
A Late-Onset and Mild Phenotype of Mitochondrial Complex I Deficiency Due to a Novel Reported Variant Within the ACAD9 Gene

 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
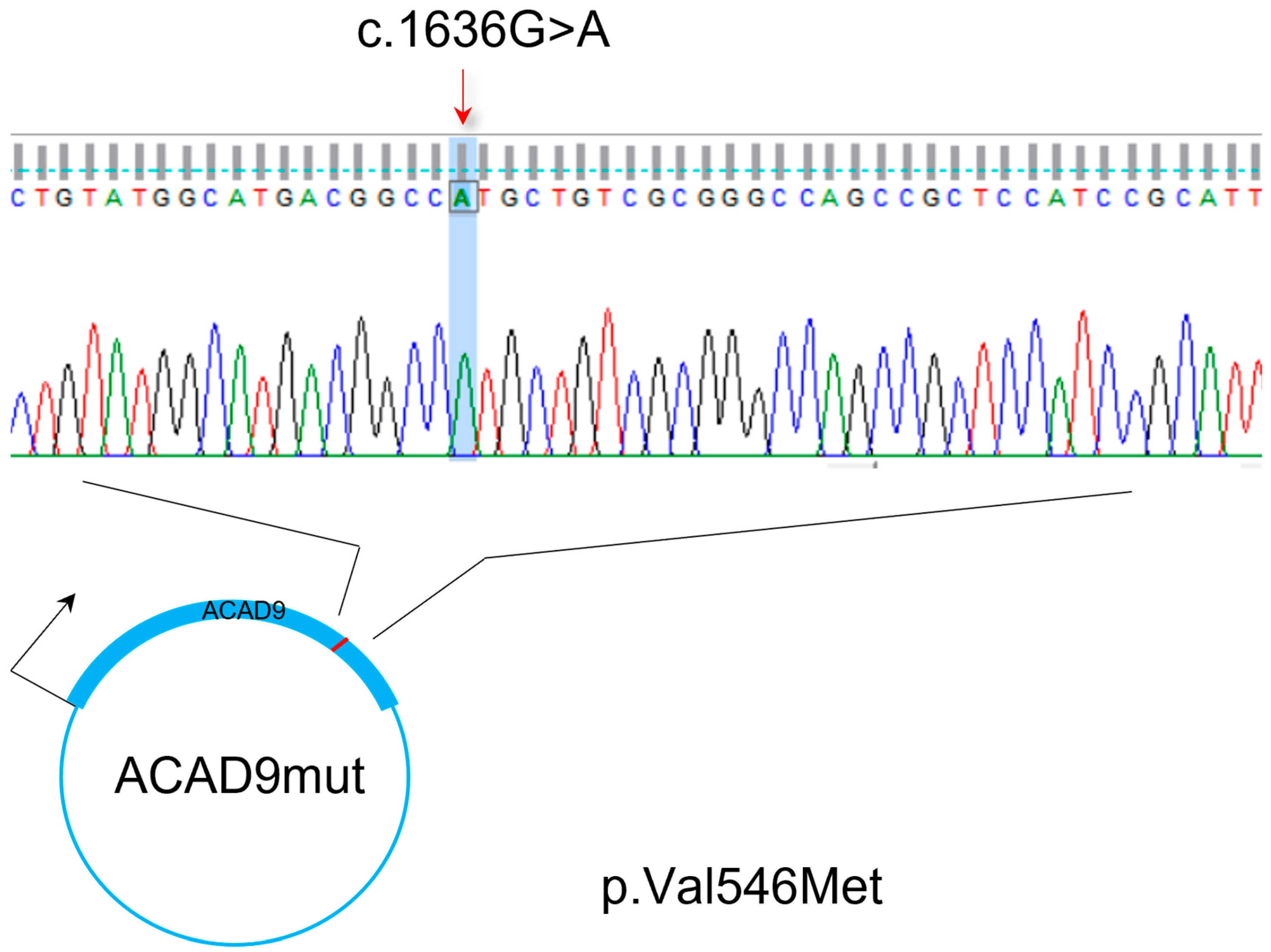
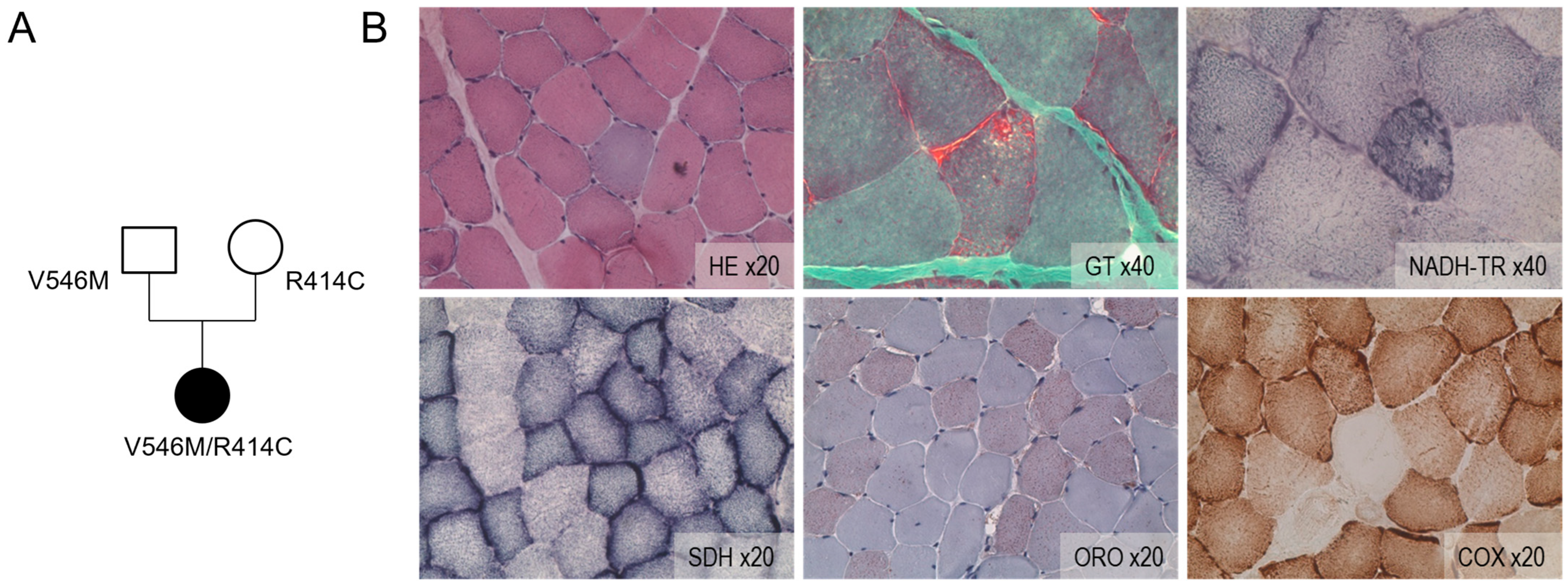
2.1. Compound Heterozygosity Is Associated with Mitochondrial Disturbance
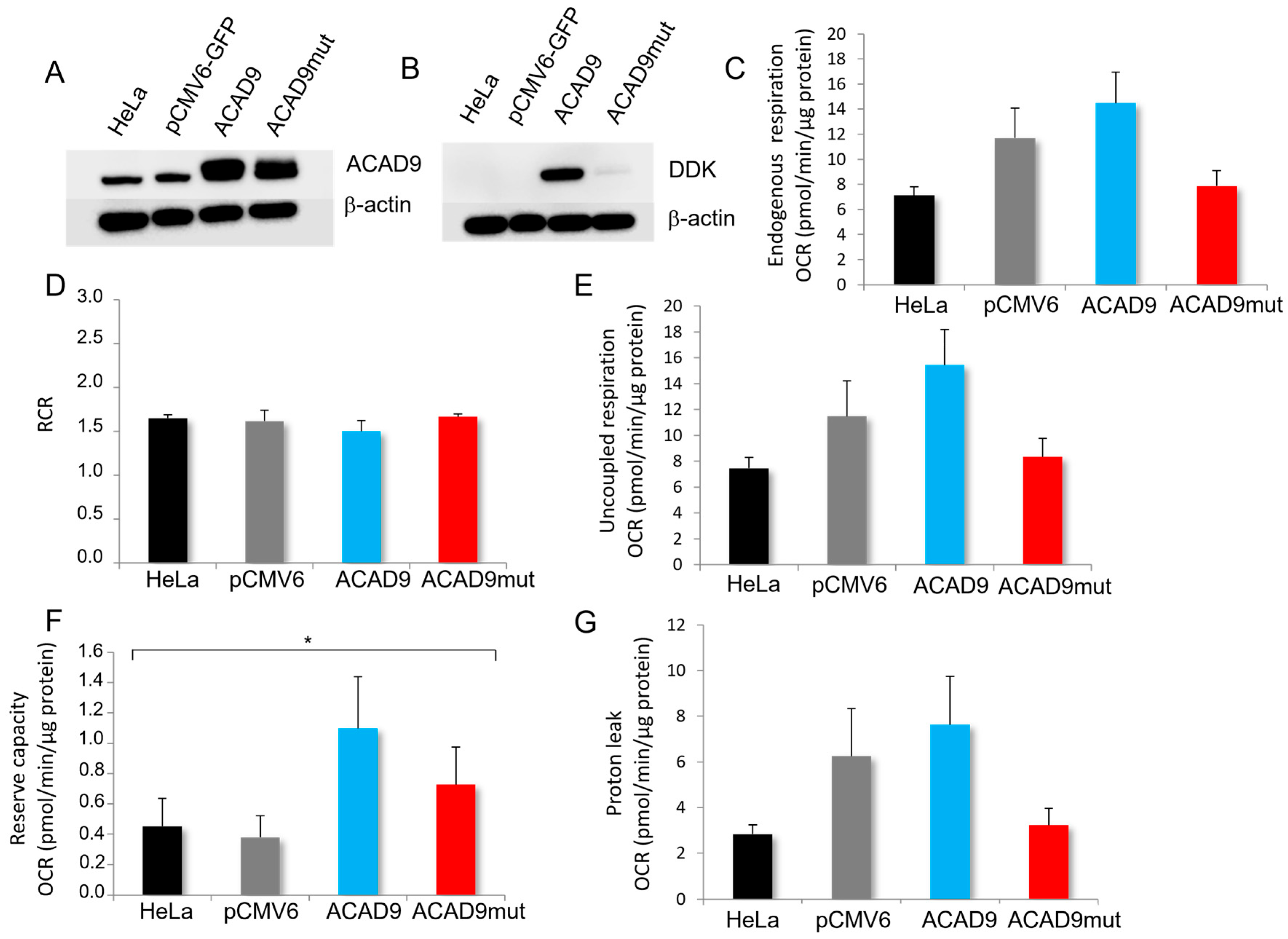
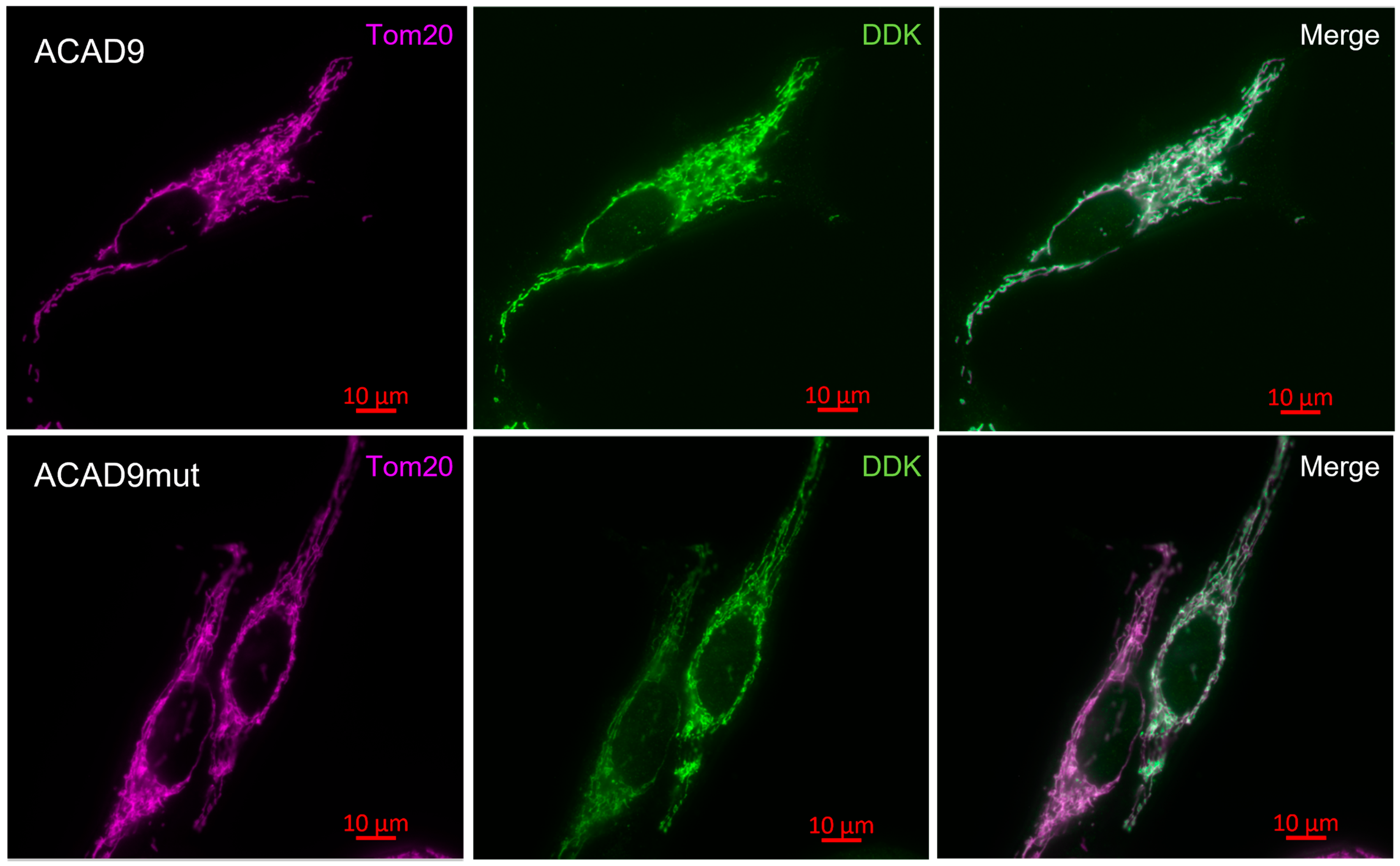
2.2. The Expression of V546M Variant Does Not Affect the Mitochondrial Efficiency
2.3. The Expression of V546M Variant Reduces the Complex I Activity
3. Discussion
4. Materials and Methods
4.1. Patient Description/Diagnosis Arguments
4.1.1. Clinical Description
4.1.2. Histological Analysis of Muscle Biopsy
4.2. In Vitro Analysis
4.2.1. Cell Culture
4.2.2. Site-Directed Mutagenesis
4.2.3. Western Blot
4.2.4. Oxygen Consumption Rate Measurements
4.2.5. ATP Assay
4.2.6. Enzymology
4.2.7. Immunocytochemistry
4.2.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACAD9 | Acyl-CoA dehydrogenase 9 |
| OMIM | Online Mendelian Inheritance in Man |
| ATP | Adenosine triphosphate |
| FAO | β-oxidation of fatty acids |
| SCAD | Short-chain fatty acid dehydrogenase |
| MCAD | Medium-chain fatty acid dehydrogenase |
| LCAD | Long-chain fatty acid dehydrogenase |
| VLCAD | Very-long-chain acyl-CoA dehydrogenase |
| FAD | Oxidized form of flavine adenine dinucleotide |
| NDUFAF1 | NADH: Ubiquinone Oxidoreductase Complex Assembly Factor 1 |
| ECSIT | Evolutionarily Conserved Signaling Intermediate in Toll Pathway |
| MCIA | Mitochondrial complex I intermediate assembly |
| TMEM126B | Transmembrane protein 126B |
| TMEM186 | Transmembrane protein 186 |
| TIMMDC1 | Translocase of inner mitochondrial membrane domain containing 1 |
| OXPHOS | Oxidative phosphorylation |
