive porphyrinogens (e.g., coproporphyrinogen, uroporphyrinogen), together with the weak accumulation of photodynamically active PpIX, may be responsible for the weak photoinactivation of
P. putida.
The dependence of aPDI efficiency based on 5-ALA has been the subject of previous analyses. For example, Fotinos et al. [
17] demonstrated a strong correlation between aPDI efficiency and the total amount of endogenous porphyrins produced in
E. coli K-12, whereas no such correlation was observed in other species (
S. aureus and
P. aeruginosa). It is believed that this phenomenon results from differences in the types of porphyrins produced and the protective mechanisms and cellular localization of porphyrins [
29]. On the other hand, a high level of photoinactivation (4-log reduction) was achieved in
P. aeruginosa but minimal concentrations of porphyrins were detected within the cells [
16]. The authors suggested that this may be due to the presence of aggregated, non-fluorescent porphyrins or porphyrinogens, which are activated during the illumination process. It has been reported that
P. aeruginosa exposed to 5-ALA generates predominantly photodynamically inactive porphyrinogens [
30].
Based on observations reported by other authors and the obtained results, it was suggested that the response to aPDI is distinctly strain-dependent in both Gram-positive and Gram-negative bacteria. Factors such as membrane composition, intracellular levels of proto- and other porphyrins, and protective mechanisms contribute to bacterial susceptibility to light-induced eradication [
31].
Our previous studies confirmed that multiple sublethal and lethal doses of light, in the presence of methylene blue as a photosensitizer, slightly increased
A. baumannii’s sensitivity to phototherapy and reduced its ability to form biofilm [
32]. It has also been demonstrated that multiple photooxidations directly affect the bacteria metabolome, leading to modifications in many metabolic pathways.
This study assessed the effect of multiple photodynamic treatments on several key virulence factors of the examined bacteria. Virulence factors, especially of
P. aeruginosa, have been extensively studied and are typically classified into three main categories: bacterial surface structures, secreted factors, and bacterial cell interactions [
3]. In this work, the effect of multiple photodynamic inactivation of bacteria on the activity of alkaline protease and elastase—both classified as secreted factors—was investigated. Alkaline protease is secreted via the type I secretion system and regulated by the quorum sensing circuit [
33] and can degrade complement components as well as IFN-γ and TNF-α, thereby counteracting host immune defenses and exacerbating infections [
34]. Elastase destroys elastin, an essential component of lung tissue and blood vessels [
35].
Lipase A, a major extracellular lipase classified as a toxin, is secreted through the type II secretion system [
36]. It can damage lung tissue by degrading the major lung surfactant, dipalmitoylphosphatidylcholine, as well as host cell membranes [
37]. Furthermore, studies have shown that this enzyme interacts with alginate in the biofilm matrix via electrostatic interactions, contributing to bacterial drug resistance [
38]. Phospholipase C is also a toxin capable of inducing host vascular permeability and damaging organs [
38].
Among the four important enzymes listed above, a significant decrease in activity due to multiple exposure to blue light was observed only for alkaline protease. The activities measured in supernatants obtained from cells incubated with 5-ALA and 2% glucose were reduced by approximately 35% and 16% (compared to pathogens not exposed to 5-ALA and light) for P. aeruginosa and P. putida, respectively.
Both examined microorganisms also exhibited a reduction in their biofilm-forming capacity on abiotic surfaces by 37–48% depending on the species and light exposure conditions. The ability to form biofilm is classified under the category of “bacterial cell interactions”. Biofilms produced by
Pseudomonas play a crucial role in protecting the bacteria from environmental stresses and antimicrobial agents. They facilitate the bacteria’s attachment to surfaces, enabling persistent colonization in various environments, including medical devices and water systems. Within the biofilm,
Pseudomonas bacteria communicate through quorum sensing (QS), coordinating their activities for survival and virulence. Overall, biofilm formation enhances the bacteria’s ability to persist, resist treatment, and cause chronic infections [
39]. It is believed that secretion of various QS-controlled extracellular enzymes (esterases, lipases, and elastases) affects EPS (Extracellular Polymeric Substances) composition and cell motility, thereby influencing the formation and architecture of mucoid
Pseudomonas biofilms [
40]. The results of our study did not confirm correlation between lipase and phospholipase activity and the administered treatment. After five cycles of phototherapy, no significant changes were observed in the activity of these enzymes. However, a reduction in the ability of pathogens to form biofilms was noted, which may suggest a potential therapeutic effect of this method in limiting biofilm formation as a bacterial resistance mechanism.
