
Assessment of Brain Morphological Abnormalities and Neurodevelopmental Risk Copy Number Variants in Individuals from the UK Biobank

 and
and
Abstract
1. Introduction
2. Results
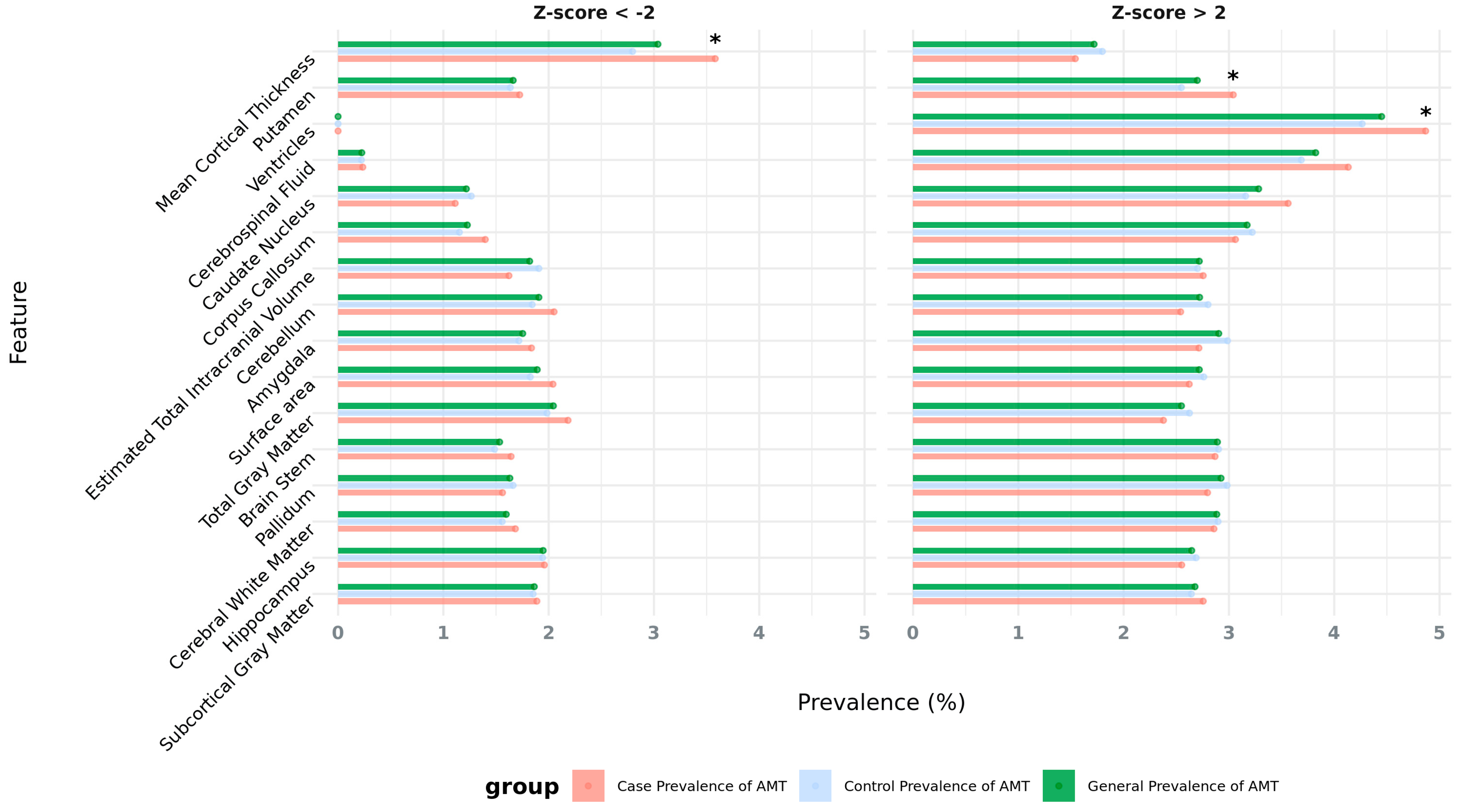
2.1. Association Between Abnormal Brain Morphological Traits and Neuropsychiatric Disorders
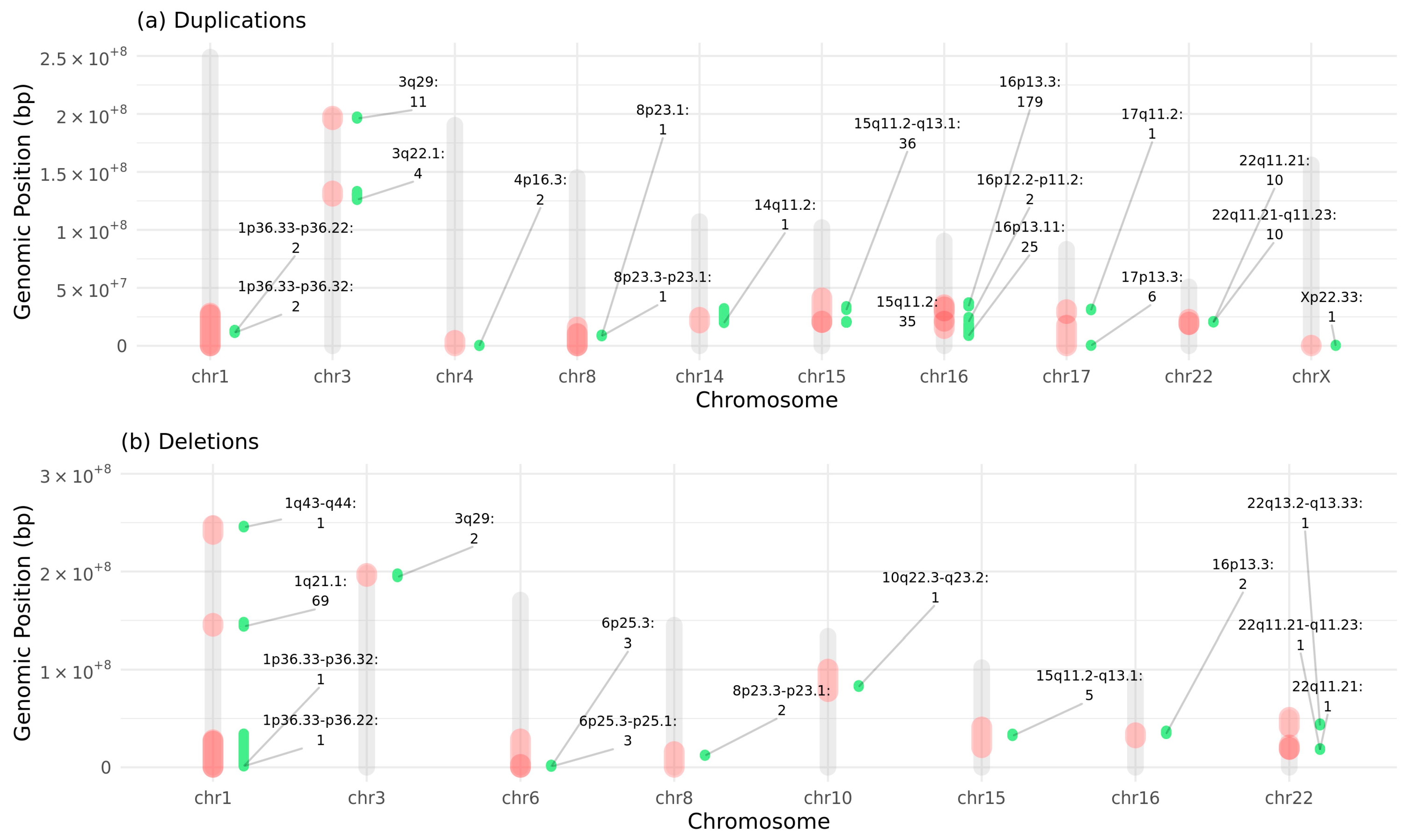
2.2. Subjects with Deletions or Duplications in NDD-Risk Regions Are Enriched for Aberrant Morphological Brain Traits
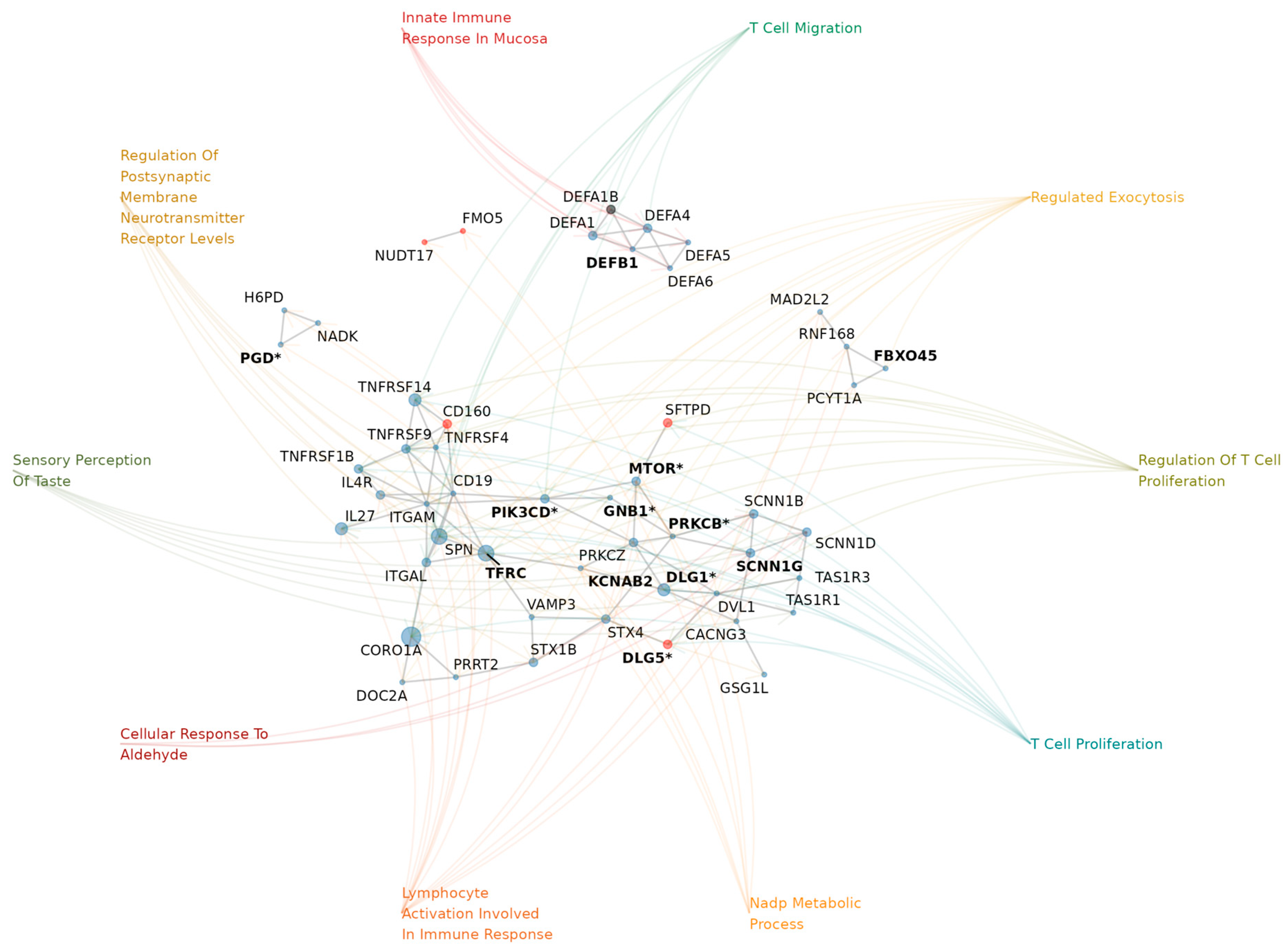
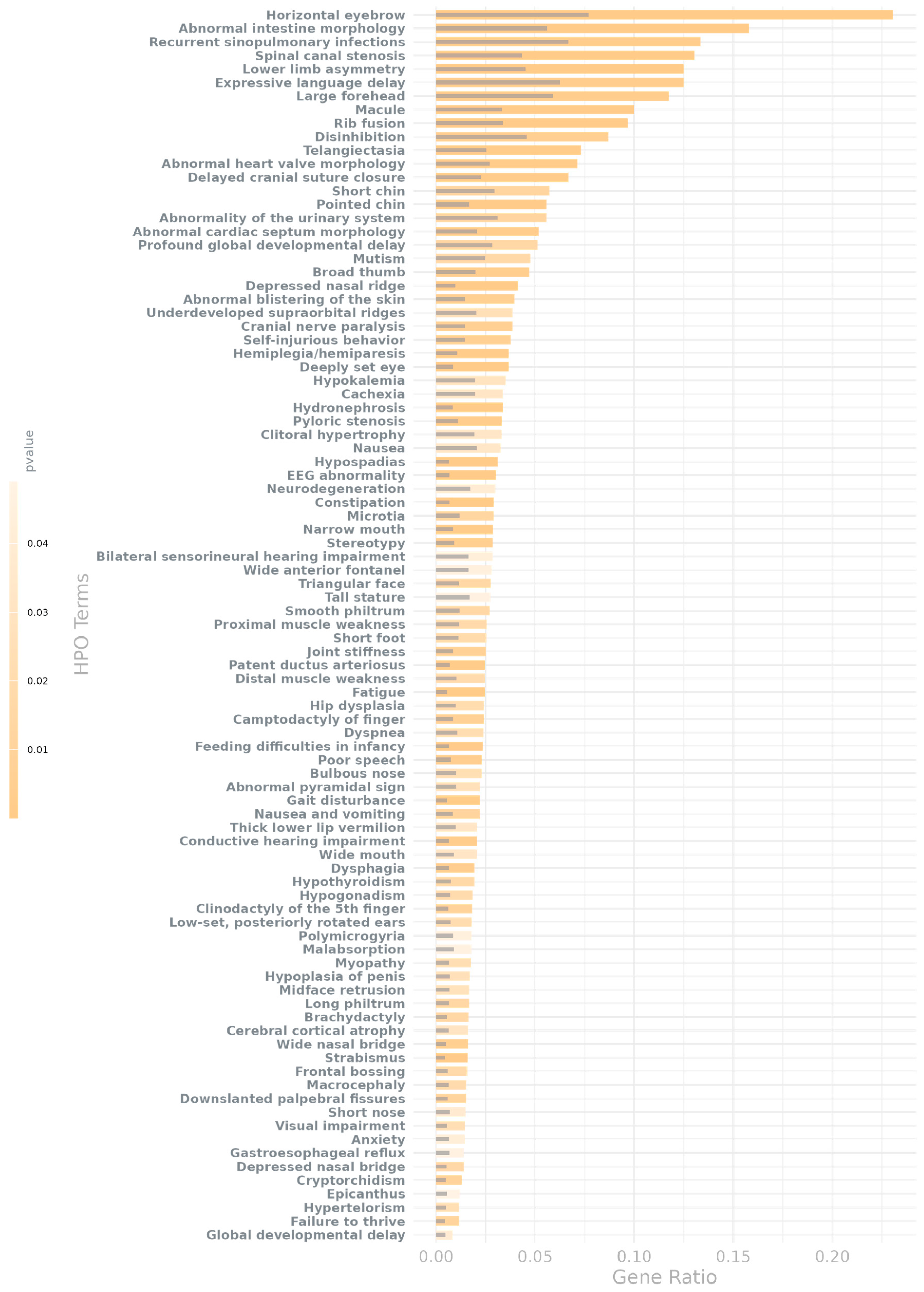
2.3. Genes in AMT-Associated CNV Regions Show HPO Enrichment for Brain Development and Hallmark NDD Traits
3. Discussion
4. Materials and Methods
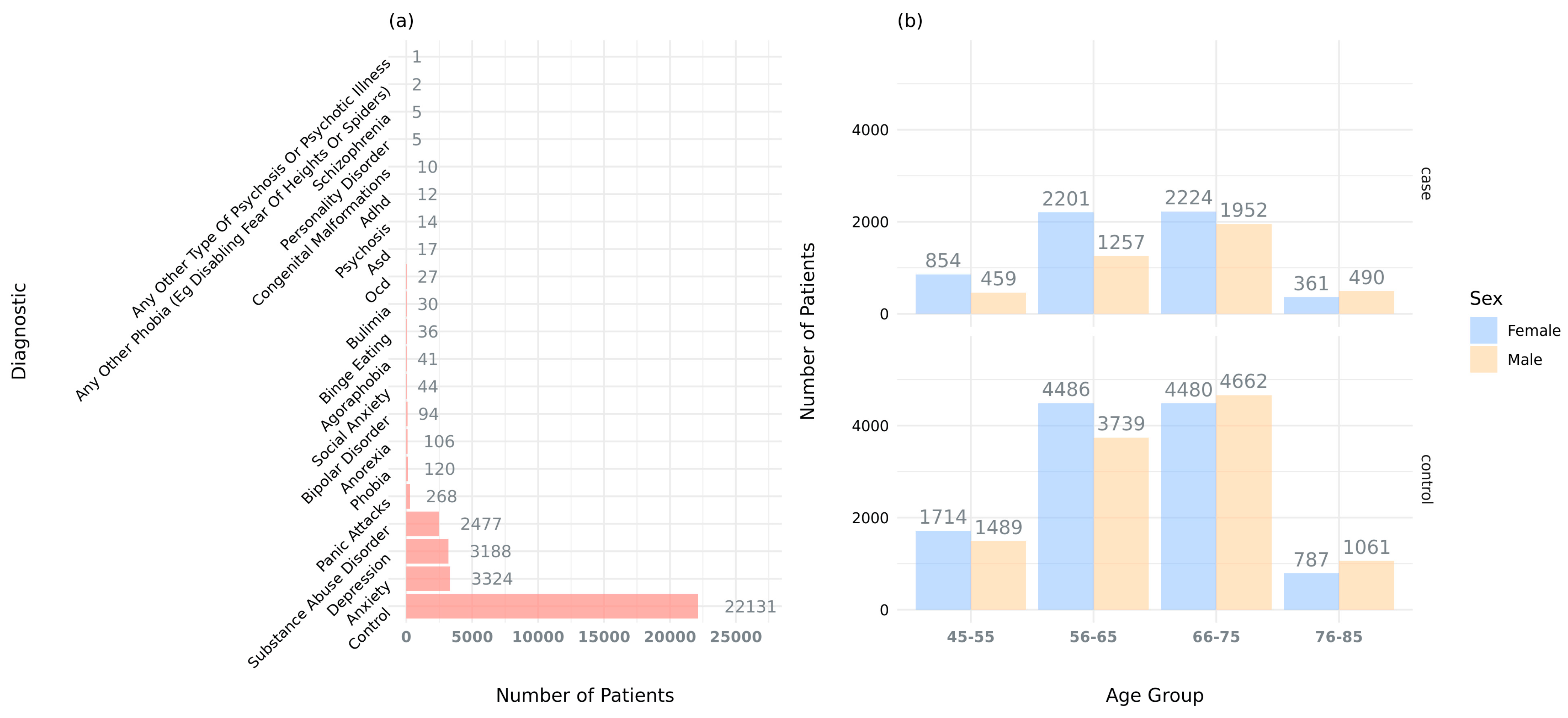
4.1. UK Biobank Data and Cohort Selection
4.2. MRI Data Analysis
4.3. Enrichment Analysis of Morphological Traits in Cases vs. Controls
4.4. Quality Control and Filtering of Genetic Data
4.5. Enrichment Analysis of Variants in Psychiatric Cases vs. Controls
4.6. Enrichment Analysis of Variants in Subjects with Abnormal Brain Morphological Traits
4.7. Adjustment for Correlated Measures of Brain Regions
4.8. Calculation of Prevalence of AMT in UKBB Subjects
4.9. Gene Set Enrichment Analysis and Functional Interpretation of Genes in NDD Risk Regions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| TIV | Total Intracranial Volume |
| SA | Surface Area |
| CT | Cortical Thickness |
| CNV | Copy Number Variant |
| AMT | Aberrant Morphological Trait |
