
TREM2 in Neurodegenerative Disorders: Mutation Spectrum, Pathophysiology, and Therapeutic Targeting

 , , and
, , and 
Abstract
1. Introduction
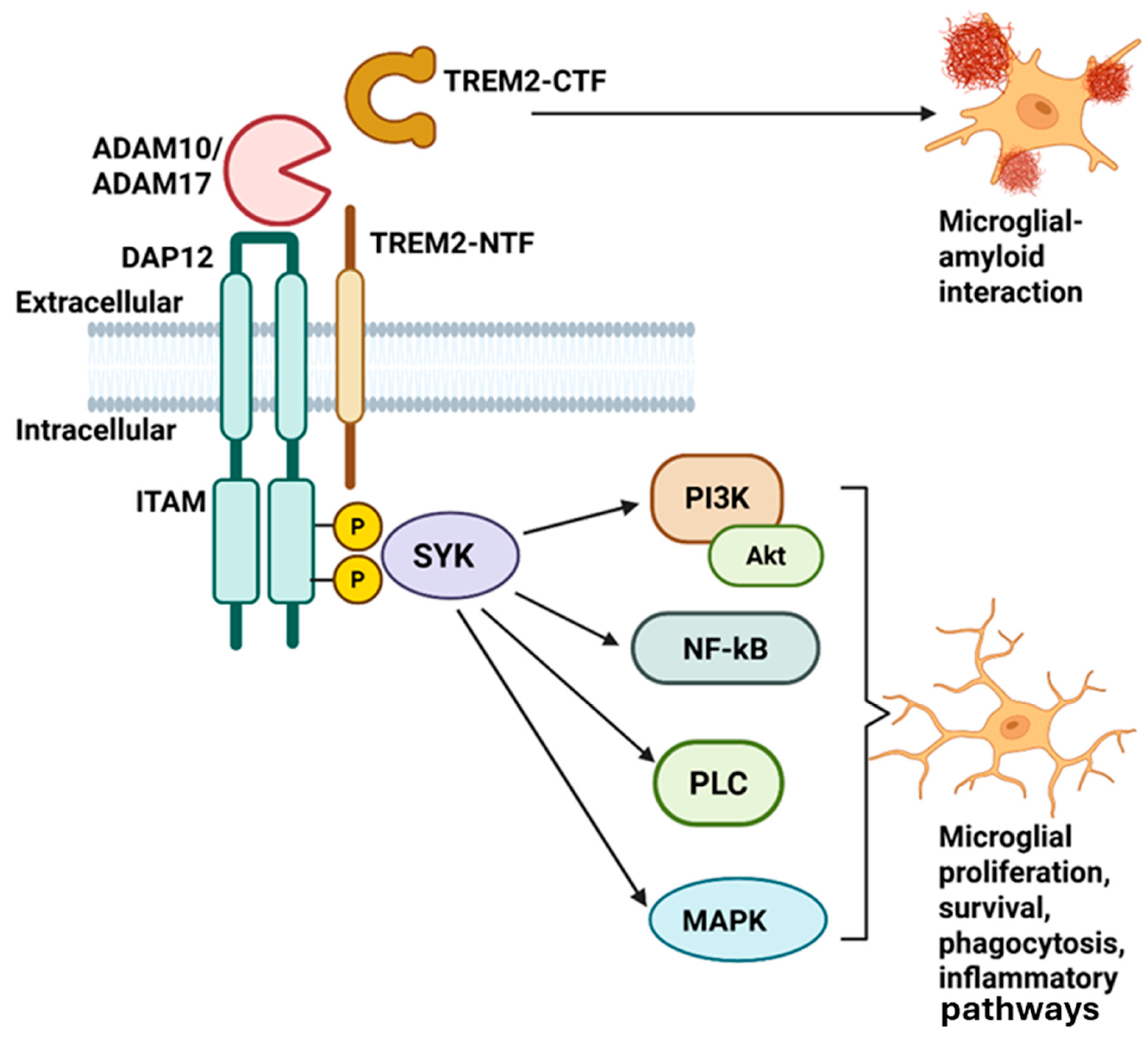
2. TREM2 Gene and TREM2 Protein Structure and Function
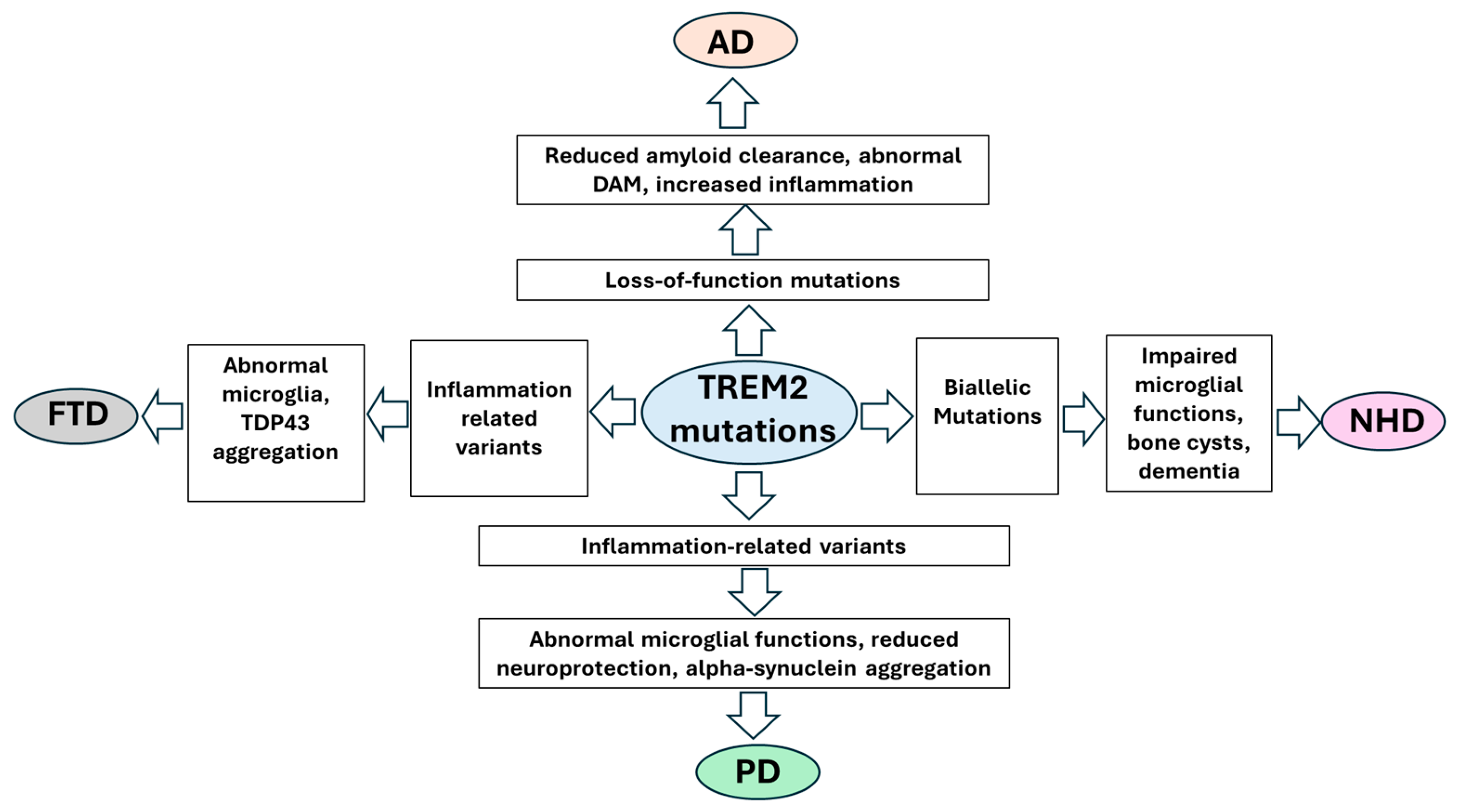
3. Neurodegenerative Disease and TREM2
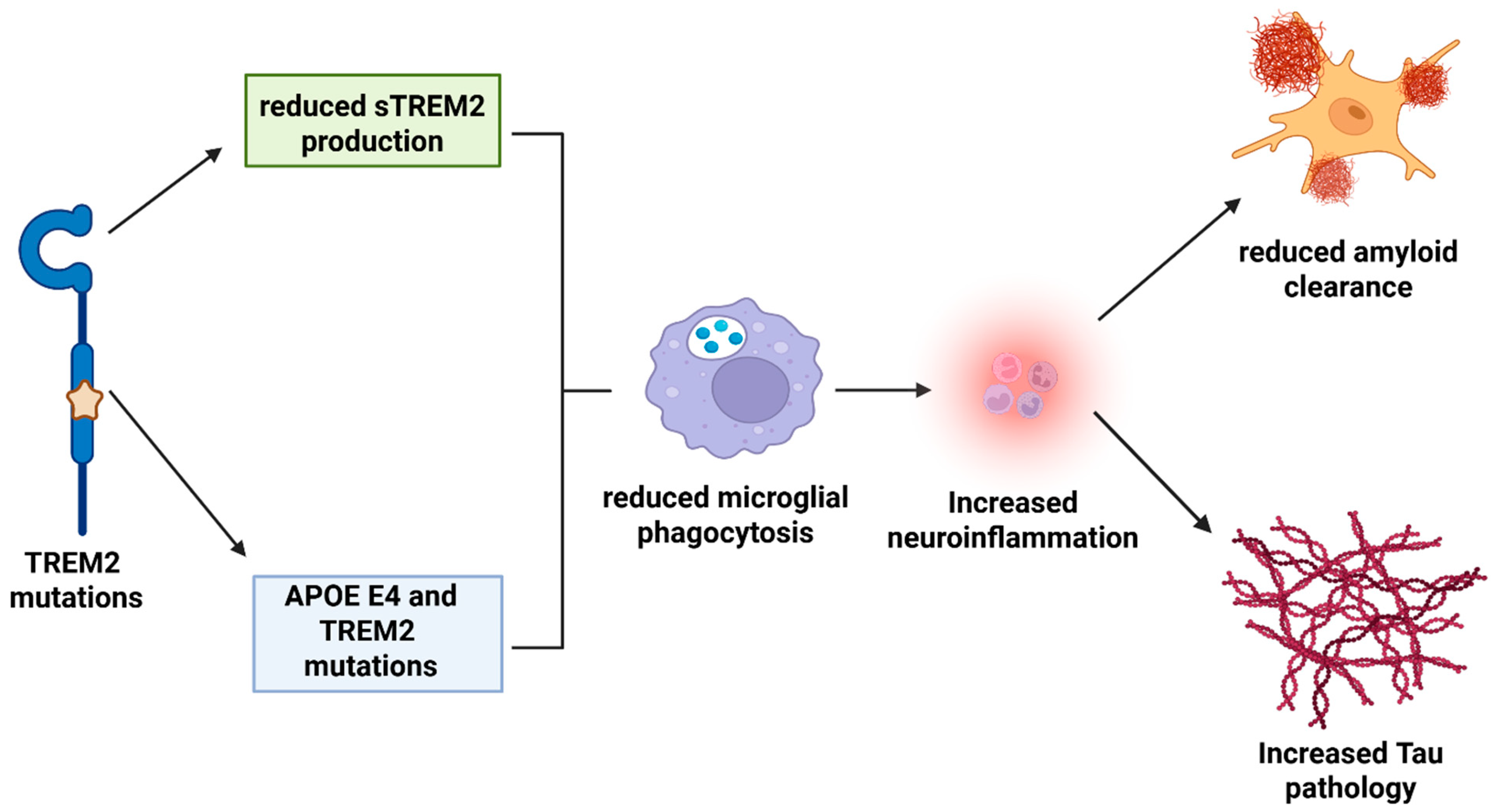
3.1. AD and TREM2
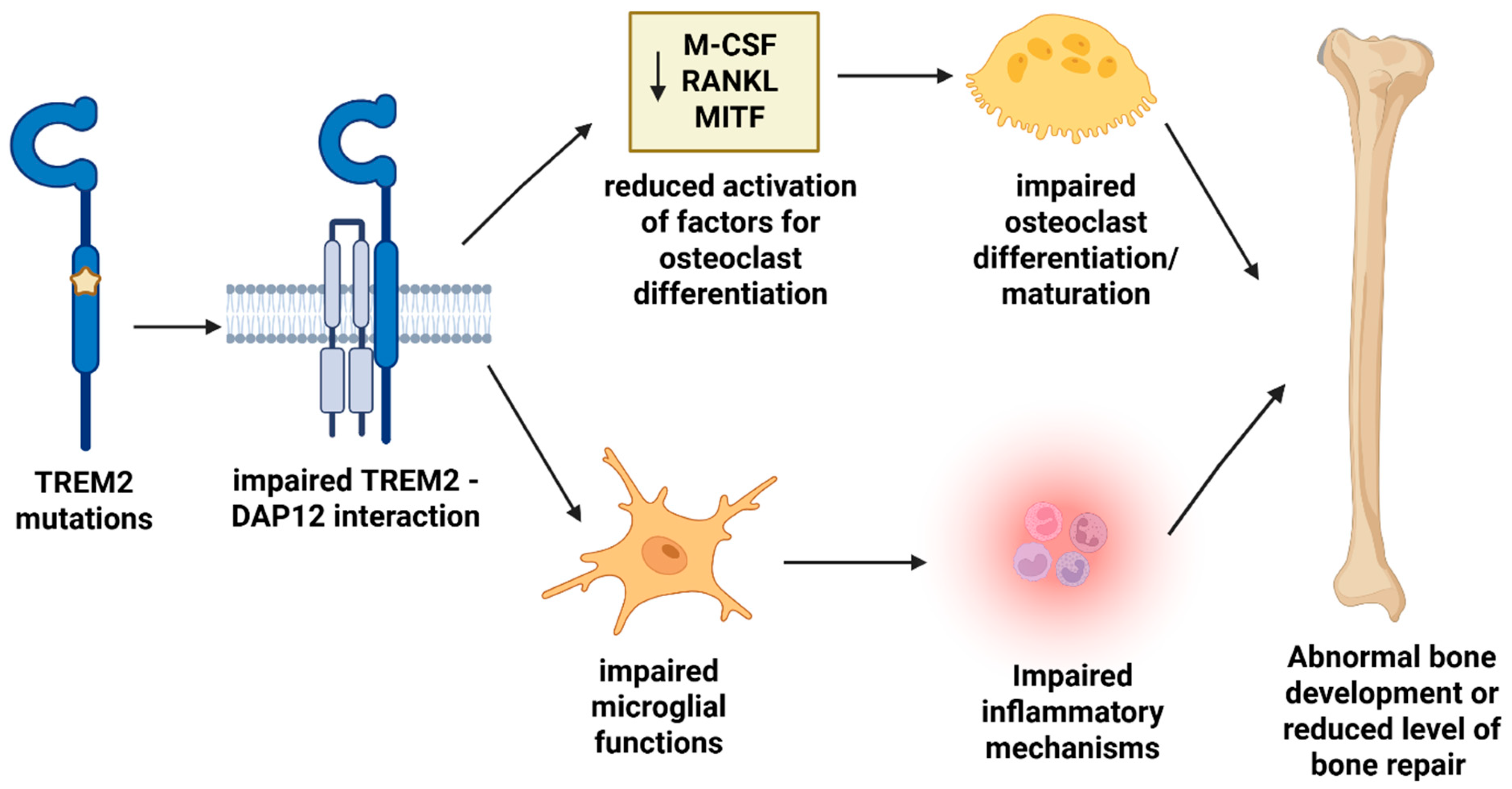
3.2. TREM2 and NHD
3.3. TREM2 and FTD
3.4. TREM2 and PD
4. TREM2 Pathological Mechanism
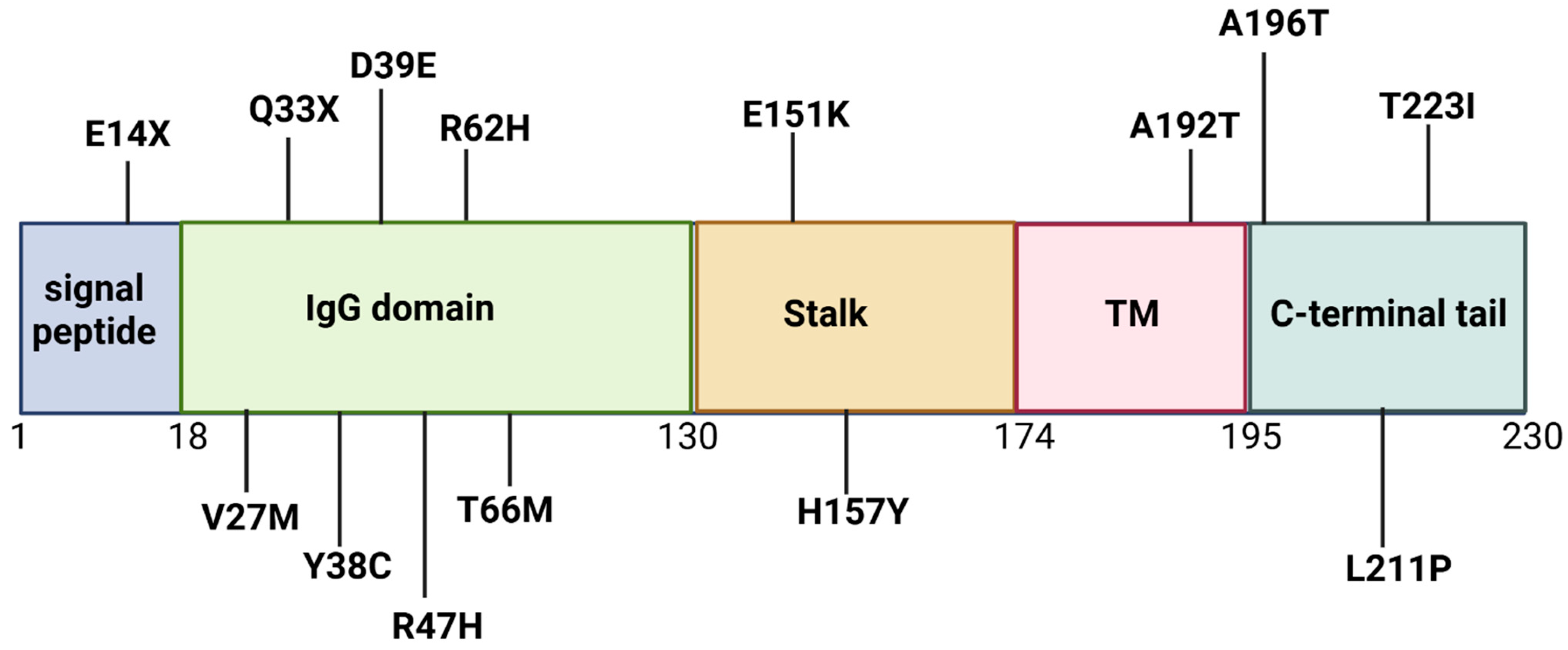
5. TREM2 Mutations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation | Gene Variant | Disease | Age of Onset | Imaging Data | Functional Data | Reference |
|---|---|---|---|---|---|---|
| Glu14Ter | 40 G > T | NHD | NA | NA | Reduced sTREM2 in blood | [55] |
| Val27Met | 79 G > A | AD | NA | NA | Not effect on TREM2 maturation, putative effects on ligand binding | [118] |
| Gln33Ter | 97 C > T | AD, NHD, FTD | 30 s–40 s | Bone cysts, cerebral atrophy: AD patient: typical AD pathology | Loss of TREM2 expression | [47,119] |
| Tyr38Cys | 113 G > A | FTD | 40 s | Cortical atrophy, white matter abnormalities | Disturbs ligand binding and TREN2 phagocytosis | [47] |
| Asp39Glu | 140 G > A | AD, FTD | NA | NA | NA | [117] |
| Arg47His | 117 C > G | AD, NHD, FTD, ALS | 50 s | Severe gray-matter loss, lower microglial coverage of plaques | Elevated CSF-Tau, reduced ligand binding and microglial activation | [11,12] |
| Arg62His | 185 C > T | AD | NA | Lower microglial coverage of plaques | Reduced ligand binding and microglial activation | [117,118] |
| Thr66Met | 197 C > T | FTD | 30 s | Frontal lobe atrophy, ventricular enlargement | Reduced cell surface expression of TREM2, impaired microglial activation | [47] |
| Glu151Lys | 451 G > A | AD | NA | NA | Reduced normal TREM2 expression | [12,119] |
| His157Tyr | 469 C > T | AD, FTD | NA | NA | Increased soluble TREM2 shedding, reduced phagocytosis | [115,116] |
| Ala192Thr | 574 G > A | AD, FTD | 50 s | hypometabolism in bilateral anterior temporal areas | Reduced cell surface expression of TREM2 | [114,115] |
| Ala196Thr | 586 G > A | AD | NA | NA | Probable reduced TREM2 cell surface expression | [111] |
| Leu211Pro | 632 T > C | AD, FTD | NA | NA | Lower TREM2 CSF levels | [117] |
| Thr223Ile | 668 C > T | AD, FTD | NA | NA | Slight changes in TREM2 maturation | [114] |
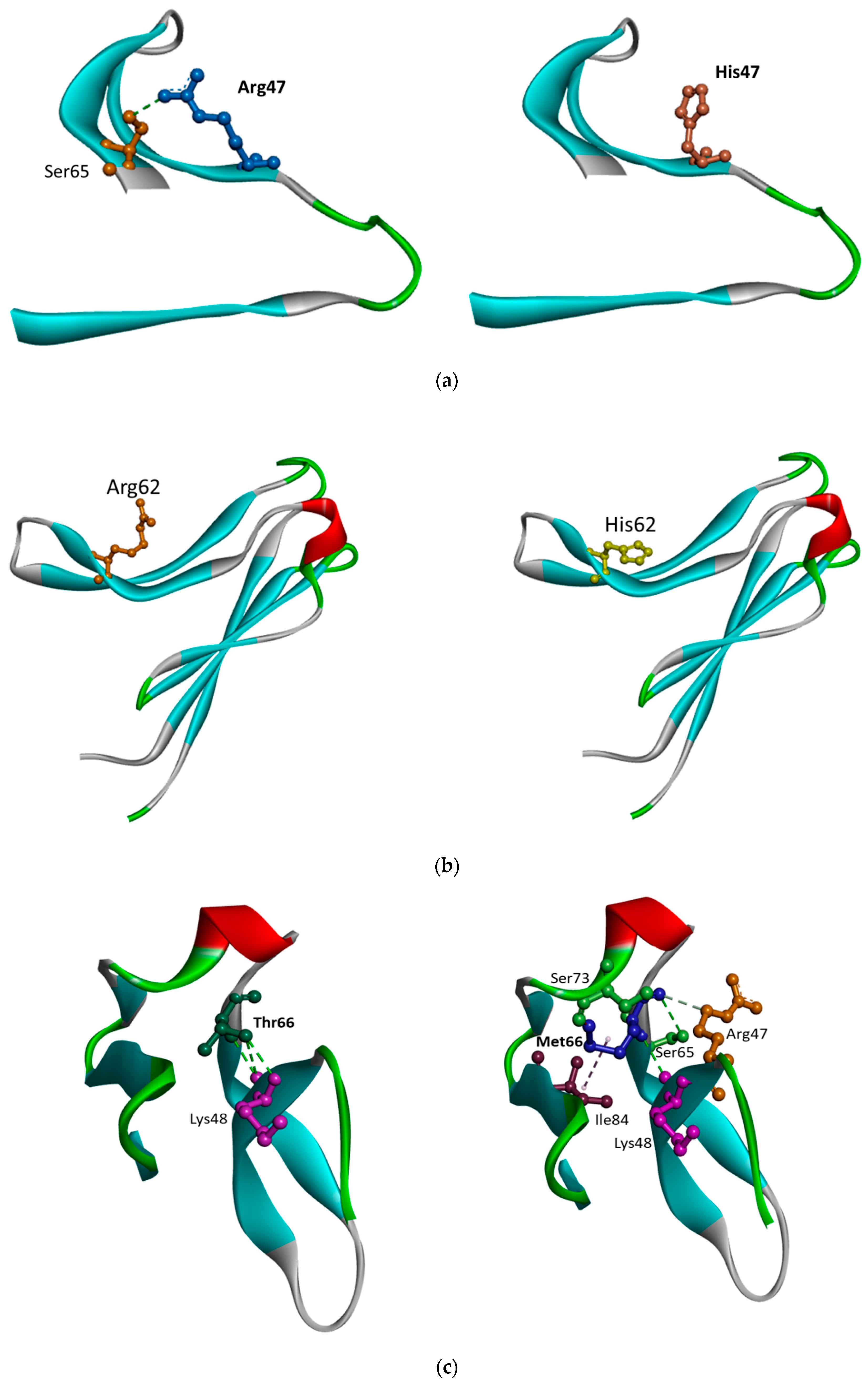
5.1. TREM2 Arg47His
5.2. TREM2 Arg62His
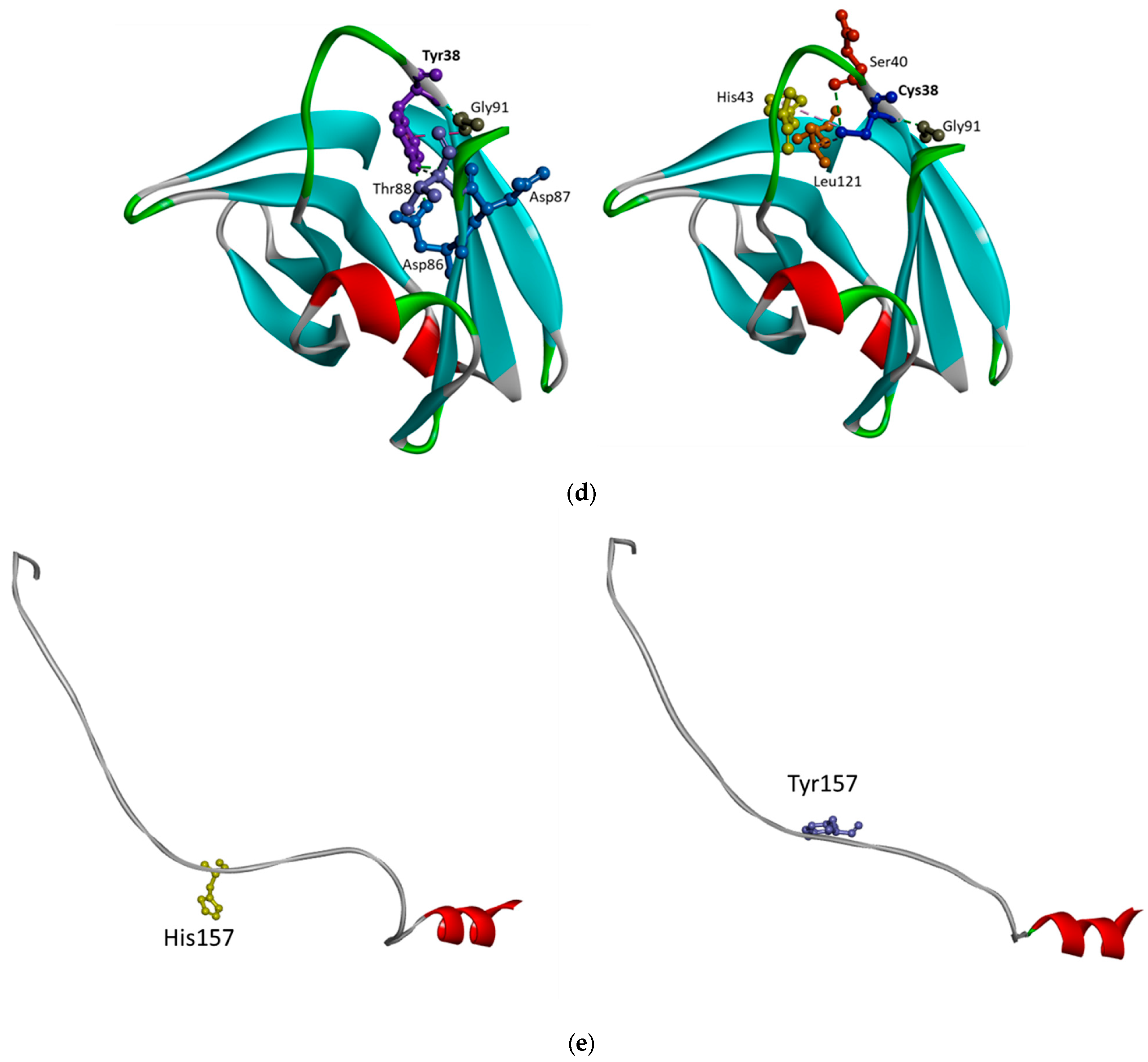
5.3. TREM2 Thr66Met and Tyr38Cys
5.4. His157Tyr
5.5. TREM2 Gln33Ter
6. TREM2 Variations and Therapeutic Targeting
7. Conclusions
Funding
Conflicts of Interest
References
- Zgorzynska, E. TREM2 in Alzheimer’s disease: Structure, function, therapeutic prospects, and activation challenges. Mol. Cell. Neurosci. 2024, 128, 103917. [Google Scholar] [CrossRef] [PubMed]
- Ulland, T.K.; Colonna, M. TREM2—A key player in microglial biology and Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Chen, Y.; Grajales-Reyes, G.; Colonna, M. TREM2 dependent and independent functions of microglia in Alzheimer’s disease. Mol. Neurodegener. 2022, 17, 84. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.-M.; Zhang, Y.-R.; Huang, Y.-Y.; Dong, Q.; Tan, L.; Yu, J.-T. The role of the immune system in Alzheimer’s disease. Ageing Res. Rev. 2021, 70, 101409. [Google Scholar] [CrossRef]
- Carmona, S.; Hardy, J.; Guerreiro, R. Chapter 26—The genetic landscape of Alzheimer disease. In Handbook of Clinical Neurology; Geschwind, D.H., Paulson, H.L., Klein, C., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 395–408. [Google Scholar]
- Deczkowska, A.; Weiner, A.; Amit, I. The physiology, pathology, and potential therapeutic applications of the TREM2 signaling pathway. Cell 2020, 181, 1207–1217. [Google Scholar] [CrossRef]
- Gratuze, M.; Leyns, C.E.; Holtzman, D.M. New insights into the role of TREM2 in Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 66. [Google Scholar] [CrossRef]
- Li, Y.; Xu, H.; Wang, H.; Yang, K.; Luan, J.; Wang, S. TREM2: Potential therapeutic targeting of microglia for Alzheimer’s disease. Biomed. Pharmacotherapy 2023, 165, 115218. [Google Scholar] [CrossRef]