A reduction in the swarming zone diameter detected in
P. aeruginosa was also observed, and this phenomenon may be connected with a decrease in the ability of this bacterium to colonize diverse environments, attach to surfaces, and form biofilms. Swarming generally refers to the coordinated, collective motility of a dense bacterial population [
41].
In summary, the results of these studies confirm the potential of 5-ALA as an effective prodrug in photodynamic therapy against Pseudomonas spp. (particularly P. aeruginosa), which may contribute to the development of novel therapeutic approaches. However, further research is necessary to optimize treatment parameters and enhance efficacy and safety, both of which are critical for combating resistant bacterial strains and safeguarding public health.
4. Materials and Methods
4.1. Microorganisms and Culture Conditions
In our experiments, two bacterial species served as test microorganisms: Pseudomonas aeruginosa ATCC 13525 and Pseudomonas putida ATCC 49128. A single colony of each strain was obtained from an agar plate (Mueller–Hinton Agar, BTL, Łódź, Poland) and inoculated into 5 mL of Mueller–Hinton Broth (BTL, Łódź, Poland). The resulting suspensions were incubated overnight at 37 °C and 30 °C for P. aeruginosa and P. putida, respectively. After incubation, 1 mL of each suspension was centrifuged separately for 4 min at 6000 rpm; the supernatant was discarded, and the pellet was resuspended in 1 mL of sterile deionized water. The bacterial suspensions were then adjusted to a McFarland 0.5 standard (optical density at 550 nm between 0.09 and 0.1). It was estimated that the inocula contained approximately 1 × 10−8 or 8 × 10−7 colony-forming units (CFU mL−1) for P. aeruginosa and P. putida, respectively.
4.2. Light Source