| NDD | Neurodevelopmental Disorder |
| UKBB | UK Biobank |
| GO | Gene Ontology |
| HPO | Human Phenotype Ontology |
| EEG | Electroencephalography |
| ICD10 | International Classification of Diseases, 10th Revision |
| MRI | Magnetic Resonance Imaging |
| IDP | Imaging-Derived Phenotype |
| FDR | False Discovery Rate |
| Kb | Kilo Bases, Measure Representing 1000 Base Pairs |
| Mb | Mega Bases, Measure Representing 1,000,000 Base Pairs |
Appendix A
Appendix A.1. Power Calculation
- p0, the proportion of controls with the variant (p0 = 254/21,877);
- RR, relative risk, representing the increased risk of NDD abnormal morphology in exposed carrying cases relative to controls (RR = (117/9,681)/p0);
- Sample sizes cases (n = 9,798);
- Sample sizes controls (n = 22,131).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of “Exposure” | Type of Cohort | No. of Unique Patients |
|---|---|---|
| No variant | case | 9,681 |
| No variant | control | 21,877 |
| Variant | case | 117 |
| Variant | control | 254 |
Appendix A.2. Mental Health Problems Ever Diagnosed by a Professional (Data Field 20544)
- Social anxiety or social phobia;
- Schizophrenia;
- Any other type of psychosis or psychotic illness;
- A personality disorder;
- Any other phobia (e.g., disabling fear of heights or spiders);
- Panic attacks;
- Obsessive–compulsive disorder (OCD);
- Mania, hypomania, bipolar, or manic depression;
- Depression;
- Bulimia nervosa;
- Psychological over-eating or binge-eating;
