
Pan-Cancer Computational Analysis of RKIP (PEBP1) and LKB1 (STK11) Co-Expression Highlights Distinct Immunometabolic Dynamics and Therapeutic Responses Within the Tumor Microenvironment


 ,
,  ,
,  ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. PEBPI/STK11 Co-Expression Favors Overall Survival in a Cancer-Type Dependent Manner
2.2. LKB1 Exhibits Broader and Stronger Functional Associations with TME Subtypes Compared to RKIP Based on a Multiple-UniReD Analysis
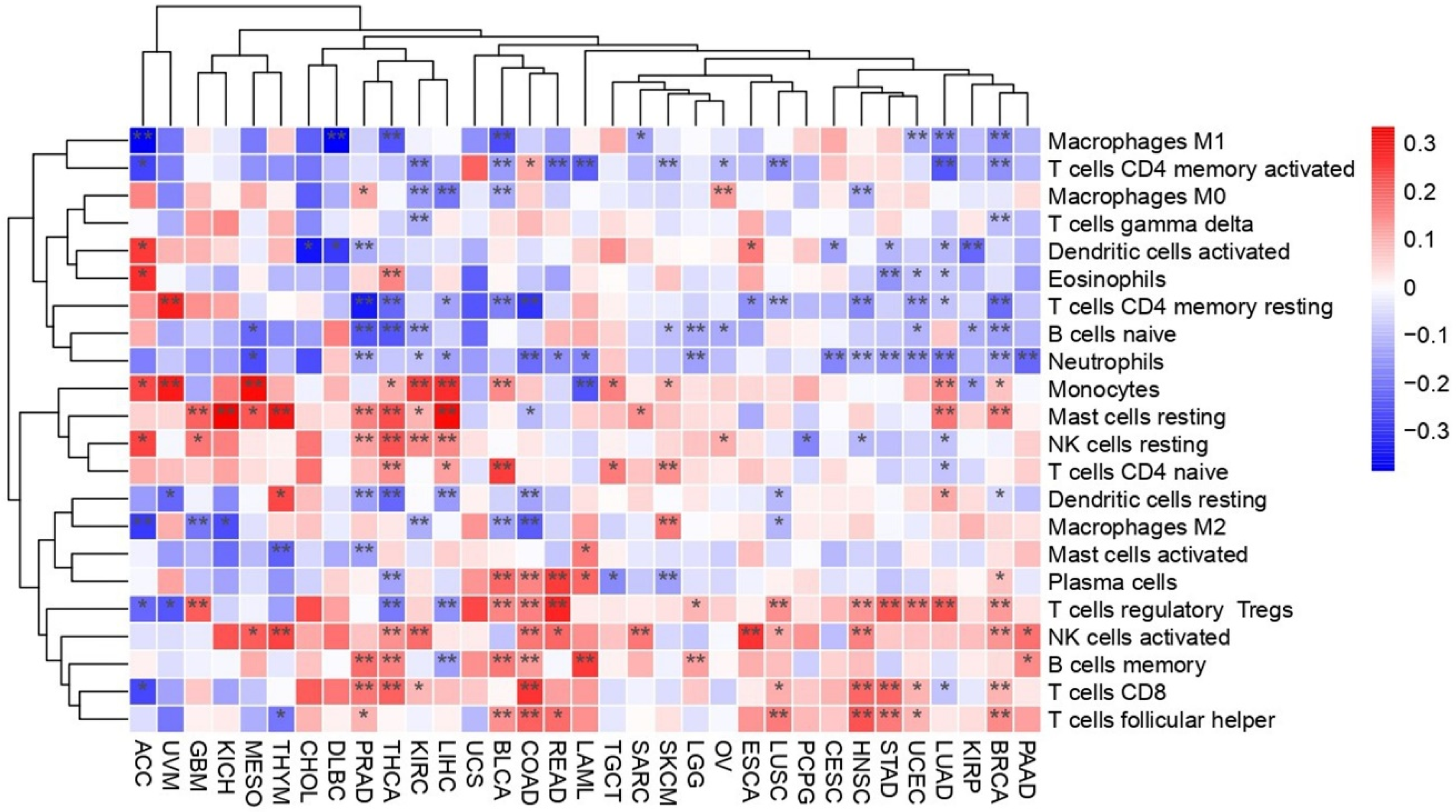
2.3. PEBP1/STK11 Co-Expression Associates with the Molecular Remodeling of Distinct TME Niches
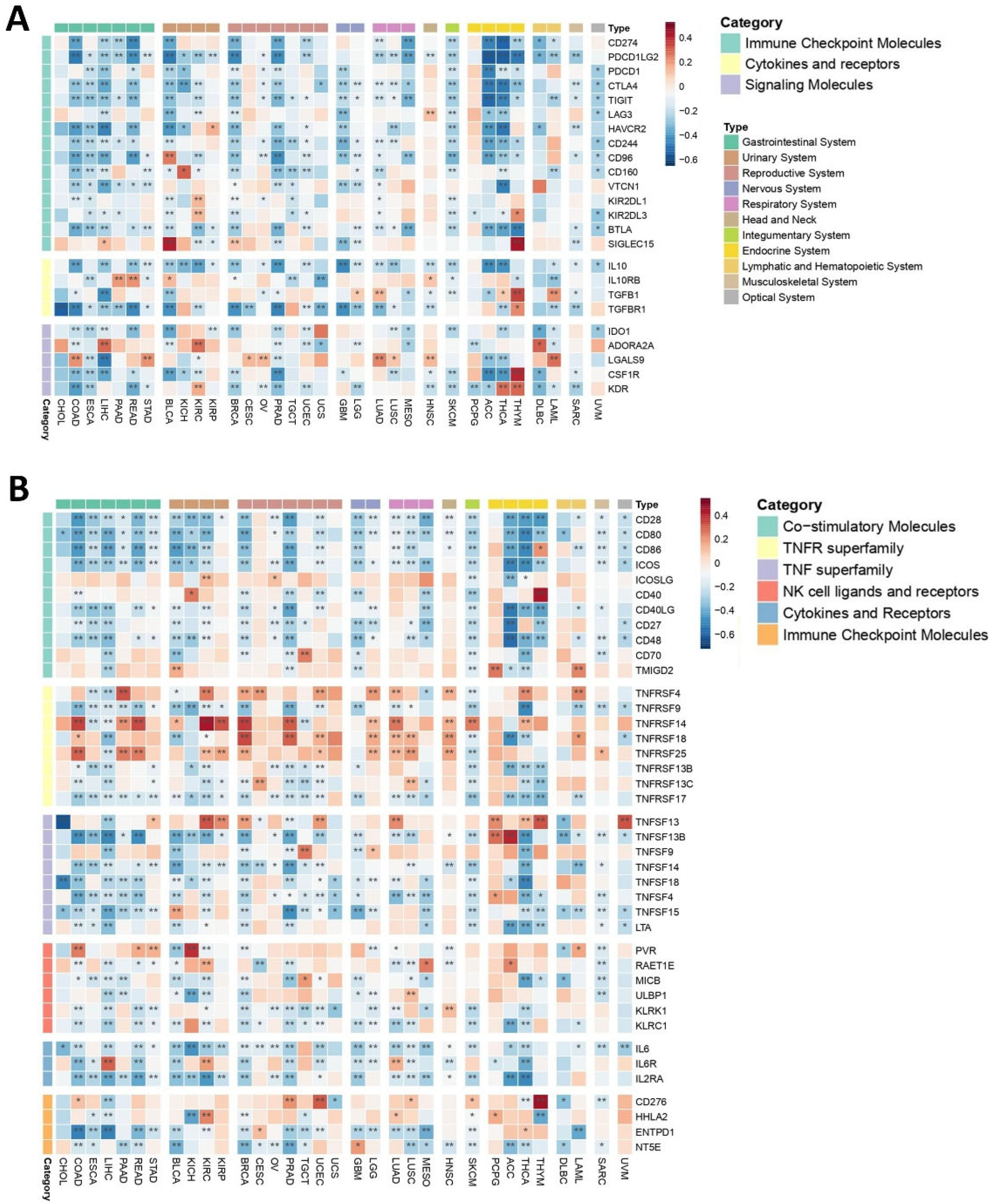
2.3.1. Immune Niche
Immune Cell Infiltration: Immune Score
Inhibitors of Immune Responses
Immune Stimulators
Chemokines and Chemokine Receptors
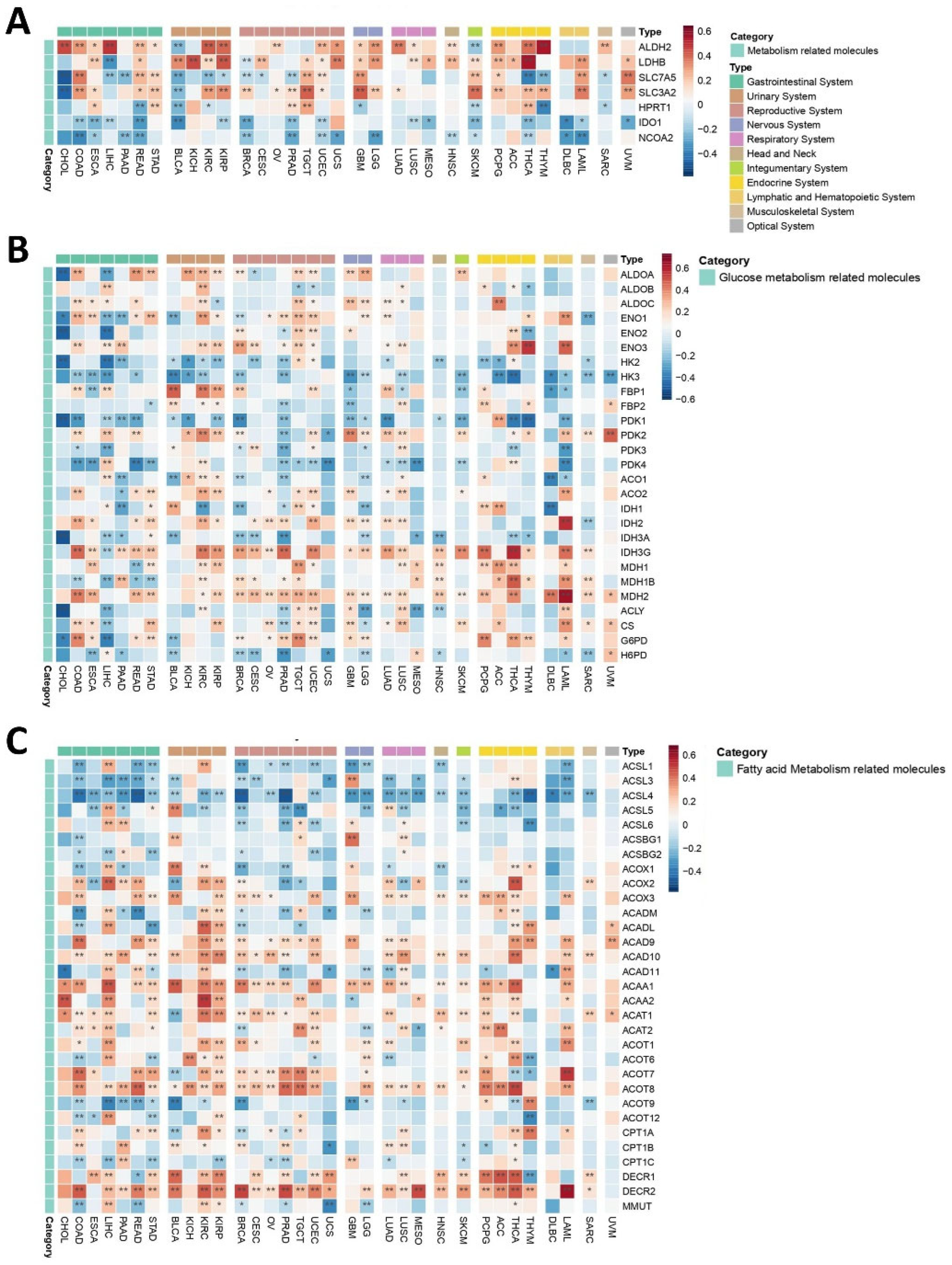
2.3.2. Metabolic Niche
Glucose Metabolism
Fatty Acid Metabolism
2.3.3. Mechanical Niche
2.3.4. Hypoxic Niche
2.4. PEBP1/STK11 Co-Expression Modulates TME Properties and Therapeutic Responses
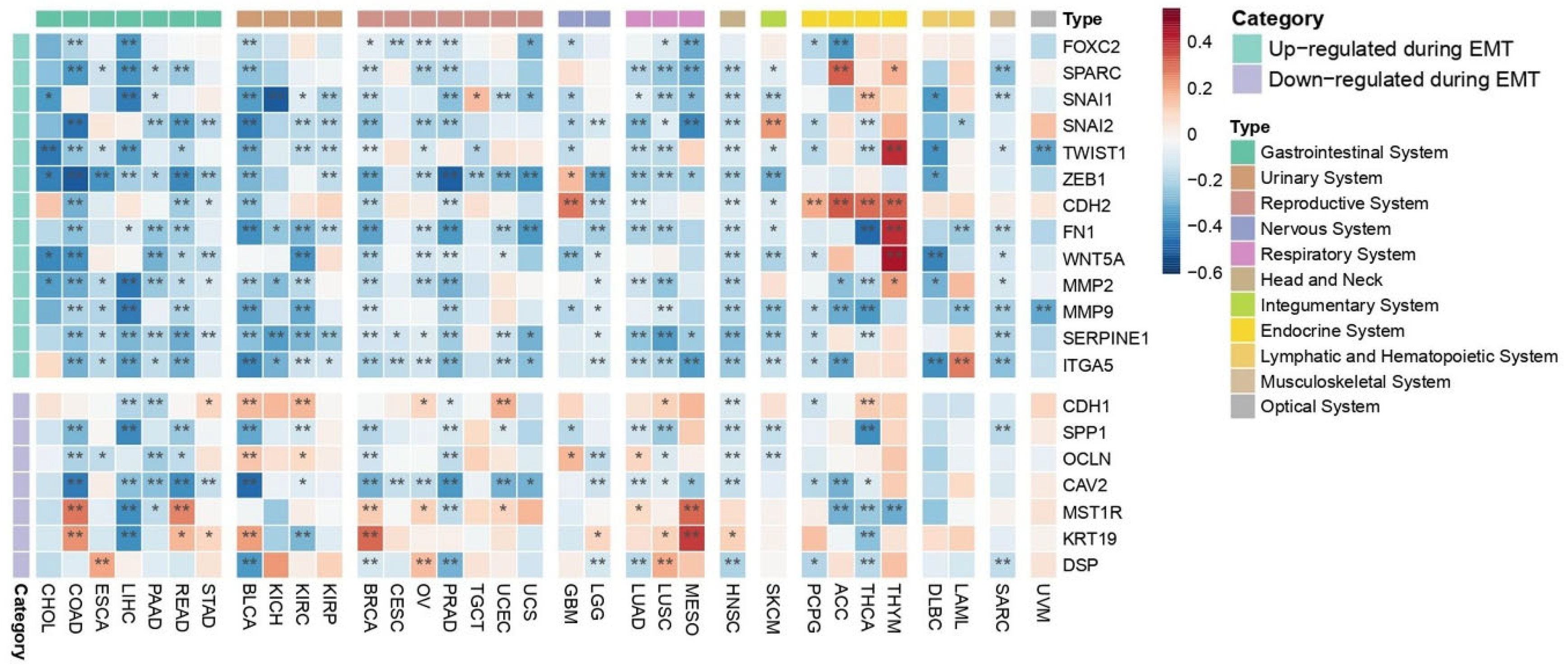
2.4.1. EMT
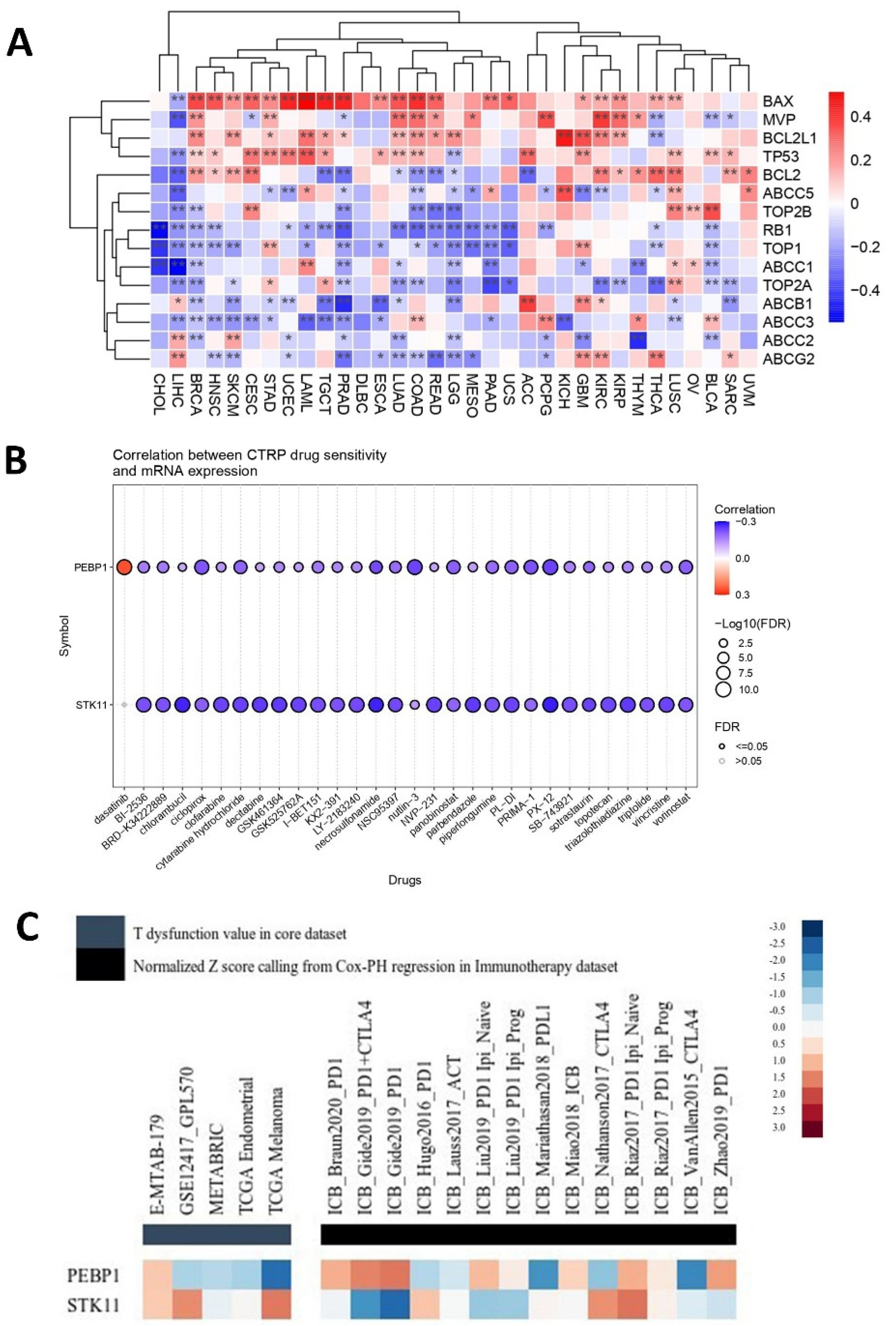
2.4.2. Predictive Response to Chemotherapy
2.4.3. Predictive Response to Immunotherapy
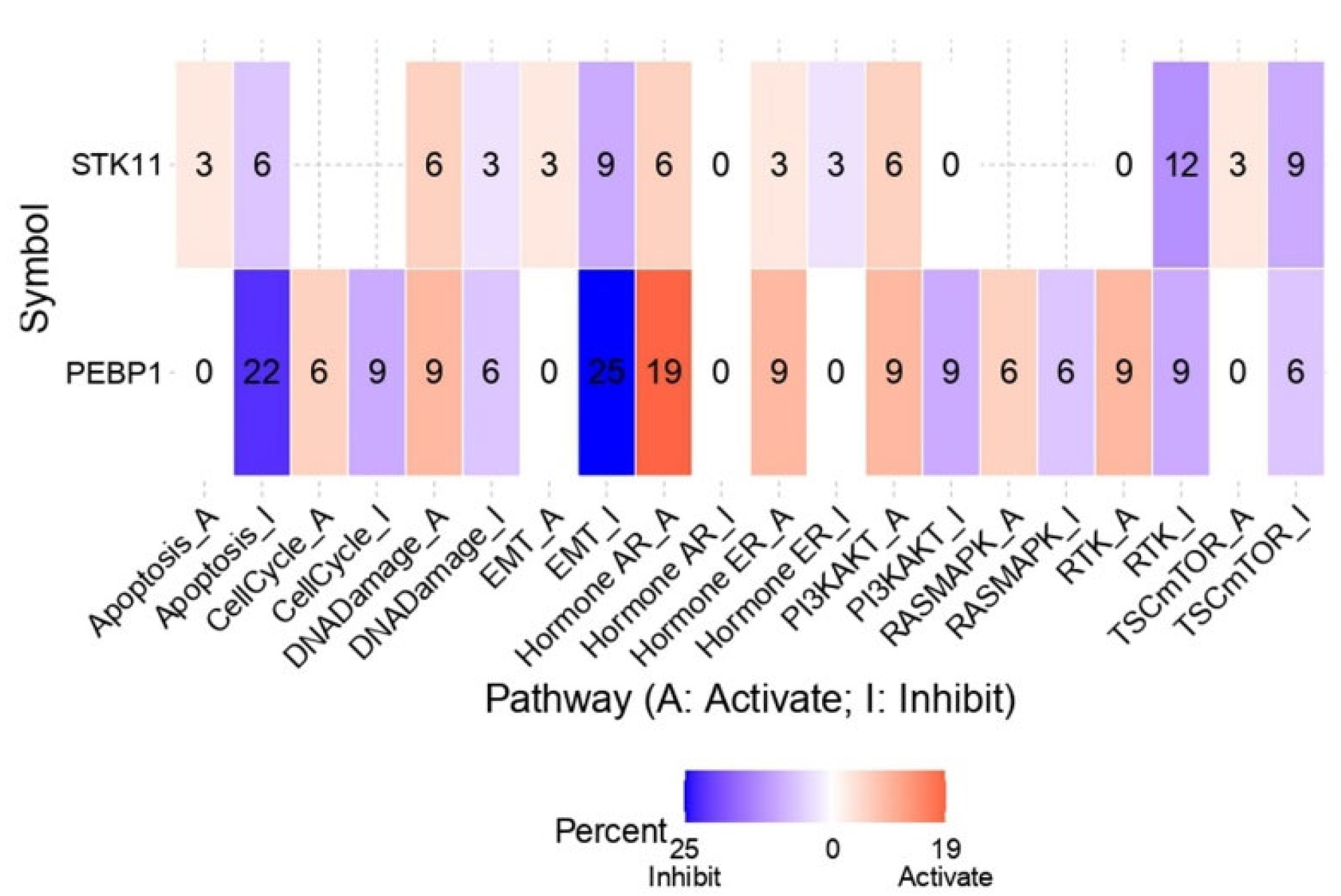
2.4.4. Pathway Analysis
3. Discussion
4. Materials and Methods
4.1. Protein Functional Analysis by the Multiple UniReD Tool
4.2. Data Acquisition and Preprocessing
4.3. Exploring PEBP1 and STK11 Expression Signature
4.4. Pathway Activity Analysis
4.5. Correlation of PEBP1/STK11 Expression with Drug Sensitivity
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Anderson, N.M.; Simon, M.C. The Tumor Microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef]
- de Visser, K.E.; Joyce, J.A. The Evolving Tumor Microenvironment: From Cancer Initiation to Metastatic Outgrowth. Cancer Cell 2023, 41, 374–403. [Google Scholar] [CrossRef]
- Wang, Q.; Shao, X.; Zhang, Y.; Zhu, M.; Wang, F.X.C.; Mu, J.; Li, J.; Yao, H.; Chen, K. Role of Tumor Microenvironment in Cancer Progression and Therapeutic Strategy. Cancer Med. 2023, 12, 11149–11165. [Google Scholar] [CrossRef]
- Jin, M.-Z.; Jin, W.-L. The Updated Landscape of Tumor Microenvironment and Drug Repurposing. Signal Transduct. Target. Ther. 2020, 5, 166. [Google Scholar] [CrossRef]
- Zhang, T.-Q.; Lv, Q.-Y.; Jin, W.-L. The Cellular-Centered View of Hypoxia Tumor Microenvironment: Molecular Mechanisms and Therapeutic Interventions. Biochim. Biophys. Acta Rev. Cancer 2024, 1879, 189137. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, A.; Trivedi, R.; Lin, S.-Y. Tumor Microenvironment: Barrier or Opportunity towards Effective Cancer Therapy. J. Biomed. Sci. 2022, 29, 83. [Google Scholar] [CrossRef] [PubMed]
- Bilotta, M.T.; Antignani, A.; Fitzgerald, D.J. Managing the TME to Improve the Efficacy of Cancer Therapy. Front. Immunol. 2022, 13, 954992. [Google Scholar] [CrossRef]
- Klemm, F.; Joyce, J.A. Microenvironmental Regulation of Therapeutic Response in Cancer. Trends Cell Biol. 2015, 25, 198–213. [Google Scholar] [CrossRef]
- Ahmed, M.; Lai, T.H.; Kim, W.; Kim, D.R. A Functional Network Model of the Metastasis Suppressor PEBP1/RKIP and Its Regulators in Breast Cancer Cells. Cancers 2021, 13, 6098. [Google Scholar] [CrossRef]
- Lorenz, K.; Rosner, M.R. Harnessing RKIP to Combat Heart Disease and Cancer. Cancers 2022, 14, 867. [Google Scholar] [CrossRef]
- Zaravinos, A.; Bonavida, B.; Chatzaki, E.; Baritaki, S. RKIP: A Key Regulator in Tumor Metastasis Initiation and Resistance to Apoptosis: Therapeutic Targeting and Impact. Cancers 2018, 10, 287. [Google Scholar] [CrossRef]
- Cessna, H.; Baritaki, S.; Zaravinos, A.; Bonavida, B. The Role of RKIP in the Regulation of EMT in the Tumor Microenvironment. Cancers 2022, 14, 4596. [Google Scholar] [CrossRef] [PubMed]