| NADH | Reduced form of nicotinamide adenine dinucleotide dehydrogenase |
| FADH2 | Reduced form of flavin adenine dinucleotide |
| RIPA | Radioimmunoprecipitation assay buffer |
| BCA | Bicinchoninic acid |
| CPK | Creatine phosphokinase |
| HE | Hematoxylin and eosin |
| GT | Gomori trichrome |
| ORO | Oil red O |
| NADH-TR | Reduced nicotinamide adenine dinucleotide dehydrogenase–tetrazolium reductase |
| SDH | Succinic dehydrogenase |
| COX | Cytochrome oxidase |
| ATCC | American Type Culture Collection |
| OCR | Oxygen consumption rates |
| CCCP | Carbonyl cyanide m-chlorophenyl hydrazone |
| GFP | Green fluorescent protein |
| PBS | Phosphate-buffered saline |
| SEM | Standard error of the mean |
| MERRF | Myoclonic epilepsy with ragged red fibers |
| MELAS | Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes |
| NARP | Neuropathy, ataxia, and retinitis pigmentosa |
| NDUFB7 | NADH: Ubiquinone Oxidoreductase Subunit B7 |
| NDUFB8 | NADH: Ubiquinone Oxidoreductase Subunit B8 (also known as CI20) |
| CI20 | Complex I subunit of 20KDa |
| NDUFS8 | NADH: Ubiquinone Oxidoreductase Subunit S8 |
| ACADVL | Gene of VLCAD protein |
| TAZ | Tafazzin |
| DNAJC19 | DNAJ heat shock protein family member C19 |
Appendix A

References
- Gerards, M.; van den Bosch, B.J.C.; Danhauser, K.; Serre, V.; van Weeghel, M.; Wanders, R.J.A.; Nicolaes, G.A.F.; Sluiter, W.; Schoonderwoerd, K.; Scholte, H.R.; et al. Riboflavin-responsive oxidative phosphorylation complex I deficiency caused by defective ACAD9: New function for an old gene. Brain 2011, 134, 210–219. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Rutledge, S.L.; Kelly, D.R.; Palmer, C.A.; Murdoch, G.; Majumder, N.; Nicholls, R.D.; Pei, Z.; Watkins, P.A.; Vockley, J. A new genetic disorder in mitochondrial fatty acid beta-oxidation: ACAD9 deficiency. Am. J. Hum. Genet. 