- Autism, Asperger’s, or autistic spectrum disorder;
- Anxiety, nerves, or generalized anxiety disorder;
- Anorexia nervosa;
- Agoraphobia;
- Attention deficit or attention deficit and hyperactivity disorder (ADD/ADHD);
- Prefer not to answer.
Appendix A.3. Diagnoses—ICD10 (Data Fields 41270, 41202, and 41204)
- Social anxiety or social phobia:
- ◦
- F40.1 → social phobias.
- ◦
- F93.2 → social anxiety disorder of childhood.
- Schizophrenia:
- ◦
- F20 → schizophrenia.
- Any other type of psychosis or psychotic illness:
- ◦
- F29 → unspecified nonorganic psychosis.
- ◦
- F10.7 → residual and late-onset psychotic disorder.
- ◦
- F10.5 → mental and behavioral disorders due to use of alcohol.
- ◦
- F11.5 → mental and behavioral disorders due to use of opioids.
- ◦
- F12.5 → mental and behavioral disorders due to use of cannabinoids.
- ◦
- F13.5 → mental and behavioral disorders due to use of sedatives or hypnotics.
- ◦
- F14.5 → mental and behavioral disorders due to use of cocaine.
- ◦
- F15.5 → mental and behavioral disorders due to use of other stimulants, including caffeine.
- ◦
- F16.5 → mental and behavioral disorders due to use of hallucinogens.
- ◦
- F17.5 → mental and behavioral disorders due to use of tobacco.
- ◦
- F18.5 → mental and behavioral disorders due to use of volatile solvents.
- ◦
- F19.5 → mental and behavioral disorders due to multiple drug use and use of other psychoactive substances.
- ◦
- F23 → acute and transient psychotic disorders.
- ◦