- Hao, C.; Wei, S.; Tong, Z.; Li, S.; Shi, Y.; Wang, X.; Zhu, Z. The Effects of RKIP Gene Expression on the Biological Characteristics of Human Triple-Negative Breast Cancer Cells in Vitro. Tumor Biol. 2012, 33, 1159–1167. [Google Scholar] [CrossRef] [PubMed]
- Hagan, S.; Al-Mulla, F.; Mallon, E.; Oien, K.; Ferrier, R.; Gusterson, B.; Curto García, J.J.; Kolch, W. Reduction of Raf-1 Kinase Inhibitor Protein Expression Correlates with Breast Cancer Metastasis. Clin. Cancer Res. 2005, 11, 7392–7397. [Google Scholar] [CrossRef] [PubMed]
- Yesilkanal, A.E.; Rosner, M.R. Raf Kinase Inhibitory Protein (RKIP) as a Metastasis Suppressor: Regulation of Signaling Networks in Cancer. Crit. Rev. Oncog. 2014, 19, 447–454. [Google Scholar] [CrossRef]
- Al-Mulla, F.; Bitar, M.S.; Feng, J.; Park, S.; Yeung, K.C. A New Model for Raf Kinase Inhibitory Protein Induced Chemotherapeutic Resistance. PLoS ONE 2012, 7, e29532. [Google Scholar] [CrossRef]
- Bustamante, A.; Baritaki, S.; Zaravinos, A.; Bonavida, B. Relationship of Signaling Pathways between RKIP Expression and the Inhibition of EMT-Inducing Transcription Factors SNAIL1/2, TWIST1/2 and ZEB1/2. Cancers 2024, 16, 3180. [Google Scholar] [CrossRef]
- Martinho, O.; Pinto, F.; Granja, S.; Miranda-Gonçalves, V.; Moreira, M.A.R.; Ribeiro, L.F.J.; di Loreto, C.; Rosner, M.R.; Longatto-Filho, A.; Reis, R.M. RKIP Inhibition in Cervical Cancer Is Associated with Higher Tumor Aggressive Behavior and Resistance to Cisplatin Therapy. PLoS ONE 2013, 8, e59104. [Google Scholar] [CrossRef]
- Beach, S.; Tang, H.; Park, S.; Dhillon, A.S.; Keller, E.T.; Kolch, W.; Yeung, K.C. Snail Is a Repressor of RKIP Transcription in Metastatic Prostate Cancer Cells. Oncogene 2008, 27, 2243–2248. [Google Scholar] [CrossRef]
- Das, S.K.; Bhutia, S.K.; Sokhi, U.K.; Azab, B.; Su, Z.; Boukerche, H.; Anwar, T.; Moen, E.L.; Chatterjee, D.; Pellecchia, M.; et al. Raf Kinase Inhibitor RKIP Inhibits MDA-9/Syntenin-Mediated Metastasis in Melanoma. Cancer Res. 2012, 72, 6217–6226. [Google Scholar] [CrossRef]
- Nie, F.; Cao, J.; Tong, J.; Zhu, M.; Gao, Y.; Ran, Z. Role of Raf-Kinase Inhibitor Protein in Colorectal Cancer and Its Regulation by Hydroxycamptothecine. J. Biomed. Sci. 2015, 22, 56. [Google Scholar] [CrossRef]
- Raquel-Cunha, A.; Cardoso-Carneiro, D.; Reis, R.M.; Martinho, O. Current Status of Raf Kinase Inhibitor Protein (RKIP) in Lung Cancer: Behind RTK Signaling. Cells 2019, 8, 442. [Google Scholar] [CrossRef]
- Dai, H.; Chen, H.; Liu, W.; You, Y.; Tan, J.; Yang, A.; Lai, X.; Bie, P. Effects of Raf Kinase Inhibitor Protein Expression on Pancreatic Cancer Cell Growth and Motility: An in Vivo and in Vitro Study. J. Cancer Res. Clin. Oncol. 2016, 142, 2107–2117. [Google Scholar] [CrossRef]
- Walker, E.J.; Rosenberg, S.A.; Wands, J.R.; Kim, M. Role of Raf Kinase Inhibitor Protein in Hepatocellular Carcinoma. For. Immunopathol. Dis. Therap 2011, 2, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.M.; Gu, H.; Yan, L.; Zhang, G.Y. RKIP Inhibits the Malignant Phenotypes of Gastric Cancer Cells. Neoplasma 2012, 60, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.; Chen, X.; Liu, X.; Wang, C.; Lv, L.; Xie, N.; Wang, T.; Huang, H. Clinicopathological Features and Prognostic Implications of Raf Kinase Inhibitor Protein Downregulation in Tongue Squamous Cell Carcinoma. Oncol. Lett. 2015, 10, 1303–1308. [Google Scholar] [CrossRef] [PubMed]
- Figy, C.; Guo, A.; Fernando, V.R.; Furuta, S.; Al-Mulla, F.; Yeung, K.C. Changes in Expression of Tumor Suppressor Gene RKIP Impact How Cancers Interact with Their Complex Environment. Cancers 2023, 15, 958. [Google Scholar] [CrossRef]