2007, 81, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Garone, C.; Donati, M.A.; Sacchini, M.; Garcia-Diaz, B.; Bruno, C.; Calvo, S.; Mootha, V.K.; Dimauro, S. Mitochondrial encephalomyopathy due to a novel mutation in ACAD9. JAMA Neurol. 2013, 70, 1177–1179. [Google Scholar] [CrossRef] [PubMed]
- Schiff, M.; Haberberger, B.; Xia, C.; Mohsen, A.-W.; Goetzman, E.S.; Wang, Y.; Uppala, R.; Zhang, Y.; Karunanidhi, A.; Prabhu, D.; et al. Complex I assembly function and fatty acid oxidation enzyme activity of ACAD9 both contribute to disease severity in ACAD9 deficiency. Hum. Mol. Genet. 2015, 24, 3238–3247. [Google Scholar] [CrossRef] [PubMed]
- Leslie, N.; Wang, X.; Peng, Y.; Valencia, C.A.; Khuchua, Z.; Hata, J.; Witte, D.; Huang, T.; Bove, K.E. Neonatal multiorgan failure due to ACAD9 mutation and complex I deficiency with mitochondrial hyperplasia in liver, cardiac myocytes, skeletal muscle, and renal tubules. Hum. Pathol. 2016, 49, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Fragaki, K.; Chaussenot, A.; Boutron, A.; Bannwarth, S.; Cochaud, C.; Richelme, C.; Sacconi, S.; Paquis-Flucklinger, V. Severe defect in mitochondrial complex I assembly with mitochondrial DNA deletions in ACAD9-deficient mild myopathy. Muscle Nerve 2017, 55, 919–922. [Google Scholar] [CrossRef] [PubMed]
- Repp, B.M.; Mastantuono, E.; Alston, C.L.; Schiff, M.; Haack, T.B.; Rötig, A.; Ardissone, A.; Lombès, A.; Catarino, C.B.; Diodato, D.; et al. Clinical, biochemical and genetic spectrum of 70 patients with ACAD9 deficiency: Is riboflavin supplementation effective? Orphanet J. Rare Dis. 2018, 13, 120. [Google Scholar] [CrossRef] [PubMed]