Diode lasers with the peak-power wavelength of ʎ = 405 nm (output power of 17 mW; radiation intensity of 45 mW cm−2) and ʎ = 635 nm (output power of 20 mW; radiation intensity of 53 mW cm−2) were used in this study.
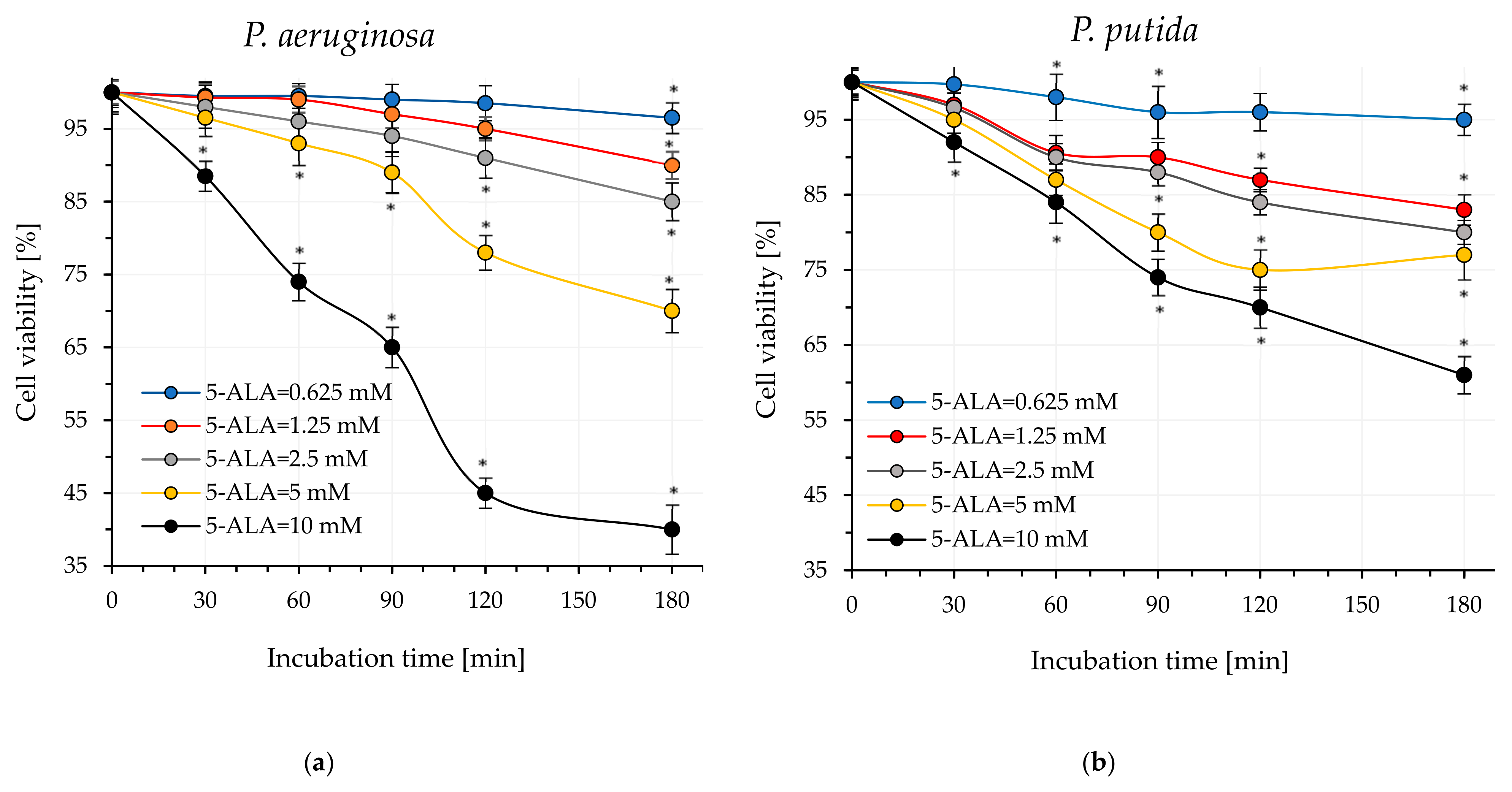
4.3. Estimation of Dark Cytotoxicity of 5-ALA
An amount of 40 µL of 5-ALA was added to the test tubes containing 450 µL of Mueller–Hinton Broth to achieve final concentrations of 10 mM, 5 mM, 2.5 mM, 1.25 mM, 1 mM, and 0.625 mM. All these samples were then inoculated with 10 µL of the standardized bacteria suspension prepared according to the procedure described in
Section 4.1. In the control sample, 5-ALA was replaced with Mueller–Hinton Broth. Subsequently, the samples were incubated in the dark at 37 °C for
P. aeruginosa and at 30 °C for
P. putida. After 60, 120, and 180 min, microbial viability was assessed using BacTiter-Glo™ reagent. The designation of live bacteria was carried out according to the procedure specified by the manufacturer (Promega Corporation, Madison, WI, USA). All experiments were conducted in three independent replicates.
4.4. Investigation of Protoporphyrin IX Level Variations in Bacterial Cells
Bacteria were prepared according to the protocol described in
Section 4.1, with a slight modification: deionized water was substituted with a 2.5 mM solution of 5-ALA. The mixtures were incubated at 37 °C (
P. aeruginosa) and 30 °C (
P. putida) for 180 min (protected from light). At 15, 30, 60, 90, 120 and 180 min of incubation, the samples (250 µL) were centrifuged for 5 min at 6000 rpm. The supernatant was carefully removed, and 250 μL of lysis solution (0.1 M NaOH/1% SDS) was added to the bacterial pellet. The samples were then placed on ice for 15 min (protected from light). After this period, the samples were centrifuged again, and the fluorescence intensities of the resulting supernatants (cell-free lysates) were measured (excitation at 410 nm and emission at 631 nm).