- Ren, G.; Baritaki, S.; Marathe, H.; Feng, J.; Park, S.; Beach, S.; Bazeley, P.S.; Beshir, A.B.; Fenteany, G.; Mehra, R.; et al. Polycomb Protein EZH2 Regulates Tumor Invasion via the Transcriptional Repression of the Metastasis Suppressor RKIP in Breast and Prostate Cancer. Cancer Res. 2012, 72, 3091–3104. [Google Scholar] [CrossRef]
- Huang, L.; Dai, T.; Lin, X.; Zhao, X.; Chen, X.; Wang, C.; Li, X.; Shen, H.; Wang, X. MicroRNA-224 Targets RKIP to Control Cell Invasion and Expression of Metastasis Genes in Human Breast Cancer Cells. Biochem. Biophys. Res. Commun. 2012, 425, 127–133. [Google Scholar] [CrossRef]
- Farooqi, A.A.; Li, Y.; Sarkar, F.H. The Biological Complexity of RKIP Signaling in Human Cancers. Exp. Mol. Med. 2015, 47, e185. [Google Scholar] [CrossRef]
- Wang, W.; Tang, X.; Qu, H.; He, Q. Translation Regulatory Long Non-coding RNA 1 Represents a Potential Prognostic Biomarker for Colorectal Cancer. Oncol. Lett. 2020, 19, 4077–4087. [Google Scholar] [CrossRef]
- Papale, M.; Netti, G.S.; Stallone, G.; Ranieri, E. Understanding Mechanisms of RKIP Regulation to Improve the Development of New Diagnostic Tools. Cancers 2022, 14, 5070. [Google Scholar] [CrossRef] [PubMed]
- Skinner, J.J.; Rosner, M.R. RKIP Structure Drives Its Function: A Three-State Model for Regulation of RKIP. Crit. Rev. Oncog. 2014, 19, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Bourouh, M.; Marignani, P.A. The Tumor Suppressor Kinase LKB1: Metabolic Nexus. Front. Cell Dev. Biol. 2022, 10, 881297. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.-X.; Xu, Z.-X. Targeting the LKB1 Tumor Suppressor. Curr. Drug Targets 2014, 15, 32–52. [Google Scholar] [CrossRef]
- Lee, S.-W.; Lin, H.-K. A New Mechanism for LKB1 Activation. Mol. Cell. Oncol. 2018, 5, e1035691. [Google Scholar] [CrossRef]
- Alessi, D.R.; Sakamoto, K.; Bayascas, J.R. LKB1-Dependent Signaling Pathways. Annu. Rev. Biochem. 2006, 75, 137–163. [Google Scholar] [CrossRef]
- Ciccarese, F.; Zulato, E.; Indraccolo, S. LKB1/AMPK Pathway and Drug Response in Cancer: A Therapeutic Perspective. Oxid. Med. Cell Longev. 2019, 2019, 8730816. [Google Scholar] [CrossRef]
- Trelford, C.B.; Shepherd, T.G. LKB1 Biology: Assessing the Therapeutic Relevancy of LKB1 Inhibitors. Cell Commun. Signal. 2024, 22, 310. [Google Scholar] [CrossRef]
- Shackelford, D.B.; Shaw, R.J. The LKB1–AMPK Pathway: Metabolism and Growth Control in Tumour Suppression. Nat. Rev. Cancer 2009, 9, 563–575. [Google Scholar] [CrossRef]
- Mirouse, V.; Billaud, M. The LKB1/AMPK Polarity Pathway. FEBS Lett. 2011, 585, 981–985. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.; Brenman, J.E. LKB1 and AMPK in Cell Polarity and Division. Trends Cell Biol. 2008, 18, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Tacheci, I.; Kopacova, M.; Bures, J. Peutz-Jeghers Syndrome. Curr. Opin. Gastroenterol. 2021, 37, 245–254. [Google Scholar] [CrossRef] [PubMed]
- McGarrity, T.J.; Kulin, H.E.; Zaino, R.J. Peutz-Jeghers Syndrome. Am. J. Gastroenterol. 2000, 95, 596–604. [Google Scholar] [CrossRef]
- Jelsig, A.M.; Karstensen, J.G.; Overeem Hansen, T.V. Progress Report: Peutz–Jeghers Syndrome. Fam. Cancer 2024, 23, 409–417. [Google Scholar] [CrossRef]
- McGarrity, T.J.; Amos, C.I.; Baker, M.J. Peutz-Jeghers Syndrome. In GeneReviews® [Internet]; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Bonanno, L.; Zulato, E.; Pavan, A.; Attili, I.; Pasello, G.; Conte, P.; Indraccolo, S. LKB1 and Tumor Metabolism: The Interplay of Immune and Angiogenic Microenvironment in Lung Cancer. Int. J. Mol. Sci. 2019, 20, 1874. [Google Scholar] [CrossRef]
- Della Corte, C.M.; Byers, L.A. Evading the STING: LKB1 Loss Leads to STING Silencing and Immune Escape in KRAS-Mutant Lung Cancers. Cancer Discov. 2019, 9, 16–18. [Google Scholar] [CrossRef]