- Telford, E.A.; Moynihan, L.M.; Markham, A.F.; Lench, N.J. Isolation and characterisation of a cDNA encoding the precursor for a novel member of the acyl-CoA dehydrogenase gene family. Biochim. Biophys. Acta 1999, 1446, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Nouws, J.; Te brinke, H.; Nijtmans, L.G.; Houten, S.M. ACAD9, a complex I assembly factor with a moonlighting function in fatty acid oxidation deficiencies. Hum. Mol. Genet. 2014, 23, 1311–1319. [Google Scholar] [CrossRef] [PubMed]
- Ensenauer, R.; He, M.; Willard, J.M.; Goetzman, E.S.; Corydon, T.J.; Vandahl, B.B.; Mohsen, A.W.; Isaya, G.; Vockley, J. Human acyl-CoA dehydrogenase-9 plays a novel role in the mitochondrial beta-oxidation of unsaturated fatty acids. J. Biol. Chem. 2005, 280, 32309–32316. [Google Scholar] [CrossRef] [PubMed]
- Chuaijit, S.; Boonyatistan, W.; Boonchuay, P.; Metheetrairut, C.; Suthammarak, W. Identification of a novel mitochondrial complex I assembly factor ACDH-12 in Caenorhabditis elegans. Mitochondrion 2019, 46, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Sinsheimer, A.; Mohsen, A.W.; Bloom, K.; Karunanidhi, A.; Bharathi, S.; Wu, Y.L.; Schiff, M.; Wang, Y.; Goetzman, E.S.; Ghaloul-Gonzalez, L.; et al. Development and characterization of a mouse model for Acad9 deficiency. Mol. Genet. Metab. 2021, 134, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, W.; Zou, D.; Chen, G.; Wan, T.; Zhang, M.; Cao, X. Cloning and functional characterization of ACAD-9, a novel member of human acyl-CoA dehydrogenase family. Biochem. Biophys. Res. Commun. 2002, 297, 1033–1042. [Google Scholar] [CrossRef] [PubMed]