- Greven, J.A.; Alexander-Brett, J.M.; Brett, T.J. Structural and functional analysis of TREM2 interactions with amyloid beta reveal molecular mechanisms that drive phagocytosis of oligomeric amyloid beta. Alzheimer’s Dement. 2024, 20, e092282. [Google Scholar] [CrossRef]
- Wang, Y.; Ulland, T.K.; Ulrich, J.D.; Song, W.; Tzaferis, J.A.; Hole, J.T.; Yuan, P.; Mahan, T.E.; Shi, Y.; Gilfillan, S.; et al. TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J. Exp. Med. 2016, 213, 667–675. [Google Scholar] [CrossRef]
- Park, J.-C.; Han, J.W.; Lee, W.; Kim, J.; Lee, S.E.; Lee, D.; Choi, H.; Han, J.; Kang, Y.J.; Diep, Y.N.; et al. Microglia Gravitate toward Amyloid Plaques Surrounded by Externalized Phosphatidylserine via TREM2. Adv. Sci. 2024, 11, 2400064. [Google Scholar] [CrossRef]
- Ulrich, J.D.; Holtzman, D.M. TREM2 function in Alzheimer’s disease and neurodegeneration. ACS Chem. Neurosci. 2016, 7, 420–427. [Google Scholar] [CrossRef]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S.; et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef]
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher, J.; Levey, A.I.; Lah, J.J.; et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 107–116. [Google Scholar] [CrossRef]
- Hickman, S.E.; El Khoury, J. TREM2 and the neuroimmunology of Alzheimer’s disease. Biochem. Pharmacol. 2014, 88, 495–498. [Google Scholar] [CrossRef]
- Dean, H.B.; Roberson, E.D.; Song, Y. Neurodegenerative Disease-Associated Variants in TREM2 Destabilize the Apical Ligand-Binding Region of the Immunoglobulin Domain. Front. Neurol 2019, 10, 1252. [Google Scholar] [CrossRef]
- Olufunmilayo, E.O.; Holsinger, R.D. Variant TREM2 signaling in Alzheimer’s disease. J. Mol. Biol. 2022, 434, 167470. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.C.; DeVaux, L.B.; Farzan, M. The Triggering Receptor Expressed on Myeloid Cells 2 Binds Apolipoprotein E. J. Biol. Chem. 2015, 290, 26033–26042. [Google Scholar] [CrossRef] [PubMed]
- Kober, D.L.; Brett, T.J. TREM2-Ligand Interactions in Health and Disease. J. Mol. Biol. 2017, 429, 1607–1629. [Google Scholar] [CrossRef] [PubMed]
- Shaw, B.C.; Snider, H.C.; Turner, A.K.; Zajac, D.J.; Simpson, J.F.; Estus, S. An Alternatively Spliced TREM2 Isoform Lacking the Ligand Binding Domain is Expressed in Human Brain. J. Alzheimer’s Dis. 2022, 87, 1647–1657. [Google Scholar] [CrossRef]
- Li, R.-Y.; Qin, Q.; Yang, H.C.; Wang, Y.Y.; Mi, Y.X.; Yin, Y.S.; Wang, M.; Yu, C.J.; Tang, Y. TREM2 in the pathogenesis of AD: A lipid metabolism regulator and potential metabolic therapeutic target. Mol. Neurodegener. 2022, 17, 40. [Google Scholar] [CrossRef]
- Zhao, Y.; Wu, X.; Li, X.; Jiang, L.L.; Gui, X.; Liu, Y.; Sun, Y.; Zhu, B.; Piña-Crespo, J.C.; Zhang, M.; et al. TREM2 is a receptor for β-amyloid that mediates microglial function. Neuron 2018, 97, 1023–1031.e7. [Google Scholar] [CrossRef]
- Poliani, P.L.; Wang, Y.; Fontana, E.; Robinette, M.L.; Yamanishi, Y.; Gilfillan, S.; Colonna, M. TREM2 sustains microglial expansion during aging and response to demyelination. J. Clin. Investig. 2015, 125, 2161–2170. [Google Scholar] [CrossRef]
- Magno, L.; Bunney, T.D.; Mead, E.; Svensson, F.; Bictash, M.N. TREM2/PLCγ2 signalling in immune cells: Function, structural insight, and potential therapeutic modulation. Mol. Neurodegener. 2021, 16, 22. [Google Scholar] [CrossRef]
- Cignarella, F.; Filipello, F.; Bollman, B.; Cantoni, C.; Locca, A.; Mikesell, R.; Manis, M.; Ibrahim, A.; Deng, L.; Benitez, B.A.; et al. TREM2 activation on microglia promotes myelin debris clearance and remyelination in a model of multiple sclerosis. Acta Neuropathol. 2020, 140, 513–534. [Google Scholar] [CrossRef]
- Yeh, F.L.; Hansen, D.V.; Sheng, M. TREM2, microglia, and neurodegenerative diseases. Trends Mol. Med. 2017, 23, 512–533. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J. The Alzheimer family of diseases: Many etiologies, one pathogenesis? Proc. Natl. Acad. Sci. USA 1997, 94, 2095–2097. [Google Scholar] [CrossRef] [PubMed]
- Small, G.W. The pathogenesis of Alzheimer’s disease. J. Clin. Psychiatry 1998, 59, 7–14. [Google Scholar] [PubMed]
- Hu, N.; Tan, M.S.; Yu, J.T.; Sun, L.; Tan, L.; Wang, Y.L.; Jiang, T.; Tan, L. Increased expression of TREM2 in peripheral blood of Alzheimer’s disease patients. J. Alzheimer’s Dis. 2013, 38, 497–501. [Google Scholar] [CrossRef]