- Kitajima, S.; Ivanova, E.; Guo, S.; Yoshida, R.; Campisi, M.; Sundararaman, S.K.; Tange, S.; Mitsuishi, Y.; Thai, T.C.; Masuda, S.; et al. Suppression of STING Associated with LKB1 Loss in KRAS-Driven Lung Cancer. Cancer Discov. 2019, 9, 34–45. [Google Scholar] [CrossRef]
- Lagoudaki, E.D.; Koutsopoulos, A.V.; Sfakianaki, M.; Papadaki, C.; Manikis, G.C.; Voutsina, A.; Trypaki, M.; Tsakalaki, E.; Fiolitaki, G.; Hatzidaki, D.; et al. LKB1 Loss Correlates with STING Loss and, in Cooperation with β-Catenin Membranous Loss, Indicates Poor Prognosis in Patients with Operable Non-Small Cell Lung Cancer. Cancers 2024, 16, 1818. [Google Scholar] [CrossRef]
- King, C.T.; Matossian, M.D.; Savoie, J.J.; Nguyen, K.; Wright, M.K.; Byrne, C.E.; Elliott, S.; Burks, H.E.; Bratton, M.R.; Pashos, N.C.; et al. Liver Kinase B1 Regulates Remodeling of the Tumor Microenvironment in Triple-Negative Breast Cancer. Front. Mol. Biosci. 2022, 9, 847505. [Google Scholar] [CrossRef]
- Chen, X.; Ye, F.; Hu, X.; Li, Z.; Jiang, B.; Fu, Y.; Cheng, X.; Shao, Z.; Zhuang, Z. Liver Kinase B1 Enhances Chemoresistance to Gemcitabine in Breast Cancer MDA-MB-231 Cells. Oncol. Lett. 2014, 8, 2086–2092. [Google Scholar] [CrossRef]
- Binkley, M.S.; Jeon, Y.-J.; Nesselbush, M.; Moding, E.J.; Nabet, B.Y.; Almanza, D.; Kunder, C.; Stehr, H.; Yoo, C.H.; Rhee, S.; et al. KEAP1/NFE2L2 Mutations Predict Lung Cancer Radiation Resistance That Can Be Targeted by Glutaminase Inhibition. Cancer Discov. 2020, 10, 1826–1841. [Google Scholar] [CrossRef] [PubMed]
- Sitthideatphaiboon, P.; Galan-Cobo, A.; Negrao, M.V.; Qu, X.; Poteete, A.; Zhang, F.; Liu, D.D.; Lewis, W.E.; Kemp, H.N.; Lewis, J.; et al. STK11/LKB1 Mutations in NSCLC Are Associated with KEAP1/NRF2-Dependent Radiotherapy Resistance Targetable by Glutaminase Inhibition. Clin. Cancer Res. 2021, 27, 1720–1733. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Liu, M.; Tang, B.; Li, Q.; Pan, B.-S.; Xu, C.; Lin, H.-K. Posttranslational Regulation of Liver Kinase B1 in Human Cancer. J. Biol. Chem. 2023, 299, 104570. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Zhu, H. LKB1 and Cancer: The Dual Role of Metabolic Regulation. Biomed. Pharmacother. 2020, 132, 110872. [Google Scholar] [CrossRef]
- Rhodes, L.V.; Tate, C.R.; Hoang, V.T.; Burks, H.E.; Gilliam, D.; Martin, E.C.; Elliott, S.; Miller, D.B.; Buechlein, A.; Rusch, D.; et al. Regulation of Triple-Negative Breast Cancer Cell Metastasis by the Tumor-Suppressor Liver Kinase B1. Oncogenesis 2015, 4, e168. [Google Scholar] [CrossRef]
- Kang, J.; Gallucci, S.; Pan, J.; Oakhill, J.S.; Sanij, E. The Role of STK11/LKB1 in Cancer Biology: Implications for Ovarian Tumorigenesis and Progression. Front. Cell Dev. Biol. 2024, 12, 1449543. [Google Scholar] [CrossRef]
- Hermanova, I.; Zúñiga-García, P.; Caro-Maldonado, A.; Fernandez-Ruiz, S.; Salvador, F.; Martín-Martín, N.; Zabala-Letona, A.; Nuñez-Olle, M.; Torrano, V.; Camacho, L.; et al. Genetic Manipulation of LKB1 Elicits Lethal Metastatic Prostate Cancer. J. Exp. Med. 2020, 217, e20191787. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Y.; Zhou, Y.; You, H. Molecular Mechanism of LKB1 in the Invasion and Metastasis of Colorectal Cancer. Oncol. Rep. 2018, 21, 1035–1044. [Google Scholar] [CrossRef]
- Sha, L.; Lian, F.; Li, K.; Chen, C.; Zhao, Y.; He, J.; Huang, S.; Wu, G. Under-Expression of LKB1 Is Associated with Enhanced P38-MAPK Signaling in Human Hepatocellular Carcinoma. Int. J. Clin. Exp. Pathol. 2018, 11, 5525–5535. [Google Scholar]
- Marcus, A.I.; Zhou, W. LKB1 Regulated Pathways in Lung Cancer Invasion and Metastasis. J. Thorac. Oncol. 2010, 5, 1883–1886. [Google Scholar] [CrossRef]