- F28 → other nonorganic psychotic disorders.
- ◦
- A personality disorder.
- ◦
- F60 → specific personality disorders.
- ◦
- F61 → mixed and other personality disorders.
- ◦
- F62 → enduring personality changes, not attributable to brain damage and disease.
- ◦
- F68 → other disorders of adult personality and behavior.
- ◦
- F69 → unspecified disorder of adult personality and behavior.
- ◦
- F07 → personality and behavioral disorders due to brain disease, damage, and dysfunction.
- Any other phobia (e.g., disabling fear of heights or spiders):
- ◦
- F40.2 → specific (isolated) phobias.
- ◦
- F40.8 → other phobic anxiety disorders.
- ◦
- F40.9 → phobic anxiety disorder, unspecified.
- Panic attacks:
- ◦
- F41.0 → panic disorder [episodic paroxysmal anxiety].
- ◦
- F43 → reaction to severe stress and adjustment disorders.
- Obsessive–compulsive disorder (OCD):
- ◦
- F42 → obsessive–compulsive disorder.
- Mania, hypomania, bipolar, or manic depression:
- ◦
- F30 → manic episode.
- ◦
- F31 → bipolar affective disorder.
- Other factors influencing mental health status:
- ◦
- Z86.4 → personal history of psychoactive substance abuse.
- ◦
- Z81.3 → family history of other psychoactive substance abuse.
- ◦
- Z71.6 → tobacco abuse counselling.
- ◦
- Z71.4 → alcohol abuse counselling and surveillance.
Appendix A.4. Excluded Diagnoses (ICD10-Data Fields 41270, 41202, and 41204)
- Dementia:
- ◦
- F01 → vascular dementia.
- ◦
- F02 → dementia in other diseases classified elsewhere.