- Prokop, S.; Miller, K.R.; Labra, S.R.; Pitkin, R.M.; Hoxha, K.; Narasimhan, S.; Changolkar, L.; Rosenbloom, A.; Lee, V.M.; Trojanowski, J.Q. Impact of TREM2 risk variants on brain region-specific immune activation and plaque microenvironment in Alzheimer’s disease patient brain samples. Acta Neuropathol. 2019, 138, 613–630. [Google Scholar] [CrossRef]
- Forabosco, P.; Ramasamy, A.; Trabzuni, D.; Walker, R.; Smith, C.; Bras, J.; Levine, A.P.; Hardy, J.; Pocock, J.M.; Guerreiro, R.; et al. Insights into TREM2 biology by network analysis of human brain gene expression data. Neurobiol. Aging 2013, 34, 2699–2714. [Google Scholar] [CrossRef]
- Heslegrave, A.; Heywood, W.; Paterson, R.; Magdalinou, N.; Svensson, J.; Johansson, P.; Öhrfelt, A.; Blennow, K.; Hardy, J.; Schott, J.; et al. Increased cerebrospinal fluid soluble TREM2 concentration in Alzheimer’s disease. Mol. Neurodegener. 2016, 11, 3. [Google Scholar] [CrossRef]
- Franzmeier, N.; Suárez-Calvet, M.; Frontzkowski, L.; Moore, A.; Hohman, T.J.; Morenas-Rodriguez, E.; Nuscher, B.; Shaw, L.; Trojanowski, J.Q.; Dichgans, M.; et al. Higher CSF sTREM2 attenuates ApoE4-related risk for cognitive decline and neurodegeneration. Mol. Neurodegener. 2020, 15, 57. [Google Scholar] [CrossRef]
- Basha Sk, C.; Mekala, J.R. Basic Science and Pathogenesis. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2024, 20 (Suppl. S1), e084634. [Google Scholar]
- Lyu, S.; Lan, Z.; Li, C. The triggering receptor expressed on myeloid cells 2–apolipoprotein E signaling pathway in diseases. Chin. Med. J. 2023, 136, 1291–1299. [Google Scholar] [CrossRef]
- Wang, Y.-C.; Huang, L.Y.; Guo, H.H.; Liu, M.; Zhang, Y.Y.; Zhang, Z.Q.; Hao, Q.; Tan, C.C.; Tan, L. Higher CSF sTREM2 attenuates APOE ε4-related risk for amyloid pathology in cognitively intact adults: The CABLE study. J. Neurochem. 2024, 169, e16273. [Google Scholar] [CrossRef] [PubMed]
- Lietzke, E.E.; Saeb, D.; Aldrich, E.C.; Bruce, K.D.; Sprenger, K.G. Synergistic reduction in interfacial flexibility of TREM2R47H and ApoE4 may underlie AD pathology. Alzheimer’s Dement. 2025, 21, e70120. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T. ApoE4 makes microglia trem2bling. Neuron 2023, 111, 142–144. [Google Scholar] [CrossRef] [PubMed]
- Knapskog, A.-B.; Henjum, K.; Idland, A.V.; Eldholm, R.S.; Persson, K.; Saltvedt, I.; Watne, L.O.; Engedal, K.; Nilsson, L.N.G. Cerebrospinal fluid sTREM2 in Alzheimer’s disease: Comparisons between clinical presentation and AT classification. Sci. Rep. 2020, 10, 15886. [Google Scholar] [CrossRef]
- Nabizadeh, F.; Seyedmirzaei, H.; Karami, S. Neuroimaging biomarkers and CSF sTREM2 levels in Alzheimer’s disease: A longitudinal study. Sci. Rep. 2024, 14, 15318. [Google Scholar] [CrossRef]
- Crook, H.; Wahdan, M.; Tuil, M.E.; Livingston, N.R.; Raza, S.; Nowell, J.; Edison, P. CSF sTREM2 is associated with neuroprotective microglial states early in Alzheimer’s disease and deleterious effects later in the disease trajectory. Alzheimer’s Dement. 2024, 20, e094051. [Google Scholar] [CrossRef]
- Španić Popovački, E.; Babić Leko, M.; Langer Horvat, L.; Brgić, K.; Vogrinc, Ž.; Boban, M.; Klepac, N.; Borovečki, F.; Šimić, G. Soluble TREM2 concentrations in the cerebrospinal fluid correlate with the severity of neurofibrillary degeneration, cognitive impairment, and inflammasome activation in Alzheimer’s disease. Neurol. Int. 2023, 15, 842–856. [Google Scholar] [CrossRef]
- Wang, S.; Chenghui, C.; Peng, D. The various roles of TREM2 in cardiovascular disease. Front. Immunol. 2025, 16, 1462508. [Google Scholar] [CrossRef]
- Li, T.; Lyu, D.; Liu, F.-Q. Cerebrospinal Fluid sTREM2 in Alzheimer’s Disease Is Associated with Both Amyloid and Tau Pathologies but not with Cognitive Status. J. Alzheimer’s Dis. 2022, 90, 1123–1138. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Lee, E.H.; Kim, H.J.; Jo, S.; Lee, S.; Seo, S.W.; Park, H.H.; Koh, S.H.; Lee, J.H. The relationship of soluble TREM2 to other biomarkers of sporadic Alzheimer’s disease. Sci. Rep. 2021, 11, 13050. [Google Scholar] [CrossRef] [PubMed]
- Paloneva, J.; Manninen, T.; Christman, G.; Hovanes, K.; Mandelin, J.; Adolfsson, R.; Bianchin, M.; Bird, T.; Miranda, R.; Salmaggi, A.; et al. Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am. J. Hum. Genet. 2002, 71, 656–662. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, R.J.; Lohmann, E.; Brás, J.M.; Gibbs, J.R.; Rohrer, J.D.; Gurunlian, N.; Dursun, B.; Bilgic, B.; Hanagasi, H.; Gurvit, H.; et al. Using exome sequencing to reveal mutations in TREM2 presenting as a frontotemporal dementia–like syndrome without bone involvement. JAMA Neurol. 2013, 70, 78–84. [Google Scholar] [CrossRef]