- Ji, H.; Ramsey, M.R.; Hayes, D.N.; Fan, C.; McNamara, K.; Kozlowski, P.; Torrice, C.; Wu, M.C.; Shimamura, T.; Perera, S.A.; et al. LKB1 Modulates Lung Cancer Differentiation and Metastasis. Nature 2007, 448, 807–810. [Google Scholar] [CrossRef] [PubMed]
- Bonanno, L.; De Paoli, A.; Zulato, E.; Esposito, G.; Calabrese, F.; Favaretto, A.; Santo, A.; Del Conte, A.; Chilosi, M.; Oniga, F.; et al. LKB1 Expression Correlates with Increased Survival in Patients with Advanced Non–Small Cell Lung Cancer Treated with Chemotherapy and Bevacizumab. Clin. Cancer Res. 2017, 23, 3316–3324. [Google Scholar] [CrossRef] [PubMed]
- Sfakianaki, M.; Papadaki, C.; Tzardi, M.; Trypaki, M.; Alam, S.; Lagoudaki, E.D.; Messaritakis, I.; Zoras, O.; Mavroudis, D.; Georgoulias, V.; et al. Loss of LKB1 Protein Expression Correlates with Increased Risk of Recurrence and Death in Patients with Resected, Stage II or III Colon Cancer. Cancer Res. Treat. 2019, 51, 1518–1526. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.-C.; Chang, Y.-C.; Lu, Y.-S.; Chung, K.-P.; Huang, C.-S.; Lu, T.-P.; Kuo, W.-H.; Wang, M.-Y.; Kuo, K.-T.; Wu, P.-F.; et al. Clinical Relevance of Liver Kinase B1(LKB1) Protein and Gene Expression in Breast Cancer. Sci. Rep. 2016, 6, 21374. [Google Scholar] [CrossRef]
- Karaglani, M.; Panagopoulou, M.; Baltsavia, I.; Apalaki, P.; Theodosiou, T.; Iliopoulos, I.; Tsamardinos, I.; Chatzaki, E. Tissue-Specific Methylation Biosignatures for Monitoring Diseases: An In Silico Approach. Int. J. Mol. Sci. 2022, 23, 2959. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. Immune Checkpoint Targeting in Cancer Therapy: Toward Combination Strategies with Curative Potential. Cell 2015, 161, 205–214. [Google Scholar] [CrossRef]
- Luciani, A.; Denley, M.C.S.; Govers, L.P.; Sorrentino, V.; Froese, D.S. Mitochondrial Disease, Mitophagy, and Cellular Distress in Methylmalonic Acidemia. Cell. Mol. Life Sci. 2021, 78, 6851–6867. [Google Scholar] [CrossRef]
- Nagelkerke, A.; Bussink, J.; Rowan, A.E.; Span, P.N. The Mechanical Microenvironment in Cancer: How Physics Affects Tumours. Semin. Cancer Biol. 2015, 35, 62–70. [Google Scholar] [CrossRef]
- Piera-Velazquez, S.; Jimenez, S.A. Endothelial to Mesenchymal Transition: Role in Physiology and in the Pathogenesis of Human Diseases. Physiol. Rev. 2019, 99, 1281–1324. [Google Scholar] [CrossRef]
- Saha, S.K.; Kim, K.; Yang, G.-M.; Choi, H.Y.; Cho, S.-G. Cytokeratin 19 (KRT19) Has a Role in the Reprogramming of Cancer Stem Cell-Like Cells to Less Aggressive and More Drug-Sensitive Cells. Int. J. Mol. Sci. 2018, 19, 1423. [Google Scholar] [CrossRef] [PubMed]
- Cazes, A.; Childers, B.G.; Esparza, E.; Lowy, A.M. The MST1R/RON Tyrosine Kinase in Cancer: Oncogenic Functions and Therapeutic Strategies. Cancers 2022, 14, 2037. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, E.; Yano, H.; Pan, C.; Komohara, Y.; Fujiwara, Y.; Zhao, S.; Shinchi, Y.; Kurotaki, D.; Suzuki, M. The Significance of SPP1 in Lung Cancers and Its Impact as a Marker for Protumor Tumor-Associated Macrophages. Cancers 2023, 15, 2250. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Y.; Liu, R.; Wang, C.; Luo, Y.; Chen, L.; He, Y.; Zhu, K.; Guo, H.; Zhang, Z.; et al. CAV2 Promotes the Invasion and Metastasis of Head and Neck Squamous Cell Carcinomas by Regulating S100 Proteins. Cell Death Discov. 2022, 8, 386. [Google Scholar] [CrossRef]
- Pajtler, K.W.; Sadowski, N.; Ackermann, S.; Althoff, K.; Schönbeck, K.; Batzke, K.; Schäfers, S.; Odersky, A.; Heukamp, L.; Astrahantseff, K.; et al. The GSK461364 PLK1 Inhibitor Exhibits Strong Antitumoral Activity in Preclinical Neuroblastoma Models. Oncotarget 2017, 8, 6730–6741. [Google Scholar] [CrossRef]