4.5. Study on the Effectiveness of aPDI
The standardized suspensions of
P. aeruginosa and
P. putida were incubated in the dark for 120 min or 30 min, respectively, with the following treatments: (1) 5-ALA at a final concentration of 2.5 mM; (2) 5-ALA + 0.5% glucose; (3) 5-ALA + 1% glucose; (4) 5-ALA + 2% glucose. Subsequently, samples of
P. aeruginosa were irradiated for durations of 1, 2, 3, 4, 5, 10, 15, 30, and 45 min using laser light at wavelengths of 410 nm and 635 nm. Samples of
P. putida were irradiated under the same experimental conditions but for durations of 10, 15, 30, and 45 min. Control samples without illumination were included to assess the initial bacterial concentration. The number of viable bacteria (colony-forming units per milliliter, CFUmL
−1) was quantified via serial dilution method. The reduction in viability of bacteria was calculated as logarithmic reduction, according to the following formula:
and percentage reduction, according to the following formula:
where the number of bacteria before experiments is defined as
, and
is the number of bacteria that remained alive after irradiation time. All experiments were conducted in triplicate.
4.6. Research on the Mechanism of Enhancing aPDI Efficiency by Glucose
The standardized suspensions of P. aeruginosa and P. putida were incubated in the dark for 120 min or 30 min, respectively, with the following treatments: (1) 5-ALA at a final concentration of 2.5 mM; (2) 5-ALA + 0.5% glucose; (3) 5-ALA + 1% glucose; (4) 5-ALA + 2% glucose. Subsequently, all samples were centrifuged for 5 min at 6000 rpm, and the obtained supernatants were carefully removed. Lysis buffer (Tris-acetate-EDTA with 2% SDS) was added to the pellets, and all samples were placed on ice for 10 min (protected from light). After this period, the samples were centrifuged again, and the resulting supernatants (cell-free lysates) were subjected to UV-Vis spectroscopy (Shimadzu UV-1650PC), fluorescence spectroscopy, and fluorescence intensity measurements (excitation at 410 nm; emission at 631 nm) (SpectraMax Gemini spectrofluorometer, ThermoFisher Scientific). The control sample was the supernatant obtained from the lysed bacterial suspension without the addition of 5-ALA or 5-ALA + glucose, prepared according to the procedure described above.
4.7. Study on the Effect of Multiple aPDI Treatments on Virulence Factors
The standardized suspensions of P. aeruginosa and P. putida were incubated in the dark for 120 min and 30 min, respectively, with 5-ALA at a final concentration of 2.5 mM. Sterilized glass slides were then inoculated with 50 µL of these bacterial suspensions and irradiated for 5 min (for P. aeruginosa) and 45 min (for P. putida) using laser light at a wavelength of 405 nm. These irradiation times were selected to ensure that a sufficient number of viable cells remained in the suspensions. Following light exposure, the treated slides were immersed in 5 mL of Mueller–Hinton II broth (BTL, Łódź, Poland) and incubated at 37 °C (for P. aeruginosa) and 30 °C (for P. putida) for 24 h. After incubation, bacterial cells were harvested by centrifugation at 6000 rpm for 5 min and resuspended in buffered saline to achieve an inoculum density of approximately 1.0 × 10−8 CFU mL−1. This bacterial suspension was then used to inoculate new slides, which were subjected again to blue light irradiation for 5 min or 45 min, following the same procedure. This cycle was repeated five times. Untreated bacterial cultures served as controls. All experiments were conducted in duplicate.
The studied bacteria, P. aeruginosa and P. putida (before and after multiple light treatments), were cultured for 24 h in Tryptic Soy Broth (Sigma-Aldrich, St. Louis, MO, USA). Following incubation, the cells were centrifuged (5 min, 6000 rpm; 4 °C), and the resulting supernatants were passed through a 0.2-micrometer filter (Minisart®, Sartorius Stedim, Bohemia, NY, USA) and subsequently dialyzed (D-Tube Dialyzer, MWCO 3.5 kDa, Merck Millipore, Burlington, MA, USA) to remove low-molecular-weight metabolic products. These cell-free supernatants were then lyophilized at temperatures below −80 °C and under a pressure of less than 40 mTorr, and subjected to further analysis. For this purpose, 10 mg of the lyophilized supernatants was dissolved in 1 mL of deionized water. The resulting solutions were used to assess the following.