- Zhou, Y.; Tada, M.; Cai, Z.; Andhey, P.S.; Swain, A.; Miller, K.R.; Gilfillan, S.; Artyomov, M.N.; Takao, M.; Kakita, A.; et al. Human early-onset dementia caused by DAP12 deficiency reveals a unique signature of dysregulated microglia. Nat. Immunol. 2023, 24, 545–557. [Google Scholar] [CrossRef]
- Williamson, J.C.; Larner, A.J. Behavioral Variant Frontotemporal Dementia-like Syndrome with Novel Heterozygous TREM2 Frameshift Mutation. Alzheimer Dis. Assoc. Disord. 2019, 33, 75–76. [Google Scholar] [CrossRef]
- Paloneva, J.; Kestilä, M.; Wu, J.; Salminen, A.; Böhling, T.; Ruotsalainen, V.; Hakola, P.; Bakker, A.B.; Phillips, J.H.; Pekkarinen, P.; et al. Loss-of-function mutations in TYROBP (DAP12) result in a presenile dementia with bone cysts. Nat. Genet. 2000, 25, 357–361. [Google Scholar] [CrossRef]
- Klunemann, H.; Ridha, B.H.; Magy, L.; Wherrett, J.R.; Hemelsoet, D.M.; Keen, R.W.; De Bleecker, J.L.; Rossor, M.N.; Marienhagen, J.; Klein, H.E.; et al. The genetic causes of basal ganglia calcification, dementia, and bone cysts: DAP12 and TREM2. Neurology 2005, 64, 1502–1507. [Google Scholar] [CrossRef]
- Xing, J.; Titus, A.R.; Humphrey, M.B. The TREM2-DAP12 signaling pathway in Nasu-Hakola disease: A molecular genetics perspective. Res. Rep. Biochem. 2015, 5, 89–100. [Google Scholar]
- Tinkler, S.M.; Linder, J.E.; Williams, D.M.; Johnson, N.W. Formation of osteoclasts from blood monocytes during 1 alpha-OH Vit D-stimulated bone resorption in mice. J. Anat. 1981, 133, 389–396. [Google Scholar] [PubMed]
- Cella, M.; Buonsanti, C.; Strader, C.; Kondo, T.; Salmaggi, A.; Colonna, M. Impaired differentiation of osteoclasts in TREM-2–deficient individuals. J. Exp. Med. 2003, 198, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Paloneva, J.; Mandelin, J.; Kiialainen, A.; Bohling, T.; Prudlo, J.; Hakola, P.; Haltia, M.; Konttinen, Y.T.; Peltonen, L.; Mandelin, J.; et al. DAP12/TREM2 deficiency results in impaired osteoclast differentiation and osteoporotic features. J. Exp. Med. 2003, 198, 669–675. [Google Scholar] [CrossRef] [PubMed]
- Konishi, H.; Kiyama, H. Microglial TREM2/DAP12 Signaling: A Double-Edged Sword in Neural Diseases. Front. Cell Neurosci. 2018, 12, 206. [Google Scholar] [CrossRef]
- Sasaki, A.; Kakita, A.; Yoshida, K.; Konno, T.; Ikeuchi, T.; Hayashi, S.; Matsuo, H.; Shioda, K. Variable expression of microglial DAP12 and TREM2 genes in Nasu-Hakola disease. Neurogenetics 2015, 16, 265–276. [Google Scholar] [CrossRef]
- Mecca, C.; Giambanco, I.; Donato, R.; Arcuri, C. Microglia and Aging: The Role of the TREM2-DAP12 and CX3CL1-CX3CR1 Axes. Int. J. Mol. Sci. 2018, 19, 318. [Google Scholar] [CrossRef]
- Cady, J.; Koval, E.D.; Benitez, B.A.; Zaidman, C.; Jockel-Balsarotti, J.; Allred, P.; Baloh, R.H.; Ravits, J.; Simpson, E.; Appel, S.H.; et al. TREM2 variant p. R47H as a risk factor for sporadic amyotrophic lateral sclerosis. JAMA Neurol. 2014, 71, 449–453. [Google Scholar] [CrossRef]
- Borroni, B.; Ferrari, F.; Galimberti, D.; Nacmias, B.; Barone, C.; Bagnoli, S.; Fenoglio, C.; Piaceri, I.; Archetti, S.; Bonvicini, C.; et al. Heterozygous TREM2 mutations in frontotemporal dementia. Neurobiol. Aging 2014, 35, 934.e7–934.e10. [Google Scholar] [CrossRef]
- Cuyvers, E.; Bettens, K.; Philtjens, S.; Van Langenhove, T.; Gijselinck, I.; van der Zee, J.; Engelborghs, S.; Vandenbulcke, M.; Van Dongen, J.; Geerts, N.; et al. Investigating the role of rare heterozygous TREM2 variants in Alzheimer’s disease and frontotemporal dementia. Neurobiol. Aging 2014, 35, 726.e11–726.e19. [Google Scholar] [CrossRef]
- Rayaprolu, S.; Mullen, B.; Baker, M.; Lynch, T.; Finger, E.; Seeley, W.W.; Hatanpaa, K.J.; Lomen-Hoerth, C.; Kertesz, A.; Bigio, E.H.; et al. TREM2 in neurodegeneration: Evidence for association of the p. R47H variant with frontotemporal dementia and Parkinson’s disease. Mol. Neurodegener. 2013, 8, 19. [Google Scholar] [CrossRef]