- Sengupta, S.; Nagalingam, A.; Muniraj, N.; Bonner, M.Y.; Mistriotis, P.; Afthinos, A.; Kuppusamy, P.; Lanoue, D.; Cho, S.; Korangath, P.; et al. Activation of Tumor Suppressor LKB1 by Honokiol Abrogates Cancer Stem-like Phenotype in Breast Cancer via Inhibition of Oncogenic Stat3. Oncogene 2017, 36, 5709–5721. [Google Scholar] [CrossRef]
- Gabriela-Freitas, M.; Pinheiro, J.; Raquel-Cunha, A.; Cardoso-Carneiro, D.; Martinho, O. RKIP as an Inflammatory and Immune System Modulator: Implications in Cancer. Biomolecules 2019, 9, 769. [Google Scholar] [CrossRef]
- Frankenberger, C.; Rabe, D.; Bainer, R.; Sankarasharma, D.; Chada, K.; Krausz, T.; Gilad, Y.; Becker, L.; Rosner, M.R. Metastasis Suppressors Regulate the Tumor Microenvironment by Blocking Recruitment of Prometastatic Tumor-Associated Macrophages. Cancer Res. 2015, 75, 4063–4073. [Google Scholar] [CrossRef]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Aref, A.R.; Skoulidis, F.; Herter-Sprie, G.S.; Buczkowski, K.A.; Liu, Y.; Awad, M.M.; Denning, W.L.; et al. STK11/LKB1 Deficiency Promotes Neutrophil Recruitment and Proinflammatory Cytokine Production to Suppress T-Cell Activity in the Lung Tumor Microenvironment. Cancer Res. 2016, 76, 999–1008. [Google Scholar] [CrossRef]
- Datar, I.; Qiu, X.; Ma, H.Z.; Yeung, M.; Aras, S.; de la Serna, I.; Al-Mulla, F.; Thiery, J.P.; Trumbly, R.; Fan, X.; et al. RKIP Regulates CCL5 Expression to Inhibit Breast Cancer Invasion and Metastasis by Controlling Macrophage Infiltration. Oncotarget 2015, 6, 39050–39061. [Google Scholar] [CrossRef]
- Viau, A.; Bienaimé, F.; Lukas, K.; Todkar, A.P.; Knoll, M.; Yakulov, T.A.; Hofherr, A.; Kretz, O.; Helmstädter, M.; Reichardt, W.; et al. Cilia-localized LKB1 Regulates Chemokine Signaling, Macrophage Recruitment, and Tissue Homeostasis in the Kidney. EMBO J. 2018, 37, e98615. [Google Scholar] [CrossRef]
- Dong, Y.; Lin, X.; Kapoor, A.; Gu, Y.; Xu, H.; Major, P.; Tang, D. Insights of RKIP-Derived Suppression of Prostate Cancer. Cancers 2021, 13, 6388. [Google Scholar] [CrossRef]
- Pons-Tostivint, E.; Lugat, A.; Fontenau, J.-F.; Denis, M.G.; Bennouna, J. STK11/LKB1 Modulation of the Immune Response in Lung Cancer: From Biology to Therapeutic Impact. Cells 2021, 10, 3129. [Google Scholar] [CrossRef]
- Lv, L.; Miao, Q.; Zhan, S.; Chen, P.; Liu, W.; Lv, J.; Yan, W.; Wang, D.; Liu, H.; Yin, J.; et al. LKB1 Dictates Sensitivity to Immunotherapy through Skp2-Mediated Ubiquitination of PD-L1 Protein in Non-Small Cell Lung Cancer. J. Immunother. Cancer 2024, 12, e009444. [Google Scholar] [CrossRef] [PubMed]
- Jing, S.-H.; Gao, X.; Yu, B.; Qiao, H. Raf Kinase Inhibitor Protein (RKIP) Inhibits Tumor Necrosis Factor-α (TNF-α) Induced Adhesion Molecules Expression in Vascular Smooth Muscle Bells by Suppressing (Nuclear Transcription Factor-ΚB (NF-KappaB) Pathway. Med. Sci. Monit. 2017, 23, 4789–4797. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-R.; Yoo, S.-J.; Kim, J.; Kang, S.W. LKB1 Regulates Inflammation of Fibroblast-like Synoviocytes from Patients with Rheumatoid Arthritis via AMPK-Dependent SLC7A11-NOX4-ROS Signaling. Cells 2023, 12, 1263. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Guan, L.; Zhou, Q.; Huang, H.; Xu, L. LKB1 Alleviates High Glucose- and High Fat-induced Inflammation and the Expression of GnRH and Sexual Precocity-related Genes, in Mouse Hypothalamic Cells by Activating the AMPK/FOXO1 Signaling Pathway. Mol. Med. Rep. 2022, 25, 143. [Google Scholar] [CrossRef]
- Feng, H.; Shen, W. ACAA1 Is a Predictive Factor of Survival and Is Correlated With T Cell Infiltration in Non-Small Cell Lung Cancer. Front. Oncol. 2020, 10, 564796. [Google Scholar] [CrossRef]