- Nouws, J.; Nijtmans, L.; Houten, S.M.; Van Den Brand, M.; Huynen, M.; Venselaar, H.; Hoefs, S.; Gloerich, J.; Kronick, J.; Hutchin, T.; et al. Acyl-CoA dehydrogenase 9 is required for the biogenesis of oxidative phosphorylation complex I. Cell Metab. 2010, 12, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.; He, J.; Ding, S.; Fearnley, I.M.; Walker, J.E. TMEM70 and TMEM242 help to assemble the rotor ring of human ATP synthase and interact with assembly factors for complex I. Proc. Natl. Acad. Sci. USA 2021, 118, e2100558118. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Lou, B.; Fu, Z.; Mohsen, A.W.; Shen, A.L.; Vockley, J.; Kim, J.J.P. Molecular mechanism of interactions between ACAD9 and binding partners in mitochondrial respiratory complex I assembly. iScience 2021, 24, 103153. [Google Scholar] [CrossRef] [PubMed]
- Heide, H.; Bleier, L.; Steger, M.; Ackermann, J.; Dröse, S.; Schwamb, B.; Zörnig, M.; Reichert, A.S.; Koch, I.; Wittig, I.; et al. Complexome profiling identifies TMEM126B as a component of the mitochondrial complex I assembly complex. Cell Metab. 2012, 16, 538–549. [Google Scholar] [CrossRef] [PubMed]
- Giachin, G.; Jessop, M.; Bouverot, R.; Acajjaoui, S.; Saïdi, M.; Chretien, A.; Bacia-Verloop, M.; Signor, L.; Mas, P.J.; Favier, A.; et al. Assembly of The Mitochondrial Complex I Assembly Complex Suggests a Regulatory Role for Deflavination. Angew. Chem.-Int. Ed. 2021, 60, 4689–4697. [Google Scholar] [CrossRef] [PubMed]
- Murgia, C.; Dehlia, A.; Guthridge, M.A. New insights into the nutritional genomics of adult-onset riboflavin-responsive diseases. Nutr. Metab. 2023, 20, 42. [Google Scholar] [CrossRef] [PubMed]
- Haack, T.B.; Danhauser, K.; Haberberger, B.; Hoser, J.; Strecker, V.; Boehm, D.; Uziel, G.; Lamantea, E.; Invernizzi, F.; Poulton, J.; et al. Exome sequencing identifies ACAD9 mutations as a cause of complex I deficiency. Nat. Genet. 2010, 42, 1131–1134. [Google Scholar] [CrossRef] [PubMed]