- Thelen, M.; Razquin, C.; Hernández, I.; Gorostidi, A.; Sánchez-Valle, R.; Ortega-Cubero, S.; Wolfsgruber, S.; Drichel, D.; Fliessbach, K.; Duenkel, T.; et al. Investigation of the role of rare TREM2 variants in frontotemporal dementia subtypes. Neurobiol. Aging 2014, 35, 2657.e13–2657.e19. [Google Scholar] [CrossRef]
- Ogonowski, N.; Santamaria-Garcia, H.; Baez, S.; Lopez, A.; Laserna, A.; Garcia-Cifuentes, E.; Ayala-Ramirez, P.; Zarante, I.; Suarez-Obando, F.; Reyes, P.; et al. Frontotemporal dementia presentation in patients with heterozygous p.H157Y variant of TREM2. J. Med. Genet. 2023, 60, 894–904. [Google Scholar] [CrossRef]
- Xie, M.; Liu, Y.U.; Zhao, S.; Zhang, L.; Bosco, D.B.; Pang, Y.P.; Zhong, J.; Sheth, U.; Martens, Y.A.; Zhao, N.; et al. TREM2 interacts with TDP-43 and mediates microglial neuroprotection against TDP-43-related neurodegeneration. Nat. Neurosci. 2022, 25, 26–38. [Google Scholar] [CrossRef]
- Xie, M. The Role of Microglia TREM2 in Tdp-43 Related Neurodegeneration. Ph.D. Thesis, College of Medicine-Mayo Clinic, Rochester, MN, USA, 2022. [Google Scholar]
- Mills, W.A., III; Eyo, U.B. TREMble before TREM2: The mighty microglial receptor conferring neuroprotective properties in TDP-43 mediated neurodegeneration. Neurosci. Bull. 2023, 39, 163–166. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hu, Y.; Xu, R. The pathogenic mechanism of TAR DNA-binding protein 43 (TDP-43) in amyotrophic lateral sclerosis. Neural Regen. Res. 2024, 19, 800–806. [Google Scholar] [CrossRef] [PubMed]
- Greven, J.A.; Wydra, J.R.; Greer, R.A.; Zhi, C.; Price, D.A.; Svoboda, J.D.; Camitta, C.L.M.; Washington, M.; Leung, D.W.; Song, Y.; et al. Biophysical mapping of TREM2-ligand interactions reveals shared surfaces for engagement of multiple Alzheimer’s disease ligands. Mol. Neurodegener. 2025, 20, 3. [Google Scholar] [CrossRef]
- Seddighi, S.; Qi, Y.A.; Brown, A.L.; Wilkins, O.G.; Bereda, C.; Belair, C.; Zhang, Y.J.; Prudencio, M.; Keuss, M.J.; Khandeshi, A.; et al. Mis-spliced transcripts generate de novo proteins in TDP-43–related ALS/FTD. Sci. Transl. Med. 2024, 16, eadg7162. [Google Scholar] [CrossRef]
- Chhangani, D.; Rincon-Limas, D.E. TDP-35, a truncated fragment of TDP-43, induces dose-dependent toxicity and apoptosis in flies. Neural Regen. Res. 2022, 17, 2441–2442. [Google Scholar] [CrossRef]
- Li, X.X.; Zhang, F. Targeting TREM2 for Parkinson’s Disease: Where to Go? Front. Immunol 2021, 12, 795036. [Google Scholar] [CrossRef]
- Huang, P.; Zhang, Z.; Zhang, P.; Feng, J.; Xie, J.; Zheng, Y.; Liang, X.; Zhu, B.; Chen, Z.; Feng, S.; et al. TREM2 Deficiency Aggravates NLRP3 Inflammasome Activation and Pyroptosis in MPTP-Induced Parkinson’s Disease Mice and LPS-Induced BV2 Cells. Mol. Neurobiol. 2024, 61, 2590–2605. [Google Scholar] [CrossRef]
- Huang, W.; Huang, W.; Lv, Q.; Xiao, Y.; Zhong, Z.; Hu, B.; Yan, S.; Yan, Y.; Zhang, J.; Shi, T.; et al. Triggering receptor expressed on myeloid cells 2 protects dopaminergic neurons by promoting autophagy in the inflammatory pathogenesis of Parkinson’s disease. Front. Neurosci. 2021, 15, 745815. [Google Scholar] [CrossRef]
- Liu, Z.; Ning, J.; Zheng, X.; Meng, J.; Han, L.; Zheng, H.; Zhong, L.; Chen, X.F.; Zhang, X.; Luo, H.; et al. TMEM59 interacts with TREM2 and modulates TREM2-dependent microglial activities. Cell Death Dis. 2020, 11, 678. [Google Scholar] [CrossRef] [PubMed]
- Lv, Q.; Zhong, Z.; Hu, B.; Yan, S.; Yan, Y.; Zhang, J.; Shi, T.; Jiang, L.; Li, W.; Huang, W. MicroRNA-3473b regulates the expression of TREM2/ULK1 and inhibits autophagy in inflammatory pathogenesis of Parkinson disease. J. Neurochem. 2021, 157, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Jay, T.R.; von Saucken, V.E.; Landreth, G.E. TREM2 in Neurodegenerative Diseases. Mol. Neurodegener. 2017, 12, 56. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Wei, X.; Yan, H.; Qin, Y.; Yan, S.; Liu, J.; Zhao, Y.; Jiang, F.; Lou, H. TREM2 deficiency aggravates α-synuclein-induced neurodegeneration and neuroinflammation in Parkinson’s disease models. FASEB J. 2019, 33, 12164–12174. [Google Scholar] [CrossRef]
- Dela Cruz, H.L.; Dela Cruz, E.L.; Zurhellen, C.J.; York, H.T.; Baun, J.A.; Dela Cruz, J.L.; Dela Cruz, J.S. New insights underlying the early events of dopaminergic dysfunction in Parkinson’s Disease. bioRxiv 2023. [Google Scholar] [CrossRef]
- Eo, H.; Kim, S.; Jung, U.J.; Kim, S.R. Alpha-Synuclein and Microglia in Parkinson’s Disease: From Pathogenesis to Therapeutic Prospects. J. Clin. Med. 2024, 13, 7243. [Google Scholar] [CrossRef]