- ◦
- F03 → unspecified dementia.
- ◦
- Alzheimer’s.
- ◦
- G30 → Alzheimer’s disease.
- ◦
- F00 → dementia in Alzheimer’s disease.
- Parkinson’s:
- ◦
- G20 → Parkinson’s disease.
- ◦
- G21 → secondary parkinsonism.
- ◦
- G22 → Parkinsonism in diseases classified elsewhere.
- ◦
- A52.1 → syphilitic parkinsonism.
- Others:
- ◦
- B22.0 → HIV disease resulting in encephalopathy (HIV dementia).
- ◦
- F05.1 → delirium superimposed on dementia.
- ◦
- G31 → other degenerative diseases of nervous system, not elsewhere classified (Senile degeneration of the brain, degeneration due to alcohol, etc.).
- ◦
- G93.4 → encephalopathy, unspecified.
- ◦
- G10 → Huntington disease.
- ◦
- C70.0→ malignant neoplasm of cerebral meninges.
- ◦
- C71 → malignant neoplasm of brain.
- ◦
- C72.8 → overlapping lesion of brain and other parts of central nervous system.
- ◦
- C79.3 → secondary malignant neoplasm of brain and cerebral meninges.
- ◦
- D33.0 → benign neoplasm of brain, supratentorial.
- ◦
- D33.1 → benign neoplasm of brain, infratentorial.
- ◦
- D33.2 → benign neoplasm of brain, unspecified.
- ◦
- D43.0 → neoplasm of uncertain or unknown behavior of brain, supratentorial.
- ◦
- D43.1 → neoplasm of uncertain or unknown behavior of brain, infratentorial.
- ◦
- D43.2 → neoplasm of uncertain or unknown behavior of brain, unspecified.
- ◦
- D32.0 → benign neoplasm of cerebral meninges.
- ◦
- D42.0 → neoplasm of uncertain or unknown behavior of cerebral meninges.
- ◦
- S06 → intracranial injury.
Appendix A.5. Limitations in the Psychiatric Inclusion Criteria
Appendix B
| Cases | Controls | |
|---|---|---|
| AMT Present | E | F |
| AMT Absent | G | H |
| Total (31,929 subjects) | E + G | F + H |
| Cases | Controls | |
|---|---|---|
| Carrier | I | J |
| Non-carrier | K | L |
| Total (31,929 subjects) | I + K | J + L |
| Subjects with AMT (Z-Score < −2 or Z-Score > 2) | Subjects Without AMT | |
|---|---|---|
| Variant Present | A | B |
| No Variant Present | C | D |
| Total (31,929 subjects) | A + C | B + D |
Appendix C. Matrix of Pearson’s Correlation for Z-Scores of Brain Measures

References
- Budday, S.; Raybaud, C.; Kuhl, E. A mechanical model predicts morphological abnormalities in the developing human brain. Sci. Rep. 2014, 4, 5644. [Google Scholar] [CrossRef] [PubMed]
- Navarri, X.; Afzali, M.H.; Lavoie, J.; Sinha, R.; Stein, D.J.; Momenan, R.; Veltman, D.J.; Korucuoglu, O.; Sjoerds, Z.; van Holst, R.J.; et al. How do substance use disorders compare to other psychiatric conditions on structural brain abnormalities? A cross-disorder meta-analytic comparison using the ENIGMA consortium findings. Hum. Brain. Mapp. 2022, 43, 399–413. [Google Scholar] [CrossRef] [PubMed]
- Scheepens, D.S.; van Waarde, J.A.; Lok, A.; de Vries, G.; Denys, D.; van Wingen, G.A. The Link Between Structural and Functional Brain Abnormalities in Depression: A Systematic Review of Multimodal Neuroimaging Studies. Front. Psychiatry 2020, 11, 485. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Hu, Y.; Yan, C.; Li, M.; Wu, Y.; Qiu, J.; Zhu, X.; Consortium, R.E.-m.-M. Brain structural abnormalities in adult major depressive disorder revealed by voxel- and source-based morphometry: Evidence from the REST-meta-MDD Consortium. Psychol. Med. 2023, 53, 3672–3682. [Google Scholar] [CrossRef] [PubMed]
- Doyle-Thomas, K.A.; Duerden, E.G.; Taylor, M.J.; Lerch, J.P.; Soorya, L.V.; Wang, A.T.; Fan, J.; Hollander, E.; Anagnostou, E. Effects of age and symptomatology on cortical thickness in autism spectrum disorders. Res. Autism. Spectr. Disord. 2013, 7, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Khundrakpam, B.S.; Lewis, J.D.; Kostopoulos, P.; Carbonell, F.; Evans, A.C. Cortical Thickness Abnormalities in Autism Spectrum Disorders Through Late Childhood, Adolescence, and Adulthood: A Large-Scale MRI Study. Cereb. Cortex. 2017, 27, 1721–1731. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Zhang, J.; Fan, S.; Ping, L.; Yu, H.; Xu, F.; Cheng, Y.; Xu, X.; Yang, C.; Zhou, C. Cortical thickness abnormalities in autism spectrum disorder. Eur. Child. Adolesc. Psychiatry 2024, 33, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Wallace, G.L.; Eisenberg, I.W.; Robustelli, B.; Dankner, N.; Kenworthy, L.; Giedd, J.N.; Martin, A. Longitudinal cortical development during adolescence and young adulthood in autism spectrum disorder: Increased cortical thinning but comparable surface area changes. J. Am. Acad. Child. Adolesc. Psychiatry 2015, 54, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Schmaal, L.; Hibar, D.P.; Samann, P.G.; Hall, G.B.; Baune, B.T.; Jahanshad, N.; Cheung, J.W.; van Erp, T.G.M.; Bos, D.; Ikram, M.A.; et al. Cortical abnormalities in adults and adolescents with major depression based on brain scans from 20 cohorts worldwide in the ENIGMA Major Depressive Disorder Working Group. Mol. Psychiatry 2017, 22, 900–909. [Google Scholar] [CrossRef] [PubMed]
- Almeida Montes, L.G.; Prado Alcantara, H.; Martinez Garcia, R.B.; De La Torre, L.B.; Avila Acosta, D.; Duarte, M.G. Brain cortical thickness in ADHD: Age, sex, and clinical correlations. J. Atten. Disord. 2013, 17, 641–654. [Google Scholar] [CrossRef] [PubMed]
- Shaw, P.; Lerch, J.; Greenstein, D.; Sharp, W.; Clasen, L.; Evans, A.; Giedd, J.; Castellanos, F.X.; Rapoport, J. Longitudinal mapping of cortical thickness and clinical outcome in children and adolescents with attention-deficit/hyperactivity disorder. Arch. Gen. Psychiatry 2006, 63, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Andrews, D.S.; Ozturk, A.; Solomon, M.; Rogers, S.; Amaral, D.G.; Nordahl, C.W. Altered Development of Amygdala-Connected Brain Regions in Males and Females with Autism. J. Neurosci. 2022, 42, 6145–6155. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.D.; Swanson, M.R.; Wolff, J.J.; Elison, J.T.; Girault, J.B.; Kim, S.H.; Smith, R.G.; Graves, M.M.; Weisenfeld, L.A.H.; Flake, L.; et al. Subcortical Brain Development in Autism and Fragile X Syndrome: Evidence for Dynamic, Age- and Disorder-Specific Trajectories in Infancy. Am. J. Psychiatry 2022, 179, 562–572. [Google Scholar] [CrossRef] [PubMed]