4.7.1. Activity of Phospholipase C
To evaluate the activity of phospholipase C, a method based on Luberto et al. [
42] was used with minor modifications. Specifically, 50 μL of p-nitrophenylphosphorylcholine (p-NPPC; 37.5 mM) (Sigma-Aldrich, USA), dissolved in 100 mM Tris-HCl buffer (pH 7.4) containing 25% glycerol, was mixed with 150 μL of cell-free supernatant from
P. aeruginosa culture, resulting in a total reaction volume of 200 μL. The activity was assessed by measuring the increase in absorbance at 405 nm, which indicates the production of p-nitrophenol, a chromogenic compound. Absorbance was recorded every 60 s at 37 °C immediately after adding p-NPPC (Shimadzu UV-1650PC). A control sample was prepared in the same way but replaced the bacterial supernatant with distilled water. The activity of this enzyme was also measured in a cell-free supernatant obtained from a 24 h culture of
P. putida, with the difference that the enzymatic reactions were conducted at 30 °C.
4.7.2. Lipase A Activity
Lipase activity was assessed using p-nitrophenyl palmitate (p-NPP) as the substrate, following the protocol outlined by [
43] with minor modifications. The assay mixture was prepared by combining 90 mL of phosphate buffer with 100 mg of gum Arabic, 207 mg of sodium deoxycholate (Sigma-Aldrich, USA), and 30 mg of p-nitrophenyl palmitate dissolved in 10 mL of isopropanol, with the pH adjusted to 8.0. To measure lipase activity, 100 μL of this assay mixture was mixed with 100 μL of cell-free filtrate from
P. aeruginosa and incubated at 37 °C for 30 min. The reaction was then stopped by adding 0.5 mL of 3 M HCl. The mixture was centrifuged at 6000 rpm for 10 min at 4 °C, and the amount of p-nitrophenol (pNP) released was quantified spectrophotometrically at 405 nm (Shimadzu UV-1650PC). One unit of lipase activity was defined as the amount of enzyme that releases 1 μmol of pNP under these standard conditions. Lipase activity was additionally measured in a cell-free supernatant derived from a 24 h culture of
P. putida, with the enzymatic reaction performed at 30 °C.
4.7.3. Activity of Alkaline Protease
The reaction mixture consisted of 500 µL of azocasein (Sigma-Aldrich, USA) at 0.05% in 0.2 M phosphate buffer (pH 7.0) and 500 µL of the cell-free supernatant derived from the P. aeruginosa culture. This mixture was incubated at 37 °C for 30 min. To stop the reaction, 1 mL of 10% (w/v) trichloroacetic acid was added. After keeping the mixture on ice for 1 h, the unreacted azocasein was precipitated by centrifugation (10 min at 6000 rpm). The resulting supernatant was then combined with an equal volume of 1 N NaOH, and its absorbance was measured at 440 nm (Shimadzu UV-1650PC). A control sample was prepared identically but replacing the enzyme or bacterial supernatant with distilled water. A value of 1 enzyme activity unit (U) was defined as the amount of enzyme that produces an increase of 0.01 in absorbance at 440 nm per minute under the assay conditions. The activity of alkaline protease was also determined in a cell-free supernatant obtained from a 24 h culture of P. putida, with the difference that the enzymatic reaction was conducted at 30 °C.
4.7.4. Elastase Activity
A volume of 1 mL of cell-free supernatant from P. aeruginosa was combined with 20 mg of Elastin-Congo red substrate (Sigma-Aldrich, USA) suspended in 2 mL of 0.2 M acid buffer (pH 7.4) and then incubated with shaking at 37 °C for 30 min. The reaction was stopped by adding 100 μL of 0.12 M EDTA (pH 8.0). After centrifugation (5 min; 6000 rpm; 4 °C), absorbance at 495 nm was measured using a spectrophotometer (Shimadzu UV-1650PC) calibrated with a control sample of Elastin-Congo red, which was incubated without supernatant. A value of 1 unit of elastase activity was defined as the amount of enzyme that caused an increase in absorbance at 495 nm of 0.01 after 1 min of incubation under standard assay conditions. Elastase activity was also determined in a cell-free supernatant obtained from a 24 h culture of P. putida, except that the enzymatic reactions were carried out at 30 °C.