- Zhuang, Z.-G.; Di, G.-H.; Shen, Z.-Z.; Ding, J.; Shao, Z.-M. Enhanced Expression of LKB1 in Breast Cancer Cells Attenuates Angiogenesis, Invasion, and Metastatic Potential. Mol. Cancer Res. 2006, 4, 843–849. [Google Scholar] [CrossRef]
- Bonavida, B. RKIP: A Pivotal Gene Product in the Pathogenesis of Cancer. Cancers 2022, 14, 6092. [Google Scholar] [CrossRef]
- Baritaki, S.; Zaravinos, A. Cross-Talks between RKIP and YY1 through a Multilevel Bioinformatics Pan-Cancer Analysis. Cancers 2023, 15, 4932. [Google Scholar] [CrossRef]
- Roy, B.C.; Kohno, T.; Iwakawa, R.; Moriguchi, T.; Kiyono, T.; Morishita, K.; Sanchez-Cespedes, M.; Akiyama, T.; Yokota, J. Involvement of LKB1 in Epithelial–Mesenchymal Transition (EMT) of Human Lung Cancer Cells. Lung Cancer 2010, 70, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Wang, J.; Wang, J.; Luh, F.; Liu, X.; Yang, L.; Liu, Y.-R.; Su, L.; Yang, Y.-C.S.; Chu, P.; et al. LKB1 Deficiency Promotes Proliferation and Invasion of Glioblastoma through Activation of MTOR and Focal Adhesion Kinase Signaling Pathways. Am. J. Cancer Res. 2019, 9, 1650–1663. [Google Scholar] [PubMed]
- He, Q.-Y.; Yi, H.-M.; Yi, H.; Xiao, T.; Qu, J.-Q.; Yuan, L.; Zhu, J.-F.; Li, J.-Y.; Wang, Y.-Y.; Li, L.-N.; et al. Reduction of RKIP Expression Promotes Nasopharyngeal Carcinoma Invasion and Metastasis by Activating Stat3 Signaling. Oncotarget 2015, 6, 16422–16436. [Google Scholar] [CrossRef]
- Janjusevic, M.; Greco, S.; Islam, M.S.; Castellucci, C.; Ciavattini, A.; Toti, P.; Petraglia, F.; Ciarmela, P. Locostatin, a Disrupter of Raf Kinase Inhibitor Protein, Inhibits Extracellular Matrix Production, Proliferation, and Migration in Human Uterine Leiomyoma and Myometrial Cells. Fertil. Steril. 2016, 106, 1530–1538.e1. [Google Scholar] [CrossRef]
- Ndembe, G.; Intini, I.; Perin, E.; Marabese, M.; Caiola, E.; Mendogni, P.; Rosso, L.; Broggini, M.; Colombo, M. LKB1: Can We Target an Hidden Target? Focus on NSCLC. Front. Oncol. 2022, 12, 889826. [Google Scholar] [CrossRef]
- Noh, H.S.; Hah, Y.-S.; Zada, S.; Ha, J.H.; Sim, G.; Hwang, J.S.; Lai, T.H.; Nguyen, H.Q.; Park, J.-Y.; Kim, H.J.; et al. PEBP1, a RAF Kinase Inhibitory Protein, Negatively Regulates Starvation-Induced Autophagy by Direct Interaction with LC3. Autophagy 2016, 12, 2183–2196. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.T.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A.; et al. Roles of the Raf/MEK/ERK Pathway in Cell Growth, Malignant Transformation and Drug Resistance. Biochim. Biophys. Acta Mol. Cell Res. 2007, 1773, 1263–1284. [Google Scholar] [CrossRef]
- Zhou, W.; Yan, L.-D.; Yu, Z.-Q.; Li, N.; Yang, Y.-H.; Wang, M.; Chen, Y.-Y.; Mao, M.-X.; Peng, X.-C.; Cai, J. Role of STK11 in ALK -positive Non-small Cell Lung Cancer (Review). Oncol. Lett. 2022, 23, 181. [Google Scholar] [CrossRef]
- Theodosiou, T.; Papanikolaou, N.; Savvaki, M.; Bonetto, G.; Maxouri, S.; Fakoureli, E.; Eliopoulos, A.G.; Tavernarakis, N.; Amoutzias, G.D.; Pavlopoulos, G.A.; et al. UniProt-Related Documents (UniReD): Assisting Wet Lab Biologists in Their Quest on Finding Novel Counterparts in a Protein Network. NAR Genom. Bioinform. 2020, 2, lqaa005. [Google Scholar] [CrossRef]
- Baltsavia, I.; Theodosiou, T.; Papanikolaou, N.; Pavlopoulos, G.A.; Amoutzias, G.D.; Panagopoulou, M.; Chatzaki, E.; Andreakos, E.; Iliopoulos, I. Prediction and Ranking of Biomarkers Using Multiple UniReD. Int. J. Mol. Sci. 2022, 23, 11112. [Google Scholar] [CrossRef]