- Tajima-Pozo, K.; Yus, M.; Ruiz-Manrique, G.; Lewczuk, A.; Arrazola, J.; Montanes-Rada, F. Amygdala Abnormalities in Adults With ADHD. J. Atten. Disord. 2018, 22, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Dager, A.D.; McKay, D.R.; Kent, J.W., Jr.; Curran, J.E.; Knowles, E.; Sprooten, E.; Goring, H.H.; Dyer, T.D.; Pearlson, G.D.; Olvera, R.L.; et al. Shared genetic factors influence amygdala volumes and risk for alcoholism. Neuropsychopharmacology 2015, 40, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Duy, P.Q.; Rakic, P.; Alper, S.L.; Butler, W.E.; Walsh, C.A.; Sestan, N.; Geschwind, D.H.; Jin, S.C.; Kahle, K.T. Brain ventricles as windows into brain development and disease. Neuron 2022, 110, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, S.; Sherrard, R.M. Cerebellum and neurodevelopmental disorders: RORalpha is a unifying force. Front. Cell. Neurosci. 2023, 17, 1108339. [Google Scholar] [CrossRef] [PubMed]
- Pierson, T.M.; Otero, M.G.; Grand, K.; Choi, A.; Graham, J.M.; Young, J.I., Jr.; Mackay, J.P. The NuRD complex and macrocephaly associated neurodevelopmental disorders. Am. J. Med. Genet. C. Semin. Med. Genet. 2019, 181, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Bahar, N.; Cler, G.J.; Krishnan, S.; Asaridou, S.S.; Smith, H.J.; Willis, H.E.; Healy, M.P.; Watkins, K.E. Differences in Cortical Surface Area in Developmental Language Disorder. Neurobiol. Lang. 2024, 5, 288–314. [Google Scholar] [CrossRef] [PubMed]
- de Mendonca Filho, E.J.; Alves, M.B.; Silveira, P.P. Brain structural abnormalities in six major psychiatric disorders: Shared variation and network perspectives. F1000Research 2021, 10, 356. [Google Scholar] [CrossRef] [PubMed]
- Sonderby, I.E.; Ching, C.R.K.; Thomopoulos, S.I.; van der Meer, D.; Sun, D.; Villalon-Reina, J.E.; Agartz, I.; Amunts, K.; Arango, C.; Armstrong, N.J.; et al. Effects of copy number variations on brain structure and risk for psychiatric illness: Large-scale studies from the ENIGMA working groups on CNVs. Hum. Brain. Mapp. 2022, 43, 300–328. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, R.; Mahjani, B.; Loos, R.J.F.; Sharp, A.J. Clinical Characterization of Copy Number Variants Associated With Neurodevelopmental Disorders in a Large-scale Multiancestry Biobank. JAMA Psychiatry 2022, 79, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Sebat, J.; Lakshmi, B.; Malhotra, D.; Troge, J.; Lese-Martin, C.; Walsh, T.; Yamrom, B.; Yoon, S.; Krasnitz, A.; Kendall, J.; et al. Strong association of de novo copy number mutations with autism. Science 2007, 316, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Collins, R.L.; Glessner, J.T.; Porcu, E.; Lepamets, M.; Brandon, R.; Lauricella, C.; Han, L.; Morley, T.; Niestroj, L.M.; Ulirsch, J.; et al. A cross-disorder dosage sensitivity map of the human genome. Cell 2022, 185, 3041–3055. [Google Scholar] [CrossRef] [PubMed]
- Azidane, S.; Gallego, X.; Durham, L.; Caceres, M.; Guney, E.; Perez-Cano, L. Identification of novel driver risk genes in CNV loci associated with neurodevelopmental disorders. Hum. Genet. Genom. Adv. 2024, 5, 100316. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.; Ching, C.R.K.; Vajdi, A.; Sun, D.; Jonas, R.K.; Jalbrzikowski, M.; Kushan-Wells, L.; Pacheco Hansen, L.; Krikorian, E.; Gutman, B.; et al. Mapping 22q11.2 Gene Dosage Effects on Brain Morphometry. J. Neurosci. 2017, 37, 6183–6199. [Google Scholar] [CrossRef] [PubMed]
- Steinman, K.J.; Spence, S.J.; Ramocki, M.B.; Proud, M.B.; Kessler, S.K.; Marco, E.J.; Green Snyder, L.; D′Angelo, D.; Chen, Q.; Chung, W.K.; et al. 16p11.2 deletion and duplication: Characterizing neurologic phenotypes in a large clinically ascertained cohort. Am. J. Med. Genet. Part A 2016, 170, 2943–2955. [Google Scholar] [CrossRef] [PubMed]
- Rogdaki, M.; Gudbrandsen, M.; McCutcheon, R.A.; Blackmore, C.E.; Brugger, S.; Ecker, C.; Craig, M.C.; Daly, E.; Murphy, D.G.M.; Howes, O. Magnitude and heterogeneity of brain structural abnormalities in 22q11.2 deletion syndrome: A meta-analysis. Mol. Psychiatry 2020, 25, 1704–1717. [Google Scholar] [CrossRef] [PubMed]
- Brunetti-Pierri, N.; Berg, J.S.; Scaglia, F.; Belmont, J.; Bacino, C.A.; Sahoo, T.; Lalani, S.R.; Graham, B.; Lee, B.; Shinawi, M.; et al. Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nat. Genet. 2008, 40, 1466–1471. [Google Scholar] [CrossRef] [PubMed]
- Sonderby, I.E.; van der Meer, D.; Moreau, C.; Kaufmann, T.; Walters, G.B.; Ellegaard, M.; Abdellaoui, A.; Ames, D.; Amunts, K.; Andersson, M.; et al. 1q21.1 distal copy number variants are associated with cerebral and cognitive alterations in humans. Transl. Psychiatry 2021, 11, 182. [Google Scholar] [CrossRef] [PubMed]
- Kopal, J.; Kumar, K.; Saltoun, K.; Modenato, C.; Moreau, C.A.; Martin-Brevet, S.; Huguet, G.; Jean-Louis, M.; Martin, C.O.; Saci, Z.; et al. Rare CNVs and phenome-wide profiling highlight brain structural divergence and phenotypical convergence. Nat. Hum. Behav. 2023, 7, 1001–1017. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.; Modenato, C.; Moreau, C.; Ching, C.R.K.; Harvey, A.; Martin-Brevet, S.; Huguet, G.; Jean-Louis, M.; Douard, E.; Martin, C.O.; et al. Subcortical Brain Alterations in Carriers of Genomic Copy Number Variants. Am. J. Psychiatry 2023, 180, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Orphanet: An Online Rare Disease and Orphan Drug Database. 1999. Available online: https://www.orpha.net/ (accessed on 18 January 2024).