All enzyme activity analyses were conducted in three independent experiments. The enzyme activity was expressed per milligram of protein present in the lyophilized supernatants. Protein concentration was determined using the Bradford assay [
44].
4.8. Study on Pyocyanin and Pyoverdine Production
To evaluate the impact of multiple aPDI-5-ALA treatments on the production of pyocyanin and pyoverdine, a 10 μL aliquot of overnight-cultured
P. aeruginosa was transferred to 1 mL of King’s B medium (BTL, Łódź, Poland) and incubated at 37 °C. The concentration of pyoverdine in the samples was quantified by measuring the absorbance of the cell-free supernatant at 405 nm, as described in reference [
45]. The pyocyanin assay involved measuring absorbance at 520 nm in an acidic solution. Specifically, a 5 mL culture sample, grown in King’s B medium to maximize pyocyanin production, was extracted with 3 mL of chloroform, followed by re-extraction into 1 mL of 0.2 N HCl, resulting in a pink to deep red solution. The absorbance of this solution was then measured at 520 nm (Shimadzu UV-1650PC).
4.9. Study on Biofilm Formation
The standardized suspensions of
P. aeruginosa and
P. putida prepared according to the procedure described in
Section 4.8 were centrifuged at 6000 rpm for 5 min. The resulting pellets were washed twice with sterile saline (0.9% NaCl) and resuspended in Mueller–Hinton Broth BTL, Łódź, Poland) to achieve an optical density at 600 nm of 1.0. A 200 µL aliquot of the bacterial suspension was then added to the wells of a sterile, black microtiter plate and incubated at 37 °C (
P. aeruginosa) or 30 °C (
P. putida) for 24 h to facilitate biofilm formation. Following incubation, the medium was carefully removed, and the biofilm was gently washed with sterile saline to eliminate planktonic cells. The biofilm was subsequently stained with 0.1% (
w/
v) crystal violet for 10 min. Excess stain was removed by rinsing with phosphate-buffered saline (PBS). The bound crystal violet was then solubilized in 5 mL of 33% glacial acetic acid, and absorbance was measured at 590 nm using a spectrophotometer. All experiments were performed in triplicate.
4.10. Study on Swarming Motility
Swarming motility of
P. aeruginosa was assessed following previously established protocols with minor modifications [
46]. Briefly, overnight cultures of these pathogens grown in minimal medium (62 mM potassium phosphate buffer (pH 7), 7 mM (NH
4)
2SO
4, 2 mM MgSO
4, 10 μM FeSO
4, 0.4% (wt/vol) glucose) were point-inoculated at the center of swarm agar plates composed of 62 mM potassium phosphate buffer (pH 7), 2 mM MgSO
4, 10 μM FeSO
4, 0.4% [wt/vol] glucose, 0.1% [wt/vol] Casamino Acids, 0.5% [wt/vol] agar-agar. The plates were incubated upright at 37 °C and the diameter of the swarming zones was subsequently measured.
4.11. Fluorescence Microscopy Studies
Pseudomonas aeruginosa cell suspensions before and after photoinactivation with blue light in the presence of 5-ALA alone or combined with 2% glucose were stained with SYTOX® Green Dead Cell Stain (ThermoFisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. Fluorescent imaging of non-viable bacterial cells was performed using an Olympus BX60 light microscope equipped with appropriate excitation/emission filter sets (excitation bandpass filter 460–490 nm; emission long-pass filter > 515 nm).
4.12. Statistical Analysis
The statistical analyses of the data obtained in the experiments were performed using the STATISTICA data analysis software package (Statistica version 13.3, StatSoft Inc., Kraków, Poland) at the significance level of p < 0.05.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}