- Deyell, J.S.; Sriparna, M.; Ying, M.; Mao, X. The Interplay between α-Synuclein and Microglia in α-Synucleinopathies. Int. J. Mol. Sci. 2023, 24, 2477. [Google Scholar] [CrossRef]
- Tsunemi, T.; Krainc, D. Zn2+ dyshomeostasis caused by loss of ATP13A2/PARK9 leads to lysosomal dysfunction and alpha-synuclein accumulation. Hum. Mol. Genet. 2014, 23, 2791–2801. [Google Scholar] [CrossRef]
- Yin, S.; Chi, X.; Wan, F.; Li, Y.; Zhou, Q.; Kou, L.; Sun, Y.; Wu, J.; Zou, W.; Wang, Y.; et al. TREM2 signaling in Parkinson’s disease: Regulation of microglial function and α-synuclein pathology. Int. Immunopharmacol. 2024, 143 Pt 2, 113446. [Google Scholar] [CrossRef]
- Shan, H.-M.; Zang, M.; Zhang, Q.; Shi, R.B.; Shi, X.J.; Mamtilahun, M.; Liu, C.; Luo, L.L.; Tian, X.; Zhang, Z.; et al. Farnesoid X receptor knockout protects brain against ischemic injury through reducing neuronal apoptosis in mice. J. Neuroinflamm. 2020, 17, 164. [Google Scholar] [CrossRef] [PubMed]
- Singaraja, R.R. TREM2: A new risk factor for Alzheimer’s disease. Clin. Genet. 2013, 83, 525–526. [Google Scholar] [CrossRef] [PubMed]
- Paradowska-Gorycka, A.; Jurkowska, M. Structure, expression pattern and biological activity of molecular complex TREM-2/DAP12. Hum. Immunol. 2013, 74, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Bouchon, A.; Hernández-Munain, C.; Cella, M.; Colonna, M. A DAP12-mediated pathway regulates expression of CC chemokine receptor 7 and maturation of human dendritic cells. J. Exp. Med. 2001, 194, 1111. [Google Scholar] [CrossRef]
- Zhong, L.; Chen, X.F.; Zhang, Z.L.; Wang, Z.; Shi, X.Z.; Xu, K.; Zhang, Y.W.; Xu, H.; Bu, G. DAP12 Stabilizes the C-terminal Fragment of the Triggering Receptor Expressed on Myeloid Cells-2 (TREM2) and Protects against LPS-induced Pro-inflammatory Response. J. Biol. Chem. 2015, 290, 15866–15877. [Google Scholar] [CrossRef]
- Ewers, M.; Franzmeier, N.; Suárez-Calvet, M.; Morenas-Rodriguez, E.; Caballero, M.A.A.; Kleinberger, G.; Piccio, L.; Cruchaga, C.; Deming, Y.; Dichgans, M.; et al. Increased soluble TREM2 in cerebrospinal fluid is associated with reduced cognitive and clinical decline in Alzheimer’s disease. Sci. Transl. Med. 2019, 11, eaav6221. [Google Scholar] [CrossRef]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms underlying inflammation in neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef]
- Chen, Y.; Yu, Y. Tau and neuroinflammation in Alzheimer’s disease: Interplay mechanisms and clinical translation. J. Neuroinflammation 2023, 20, 165. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Y.; Wang, J.; Xia, Y.; Zhang, J.; Chen, L. Recent advances in Alzheimer’s disease: Mechanisms, clinical trials and new drug development strategies. Signal Transduct. Target. Ther. 2024, 9, 211. [Google Scholar] [CrossRef]
- Amos, P.J.; Fung, S.; Case, A.; Kifelew, J.; Osnis, L.; Smith, C.L.; Green, K.; Naydenov, A.; Aloi, M.; Hubbard, J.J.; et al. Modulation of hematopoietic lineage specification impacts TREM2 expression in microglia-like cells derived from human stem cells. ASN Neuro 2017, 9, 1759091417716610. [Google Scholar] [CrossRef]
- Wang, M.; Gao, X.; Zhao, K.; Chen, H.; Xu, M.; Wang, K. Effect of TREM2 on release of inflammatory factor from LPS-stimulated microglia and its possible mechanism. Ann. Clin. Lab. Sci. 2019, 49, 249–256. [Google Scholar]
- Novoa, C.; Salazar, P.; Cisternas, P.; Gherardelli, C.; Vera-Salazar, R.; Zolezzi, J.M.; Inestrosa, N.C. Inflammation context in Alzheimer’s disease, a relationship intricate to define. Biol. Res. 2022, 55, 39. [Google Scholar] [CrossRef] [PubMed]
- Miao, J.; Ma, H.; Yang, Y.; Liao, Y.; Lin, C.; Zheng, J.; Yu, M.; Lan, J. Microglia in Alzheimer’s disease: Pathogenesis, mechanisms, and therapeutic potentials. Front. Aging Neurosci. 2023, 15, 1201982. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Xiao, D.; Mao, Q.; Xia, H.R. Role of neuroinflammation in neurodegeneration development. Signal Transduct. Target. Ther. 2023, 8, 267. [Google Scholar] [CrossRef]
- Lin, M.; Yu, J.X.; Zhang, W.X.; Lao, F.X.; Huang, H.C. Roles of TREM2 in the Pathological Mechanism and the Therapeutic Strategies of Alzheimer’s Disease. J. Prev. Alzheimer’s Dis. 2024, 11, 1682–1695. [Google Scholar] [CrossRef] [PubMed]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef]