- Lopez-Arango, G.; Deguire, F.; Agbogba, K.; Boucher, M.A.; Knoth, I.S.; El-Jalbout, R.; Cote, V.; Damphousse, A.; Kadoury, S.; Lippe, S. Impact of brain overgrowth on sensorial learning processing during the first year of life. Front. Hum. Neurosci. 2022, 16, 928543. [Google Scholar] [CrossRef] [PubMed]
- Zaqout, S.; Kaindl, A.M. Autosomal Recessive Primary Microcephaly: Not Just a Small Brain. Front. Cell Dev. Biol. 2021, 9, 784700. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Pineiro, D.; Parker, N.; Shin, J.; French, L.; Grydeland, H.; Jackowski, A.P.; Mowinckel, A.M.; Patel, Y.; Pausova, Z.; Salum, G.; et al. Cellular correlates of cortical thinning throughout the lifespan. Sci. Rep. 2020, 10, 21803. [Google Scholar] [CrossRef] [PubMed]
- van Haren, N.E.; Schnack, H.G.; Cahn, W.; van den Heuvel, M.P.; Lepage, C.; Collins, L.; Evans, A.C.; Hulshoff Pol, H.E.; Kahn, R.S. Changes in cortical thickness during the course of illness in schizophrenia. Arch. Gen. Psychiatry 2011, 68, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Dowling, G.J.; Weiss, S.R.; Condon, T.P. Drugs of abuse and the aging brain. Neuropsychopharmacology 2008, 33, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Pink, A.; Przybelski, S.A.; Krell-Roesch, J.; Stokin, G.B.; Roberts, R.O.; Mielke, M.M.; Knopman, D.S.; Jack, C.R.; Petersen, R.C.; Geda, Y.E. Cortical Thickness and Depressive Symptoms in Cognitively Normal Individuals: The Mayo Clinic Study of Aging. J. Alzheimers. Dis. 2017, 58, 1273–1281. [Google Scholar] [CrossRef] [PubMed]
- de la Cruz, F.; Schumann, A.; Suttkus, S.; Helbing, N.; Zopf, R.; Bar, K.J. Cortical thinning and associated connectivity changes in patients with anorexia nervosa. Transl. Psychiatry 2021, 11, 95. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.; Banaschewski, T.; Bokde, A.L.W.; Desrivieres, S.; Flor, H.; Grigis, A.; Garavan, H.; Gowland, P.; Heinz, A.; Ittermann, B.; et al. Hemispheric asymmetry in cortical thinning reflects intrinsic organization of the neurotransmitter systems and homotopic functional connectivity. Proc. Natl. Acad. Sci. USA 2023, 120, e2306990120. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Mao, Q.; Shi, J.; Wang, X.; Li, C.R. Putamen gray matter volumes in neuropsychiatric and neurodegenerative disorders. World J. Psychiatry Ment. Health Res. 2019, 3, 1020. [Google Scholar] [PubMed]
- Todd, K.L.; Brighton, T.; Norton, E.S.; Schick, S.; Elkins, W.; Pletnikova, O.; Fortinsky, R.H.; Troncoso, J.C.; Molfese, P.J.; Resnick, S.M.; et al. Ventricular and Periventricular Anomalies in the Aging and Cognitively Impaired Brain. Front. Aging Neurosci. 2017, 9, 445. [Google Scholar] [CrossRef] [PubMed]
- Gaser, C.; Nenadic, I.; Buchsbaum, B.R.; Hazlett, E.A.; Buchsbaum, M.S. Ventricular enlargement in schizophrenia related to volume reduction of the thalamus, striatum, and superior temporal cortex. Am. J. Psychiatry 2004, 161, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Fannon, D.; Tennakoon, L.; Sumich, A.; O’Ceallaigh, S.; Doku, V.; Chitnis, X.; Lowe, J.; Soni, W.; Sharma, T. Third ventricle enlargement and developmental delay in first-episode psychosis: Preliminary findings. Br. J. Psychiatry 2000, 177, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Mannik, K.; Magi, R.; Mace, A.; Cole, B.; Guyatt, A.L.; Shihab, H.A.; Maillard, A.M.; Alavere, H.; Kolk, A.; Reigo, A.; et al. Copy number variations and cognitive phenotypes in unselected populations. JAMA 2015, 313, 2044–2054. [Google Scholar] [CrossRef] [PubMed]
- Modenato, C.; Kumar, K.; Moreau, C.; Martin-Brevet, S.; Huguet, G.; Schramm, C.; Jean-Louis, M.; Martin, C.O.; Younis, N.; Tamer, P.; et al. Effects of eight neuropsychiatric copy number variants on human brain structure. Transl. Psychiatry 2021, 11, 399. [Google Scholar] [CrossRef] [PubMed]
- Fry, A.; Littlejohns, T.J.; Sudlow, C.; Doherty, N.; Adamska, L.; Sprosen, T.; Collins, R.; Allen, N.E. Comparison of Sociodemographic and Health-Related Characteristics of UK Biobank Participants With Those of the General Population. Am. J. Epidemiol. 2017, 186, 1026–1034. [Google Scholar] [CrossRef] [PubMed]
- Sears, L.L.; Vest, C.; Mohamed, S.; Bailey, J.; Ranson, B.J.; Piven, J. An MRI study of the basal ganglia in autism. Prog. Neuropsychopharmacol. Biol. Psychiatry 1999, 23, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Wolff, J.J.; Hazlett, H.C.; Lightbody, A.A.; Reiss, A.L.; Piven, J. Repetitive and self-injurious behaviors: Associations with caudate volume in autism and fragile X syndrome. J. Neurodev. Disord. 2013, 5, 12. [Google Scholar] [CrossRef] [PubMed]