- Dewulf, J.P.; Barrea, C.; Vincent, M.F.; De Laet, C.; Van Coster, R.; Seneca, S.; Marie, S.; Nassogne, M.C. Evidence of a wide spectrum of cardiac involvement due to ACAD9 mutations: Report on nine patients. Mol. Genet. Metab. 2016, 118, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Haack, T.B.; Haberberger, B.; Frisch, E.M.; Wieland, T.; Iuso, A.; Gorza, M.; Strecker, V.; Graf, E.; Mayr, J.A.; Herberg, U.; et al. Molecular diagnosis in mitochondrial complex I deficiency using exome sequencing. J. Med. Genet. 2012, 49, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Riley, L.G.; Cowley, M.J.; Gayevskiy, V.; Minoche, A.E.; Puttick, C.; Thorburn, D.R.; Rius, R.; Compton, A.G.; Menezes, M.J.; Bhattacharya, K.; et al. The diagnostic utility of genome sequencing in a pediatric cohort with suspected mitochondrial disease. Genet. Med. 2020, 22, 1254–1261. [Google Scholar] [CrossRef] [PubMed]
- Schrank, B.; Schoser, B.; Klopstock, T.; Schneiderat, P.; Horvath, R.; Abicht, A.; Holinski-Feder, E.; Augustis, S. Lifetime exercise intolerance with lactic acidosis as key manifestation of novel compound heterozygous ACAD9 mutations causing complex I deficiency. Neuromuscul. Disord. 2017, 27, 473–476. [Google Scholar] [CrossRef] [PubMed]
- Gerull, B.; Klaassen, S.; Brodehl, A. The genetic Landscape of cardiomyopathies. In Genetic Causes of Cardiac Disease; Erdmann, J., Moretti, A., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 45–94. [Google Scholar]
- Aintablian, H.K.; Narayanan, V.; Belnap, N.; Ramsey, K.; Grebe, T.A. An atypical presentation of ACAD9 deficiency: Diagnosis by whole exome sequencing broadens the phenotypic spectrum and alters treatment approach. Mol. Genet. Metab. Rep. 2017, 10, 38. [Google Scholar] [CrossRef] [PubMed]