- Zhao, Y.; Guo, Q.; Tian, J.; Liu, W.; Wang, X. TREM2 bridges microglia and extracellular microenvironment: Mechanistic landscape and therapeutical prospects on Alzheimer’s disease. Ageing Res. Rev. 2025, 103, 102596. [Google Scholar] [CrossRef]
- Simpson, D.S.; Oliver, P.L. ROS generation in microglia: Understanding oxidative stress and inflammation in neurodegenerative disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef]
- Wang, X.; Lopez, O.L.; Sweet, R.A.; Becker, J.T.; DeKosky, S.T.; Barmada, M.M.; Demirci, F.Y.; Kamboh, M.I. Genetic determinants of disease progression in Alzheimer’s disease. J. Alzheimer’s Dis. 2014, 43, 649–655. [Google Scholar] [CrossRef]
- Wang, Y.; Cella, M.; Mallinson, K.; Ulrich, J.D.; Young, K.L.; Robinette, M.L.; Gilfillan, S.; Krishnan, G.M.; Sudhakar, S.; Zinselmeyer, B.H.; et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 2015, 160, 1061–1071. [Google Scholar] [CrossRef] [PubMed]
- Atagi, Y.; Liu, C.C.; Painter, M.M.; Chen, X.F.; Verbeeck, C.; Zheng, H.; Li, X.; Rademakers, R.; Kang, S.S.; Xu, H.; et al. Apolipoprotein E is a ligand for triggering receptor expressed on myeloid cells 2 (TREM2). J. Biol. Chem. 2015, 290, 26043–26050. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, C.M.; Fitz, N.F.; Nam, K.N.; Lefterov, I.; Koldamova, R. The Role of APOE and TREM2 in Alzheimer’s Disease-Current Understanding and Perspectives. Int. J. Mol. Sci. 2018, 20, 81. [Google Scholar] [CrossRef] [PubMed]
- Jendresen, C.; Årskog, V.; Daws, M.R.; Nilsson, L.N. The Alzheimer’s disease risk factors apolipoprotein E and TREM2 are linked in a receptor signaling pathway. J. Neuroinflamm. 2017, 14, 59. [Google Scholar] [CrossRef]
- Yeh, F.L.; Wang, Y.; Tom, I.; Gonzalez, L.C.; Sheng, M. TREM2 Binds to Apolipoproteins, Including APOE and CLU/APOJ, and Thereby Facilitates Uptake of Amyloid-Beta by Microglia. Neuron 2016, 91, 328–340. [Google Scholar] [CrossRef]
- Song, W.; Hooli, B.; Mullin, K.; Jin, S.C.; Cella, M.; Ulland, T.K.; Wang, Y.; Tanzi, R.E.; Colonna, M. Alzheimer’s disease-associated TREM2 variants exhibit either decreased or increased ligand-dependent activation. Alzheimer’s Dement. 2017, 13, 381–387. [Google Scholar] [CrossRef]
- Shirotani, K.; Hori, Y.; Yoshizaki, R.; Higuchi, E.; Colonna, M.; Saito, T.; Hashimoto, S.; Saito, T.; Saido, T.C.; Iwata, N. Aminophospholipids are signal-transducing TREM2 ligands on apoptotic cells. Sci. Rep. 2019, 9, 7508. [Google Scholar] [CrossRef]
- Kiialainen, A.; Hovanes, K.; Paloneva, J.; Kopra, O.; Peltonen, L. Dap12 and Trem2, molecules involved in innate immunity and neurodegeneration, are co-expressed in the CNS. Neurobiol. Dis. 2005, 18, 314–322. [Google Scholar] [CrossRef]
- Sirkis, D.W.; Bonham, L.W.; Aparicio, R.E.; Geier, E.G.; Ramos, E.M.; Wang, Q.; Karydas, A.; Miller, Z.A.; Miller, B.L.; Coppola, G.; et al. Rare TREM2 variants associated with Alzheimer’s disease display reduced cell surface expression. Acta Neuropathol. Commun. 2016, 4, 98. [Google Scholar] [CrossRef]
- Soragna, D.; Papi, L.; Ratti, M.T.; Sestini, R.; Tupler, R.; Montalbetti, L. An Italian family affected by Nasu-Hakola disease with a novel genetic mutation in the TREM2 gene. J. Neurology. Neurosurg. Psychiatry 2003, 74, 825–826. [Google Scholar] [CrossRef]
- Sims, R.; van der Lee, S.J.; Naj, A.C.; Bellenguez, C.; Badarinarayan, N.; Jakobsdottir, J.; Kunkle, B.W.; Boland, A.; Raybould, R.; Bis, J.C.; et al. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat. Genet. 2017, 49, 1373–1384. [Google Scholar] [CrossRef]
- Jin, S.C.; Benitez, B.A.; Karch, C.M.; Cooper, B.; Skorupa, T.; Carrell, D.; Norton, J.B.; Hsu, S.; Harari, O.; Cai, Y.; et al. Coding variants in TREM2 increase risk for Alzheimer’s disease. Hum. Mol. Genet. 2014, 23, 5838–5846. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Tan, L.; Chen, Q.; Tan, M.S.; Zhou, J.S.; Zhu, X.C.; Lu, H.; Wang, H.F.; Zhang, Y.D.; Yu, J.T. A rare coding variant in TREM2 increases risk for Alzheimer’s disease in Han Chinese. Neurobiol. Aging 2016, 42, 217.e1–217.e3. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Hou, J.K.; Gao, Q.; Yu, J.T.; Zhou, J.S.; Zhao, H.D.; Zhang, Y.D. TREM2 p. H157Y variant and the risk of Alzheimer’s disease: A meta-analysis involving 14,510 subjects. Curr. Neurovascular Res. 2016, 13, 318–320. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.C.; Carrasquillo, M.M.; Benitez, B.A.; Skorupa, T.; Carrell, D.; Patel, D.; Lincoln, S.; Krishnan, S.; Kachadoorian, M.; Reitz, C.; et al. TREM2 is associated with increased risk for Alzheimer’s disease in African Americans. Mol. Neurodegener. 2015, 10, 19. [Google Scholar] [CrossRef]