- Andrews, D.S.; Aksman, L.; Kerns, C.M.; Lee, J.K.; Winder-Patel, B.M.; Harvey, D.J.; Waizbard-Bartov, E.; Heath, B.; Solomon, M.; Rogers, S.J.; et al. Association of Amygdala Development With Different Forms of Anxiety in Autism Spectrum Disorder. Biol. Psychiatry 2022, 91, 977–987. [Google Scholar] [CrossRef] [PubMed]
- Schumann, C.M.; Hamstra, J.; Goodlin-Jones, B.L.; Lotspeich, L.J.; Kwon, H.; Buonocore, M.H.; Lammers, C.R.; Reiss, A.L.; Amaral, D.G. The amygdala is enlarged in children but not adolescents with autism; the hippocampus is enlarged at all ages. J. Neurosci. 2004, 24, 6392–6401. [Google Scholar] [CrossRef] [PubMed]
- Okada, N.; Yahata, N.; Koshiyama, D.; Morita, K.; Sawada, K.; Kanata, S.; Fujikawa, S.; Sugimoto, N.; Toriyama, R.; Masaoka, M.; et al. Abnormal asymmetries in subcortical brain volume in early adolescents with subclinical psychotic experiences. Transl. Psychiatry 2018, 8, 254. [Google Scholar] [CrossRef] [PubMed]
- Turner, A.H.; Greenspan, K.S.; van Erp, T.G.M. Pallidum and lateral ventricle volume enlargement in autism spectrum disorder. Psychiatry Res. Neuroimaging 2016, 252, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Jordan, V.K.; Zaveri, H.P.; Scott, D.A. 1p36 deletion syndrome: An update. Appl. Clin. Genet. 2015, 8, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Perkowski, J.J.; Murphy, G.G. Deletion of the mouse homolog of KCNAB2, a gene linked to monosomy 1p36, results in associative memory impairments and amygdala hyperexcitability. J. Neurosci. 2011, 31, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Zubova, A.V.; Groshkov, A.A.; Berdnikov, A.K.; Novikova, S.V.; Rozanova, N.A.; Nikolaeva, L.V.; Salmin, V.V.; Kolotyeva, N.A.; Khaspekov, L.G.; Salmina, A.B.; et al. Evolution, Possibilities, and Prospects for Application of the Methods of Assessment of Pyridine Nucleotides Pool for Studying Mechanisms of Brain Plasticity in Normal and Pathological Conditions. Biochemistry 2025, 90, 231–246. [Google Scholar] [CrossRef] [PubMed]
- Alfaro-Almagro, F.; Jenkinson, M.; Bangerter, N.K.; Andersson, J.L.R.; Griffanti, L.; Douaud, G.; Sotiropoulos, S.N.; Jbabdi, S.; Hernandez-Fernandez, M.; Vallee, E.; et al. Image processing and Quality Control for the first 10,000 brain imaging datasets from UK Biobank. Neuroimage 2018, 166, 400–424. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.L.; Alfaro-Almagro, F.; Bangerter, N.K.; Thomas, D.L.; Yacoub, E.; Xu, J.; Bartsch, A.J.; Jbabdi, S.; Sotiropoulos, S.N.; Andersson, J.L.; et al. Multimodal population brain imaging in the UK Biobank prospective epidemiological study. Nat. Neurosci. 2016, 19, 1523–1536. [Google Scholar] [CrossRef] [PubMed]
- Halldorsson, B.V.; Eggertsson, H.P.; Moore, K.H.S.; Hauswedell, H.; Eiriksson, O.; Ulfarsson, M.O.; Palsson, G.; Hardarson, M.T.; Oddsson, A.; Jensson, B.O.; et al. The sequences of 150,119 genomes in the UK Biobank. Nature 2022, 607, 732–740. [Google Scholar] [CrossRef] [PubMed]
- Cisterna, A.; Gonzalez-Vidal, A.; Ruiz, D.; Ortiz, J.; Gomez-Pascual, A.; Chen, Z.; Nalls, M.; Faghri, F.; Hardy, J.; Diez, I.; et al. PhenoExam: Gene set analyses through integration of different phenotype databases. BMC Bioinform. 2022, 23, 567. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azidane, S.; Eizaguerri, S.; Gallego, X.; Durham, L.; Guney, E.; Pérez-Cano, L. Assessment of Brain Morphological Abnormalities and Neurodevelopmental Risk Copy Number Variants in Individuals from the UK Biobank. Int. J. Mol. Sci. 2025, 26, 7062. https://doi.org/10.3390/ijms26157062
Azidane S, Eizaguerri S, Gallego X, Durham L, Guney E, Pérez-Cano L. Assessment of Brain Morphological Abnormalities and Neurodevelopmental Risk Copy Number Variants in Individuals from the UK Biobank. International Journal of Molecular Sciences. 2025; 26(15):7062. https://doi.org/10.3390/ijms26157062
Chicago/Turabian StyleAzidane, Sara, Sandra Eizaguerri, Xavier Gallego, Lynn Durham, Emre Guney, and Laura Pérez-Cano. 2025. "Assessment of Brain Morphological Abnormalities and Neurodevelopmental Risk Copy Number Variants in Individuals from the UK Biobank" International Journal of Molecular Sciences 26, no. 15: 7062. https://doi.org/10.3390/ijms26157062
APA StyleAzidane, S., Eizaguerri, S., Gallego, X., Durham, L., Guney, E., & Pérez-Cano, L. (2025). Assessment of Brain Morphological Abnormalities and Neurodevelopmental Risk Copy Number Variants in Individuals from the UK Biobank. International Journal of Molecular Sciences, 26(15), 7062. https://doi.org/10.3390/ijms26157062