- Akbani, R.; Ng, P.K.S.; Werner, H.M.J.; Shahmoradgoli, M.; Zhang, F.; Ju, Z.; Liu, W.; Yang, J.-Y.; Yoshihara, K.; Li, J.; et al. A Pan-Cancer Proteomic Perspective on The Cancer Genome Atlas. Nat. Commun. 2014, 5, 3887. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Zhang, Q.; Lei, Q.; Luo, M.; Xie, G.; Wang, H.; Guo, A. ImmuCellAI: A Unique Method for Comprehensive T-Cell Subsets Abundance Prediction and Its Application in Cancer Immunotherapy. Adv. Sci. 2020, 7, 1902880. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Xiang, Y.; Ozguc, F.M.; Kim, Y.; Liu, C.-J.; Park, P.K.; Hu, Q.; Diao, L.; Lou, Y.; Lin, C.; et al. The Genomic Landscape and Pharmacogenomic Interactions of Clock Genes in Cancer Chronotherapy. Cell Syst. 2018, 6, 314–328.e2. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Bodycombe, N.E.; Cheah, J.H.; Price, E.V.; Liu, K.; Schaefer, G.I.; Ebright, R.Y.; Stewart, M.L.; Ito, D.; Wang, S.; et al. An Interactive Resource to Identify Cancer Genetic and Lineage Dependencies Targeted by Small Molecules. Cell 2013, 154, 1151–1161. [Google Scholar] [CrossRef]
- Rees, M.G.; Seashore-Ludlow, B.; Cheah, J.H.; Adams, D.J.; Price, E.V.; Gill, S.; Javaid, S.; Coletti, M.E.; Jones, V.L.; Bodycombe, N.E.; et al. Correlating Chemical Sensitivity and Basal Gene Expression Reveals Mechanism of Action. Nat. Chem. Biol. 2016, 12, 109–116. [Google Scholar] [CrossRef]
- Seashore-Ludlow, B.; Rees, M.G.; Cheah, J.H.; Cokol, M.; Price, E.V.; Coletti, M.E.; Jones, V.; Bodycombe, N.E.; Soule, C.K.; Gould, J.; et al. Harnessing Connectivity in a Large-Scale Small-Molecule Sensitivity Dataset. Cancer Discov. 2015, 5, 1210–1223. [Google Scholar] [CrossRef]
- Luna, A.; Elloumi, F.; Varma, S.; Wang, Y.; Rajapakse, V.N.; Aladjem, M.I.; Robert, J.; Sander, C.; Pommier, Y.; Reinhold, W.C. CellMiner Cross-Database (CellMinerCDB) Version 1.2: Exploration of Patient-Derived Cancer Cell Line Pharmacogenomics. Nucleic Acids Res. 2021, 49, D1083–D1093. [Google Scholar] [CrossRef]
- Fu, J.; Li, K.; Zhang, W.; Wan, C.; Zhang, J.; Jiang, P.; Liu, X.S. Large-Scale Public Data Reuse to Model Immunotherapy Response and Resistance. Genome Med. 2020, 12, 21. [Google Scholar] [CrossRef]
- Jiang, P.; Gu, S.; Pan, D.; Fu, J.; Sahu, A.; Hu, X.; Li, Z.; Traugh, N.; Bu, X.; Li, B.; et al. Signatures of T Cell Dysfunction and Exclusion Predict Cancer Immunotherapy Response. Nat. Med. 2018, 24, 1550–1558. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skouradaki, E.; Zaravinos, A.; Panagopoulou, M.; Chatzaki, E.; Dovrolis, N.; Baritaki, S. Pan-Cancer Computational Analysis of RKIP (PEBP1) and LKB1 (STK11) Co-Expression Highlights Distinct Immunometabolic Dynamics and Therapeutic Responses Within the Tumor Microenvironment. Int. J. Mol. Sci. 2025, 26, 7145. https://doi.org/10.3390/ijms26157145
Skouradaki E, Zaravinos A, Panagopoulou M, Chatzaki E, Dovrolis N, Baritaki S. Pan-Cancer Computational Analysis of RKIP (PEBP1) and LKB1 (STK11) Co-Expression Highlights Distinct Immunometabolic Dynamics and Therapeutic Responses Within the Tumor Microenvironment. International Journal of Molecular Sciences. 2025; 26(15):7145. https://doi.org/10.3390/ijms26157145
Chicago/Turabian StyleSkouradaki, Evangelia, Apostolos Zaravinos, Maria Panagopoulou, Ekaterini Chatzaki, Nikolas Dovrolis, and Stavroula Baritaki. 2025. "Pan-Cancer Computational Analysis of RKIP (PEBP1) and LKB1 (STK11) Co-Expression Highlights Distinct Immunometabolic Dynamics and Therapeutic Responses Within the Tumor Microenvironment" International Journal of Molecular Sciences 26, no. 15: 7145. https://doi.org/10.3390/ijms26157145
APA StyleSkouradaki, E., Zaravinos, A., Panagopoulou, M., Chatzaki, E., Dovrolis, N., & Baritaki, S. (2025). Pan-Cancer Computational Analysis of RKIP (PEBP1) and LKB1 (STK11) Co-Expression Highlights Distinct Immunometabolic Dynamics and Therapeutic Responses Within the Tumor Microenvironment. International Journal of Molecular Sciences, 26(15), 7145. https://doi.org/10.3390/ijms26157145