- Collet, M.; Assouline, Z.; Bonnet, D.; Rio, M.; Iserin, F.; Sidi, D.; Goldenberg, A.; Lardennois, C.; Metodiev, M.D.; Haberberger, B.; et al. High incidence and variable clinical outcome of cardiac hypertrophy due to ACAD9 mutations in childhood. Eur. J. Hum. Genet. EJHG 2016, 24, 1112–1116. [Google Scholar] [CrossRef] [PubMed]
- Fragaki, K.; Chaussenot, A.; Boutron, A.; Bannwarth, S.; Rouzier, C.; Chabrol, B.; Paquis-Flucklinger, V. Assembly defects of multiple respiratory chain complexes in a child with cardiac hypertrophy associated with a novel ACAD9 mutation. Mol. Genet. Metab. 2017, 121, 224–226. [Google Scholar] [CrossRef] [PubMed]
- McGregor, L.; Acajjaoui, S.; Desfosses, A.; Saïdi, M.; Bacia-Verloop, M.; Schwarz, J.J.; Juyoux, P.; von Velsen, J.; Bowler, M.W.; McCarthy, A.A.; et al. The assembly of the Mitochondrial Complex I Assembly complex uncovers a redox pathway coordination. Nat. Commun. 2023, 14, 8248. [Google Scholar] [CrossRef] [PubMed]
- Formosa, L.E.; Dibley, M.G.; Stroud, D.A.; Ryan, M.T. Building a complex complex: Assembly of mitochondrial respiratory chain complex I. Semin. Cell Dev. Biol. 2018, 76, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Brauer, N.; Maruta, Y.; Lisci, M.; Strege, K.; Oschlies, I.; Nakamura, H.; Böhm, S.; Lehmberg, K.; Brandhoff, L.; Ehl, S.; et al. Immunodeficiency with susceptibility to lymphoma with complex genotype affecting energy metabolism (FBP1, ACAD9) and vesicle trafficking (RAB27A). Front. Immunol. 2023, 14, 1151166. [Google Scholar] [CrossRef] [PubMed]
- Cogburn, L.A.; Smarsh, D.N.; Wang, X.; Trakooljul, N.; Carré, W.; White, H.B., III. Transcriptional profiling of liver in riboflavin-deficient chicken embryos explains impaired lipid utilization, energy depletion, massive hemorrhaging, and delayed feathering. BMC Genom. 2018, 19, 177. [Google Scholar] [CrossRef] [PubMed]
- Bourgeois, J.M.; Tarnopolsky, M.A. Pathology of skeletal muscle in mitochondrial disorders. Mitochondrion 2004, 4, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, H.M.; Carl, S.M.; Swerdlow, R.H. Cytoplasmic hybrid (cybrid) cell lines as a practical model for mitochondriopathies. Redox Biol. 2014, 2, 619–631. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giguet-Valard, A.G.; Ait-El-Mkadem Saadi, S.; Duclos, S.; Lacombe, D.; Bellance, R.; Bellance, N. A Late-Onset and Mild Phenotype of Mitochondrial Complex I Deficiency Due to a Novel Reported Variant Within the ACAD9 Gene. Int. J. Mol. Sci. 2025, 26, 7128. https://doi.org/10.3390/ijms26157128
Giguet-Valard AG, Ait-El-Mkadem Saadi S, Duclos S, Lacombe D, Bellance R, Bellance N. A Late-Onset and Mild Phenotype of Mitochondrial Complex I Deficiency Due to a Novel Reported Variant Within the ACAD9 Gene. International Journal of Molecular Sciences. 2025; 26(15):7128. https://doi.org/10.3390/ijms26157128
Chicago/Turabian StyleGiguet-Valard, Anna Gaelle, Samira Ait-El-Mkadem Saadi, Sophie Duclos, Didier Lacombe, Rémi Bellance, and Nadège Bellance. 2025. "A Late-Onset and Mild Phenotype of Mitochondrial Complex I Deficiency Due to a Novel Reported Variant Within the ACAD9 Gene" International Journal of Molecular Sciences 26, no. 15: 7128. https://doi.org/10.3390/ijms26157128
APA StyleGiguet-Valard, A. G., Ait-El-Mkadem Saadi, S., Duclos, S., Lacombe, D., Bellance, R., & Bellance, N. (2025). A Late-Onset and Mild Phenotype of Mitochondrial Complex I Deficiency Due to a Novel Reported Variant Within the ACAD9 Gene. International Journal of Molecular Sciences, 26(15), 7128. https://doi.org/10.3390/ijms26157128