- Jiang, T.; Carrasquillo, M.M.; Benitez, B.A.; Skorupa, T.; Carrell, D.; Patel, D.; Lincoln, S.; Krishnan, S.; Kachadoorian, M.; Reitz, C.; et al. TREM2 in Alzheimer’s disease. Mol. Neurobiol. 2013, 48, 180–185. [Google Scholar] [CrossRef]
- Ulrich, J.D.; Ulland, T.K.; Colonna, M.; Holtzman, D.M. Elucidating the Role of TREM2 in Alzheimer’s Disease. Neuron 2017, 94, 237–248. [Google Scholar] [CrossRef]
- Song, W.M.; Joshita, S.; Zhou, Y.; Ulland, T.K.; Gilfillan, S.; Colonna, M. Humanized TREM2 mice reveal microglia-intrinsic and-extrinsic effects of R47H polymorphism. J. Exp. Med. 2018, 215, 745–760. [Google Scholar] [CrossRef]
- Dodd, R.B. An exTREMe disruption in Alzheimer’s cleanup. J. Biol. Chem. 2018, 293, 12647–12648. [Google Scholar] [CrossRef]
- Gratuze, M.; Leyns, C.E.; Sauerbeck, A.D.; St-Pierre, M.K.; Xiong, M.; Kim, N.; Serrano, J.R.; Tremblay, M.È.; Kummer, T.T.; Colonna, M.; et al. Impact of TREM2 R47H variant on tau pathology–induced gliosis and neurodegeneration. J. Clin. Investig. 2020, 130, 4954–4968. [Google Scholar] [CrossRef]
- Hall-Roberts, H.; Agarwal, D.; Obst, J.; Smith, T.B.; Monzón-Sandoval, J.; Di Daniel, E.; Webber, C.; James, W.S.; Mead, E.; Davis, J.B.; et al. TREM2 Alzheimer’s variant R47H causes similar transcriptional dysregulation to knockout, yet only subtle functional phenotypes in human iPSC-derived macrophages. Alzheimer’s Res. Ther. 2020, 12, 151. [Google Scholar] [CrossRef]
- Park, J.-S.; Ji, I.J.; Kim, D.H.; An, H.J.; Yoon, S.Y. The Alzheimer’s disease-associated R47H variant of TREM2 has an altered glycosylation pattern and protein stability. Front. Neurosci. 2017, 10, 618. [Google Scholar] [CrossRef]
- Zhu, B.; Liu, Y.; Hwang, S.; Archuleta, K.; Huang, H.; Campos, A.; Murad, R.; Piña-Crespo, J.; Xu, H.; Huang, T.Y. Trem2 deletion enhances tau dispersion and pathology through microglia exosomes. Mol. Neurodegener. 2022, 17, 58. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Lewis, C.A.; Ulrich, J.D.; Holtzman, D.M. Chronic TREM2 activation exacerbates Aβ-associated tau seeding and spreading. J. Exp. Med. 2023, 220, e20220654. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Huang, J.; Huang, N.; Luo, Y. The role of TREM2 in Alzheimer’s disease: From the perspective of Tau. Front. Cell Dev. Biol. 2023, 11, 1280257. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Song, W.M.; Andhey, P.S.; Swain, A.; Levy, T.; Miller, K.R.; Poliani, P.L.; Cominelli, M.; Grover, S.; Gilfillan, S.; et al. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat. Med. 2020, 26, 131–142. [Google Scholar] [CrossRef]
- Xiang, X.; Kawauchi, S.; Kramár, E.A.; Rezaie, N.; Liang, H.Y.; Sakr, J.S.; Gomez-Arboledas, A.; Arreola, M.A.; Cunha, C.D.; Phan, J.; et al. The Trem2 R47H Alzheimer’s risk variant impairs splicing and reduces Trem2 mRNA and protein in mice but not in humans. Mol. Neurodegener. 2018, 13, 49. [Google Scholar] [CrossRef]
- Cheng-Hathaway, P.J.; Reed-Geaghan, E.G.; Jay, T.R.; Casali, B.T.; Bemiller, S.M.; Puntambekar, S.S.; von Saucken, V.E.; Williams, R.Y.; Karlo, J.C.; Moutinho, M.; et al. The T rem 2 R47H variant confers loss-of-function-like phenotypes in Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 29. [Google Scholar] [CrossRef]
- Budyak, E.I.; Kwon, J.; Messenger, E.J.; Maharjan, S.; Koothur, J.J. TREM2 Alteration Increases AD Biomarkers and is Associated with Key Genes with 5xFAD Mice Model Analysis on MODEL-AD Database. bioRxiv 2023. [Google Scholar] [CrossRef]
- Menzies, G.E.; Sims, R.; Williams, J. Molecular Dynamics simulations of Alzheimer’s variants, R47H and R62H, in TREM2 provide evidence for structural alterations behind functional changes. bioRxiv 2019. [Google Scholar] [CrossRef]
- Kober, D.L.; Alexander-Brett, J.M.; Karch, C.M.; Cruchaga, C.; Colonna, M.; Holtzman, M.J.; Brett, T.J. Neurodegenerative disease mutations in TREM2 reveal a functional surface and distinct loss-of-function mechanisms. Elife 2016, 5, e20391. [Google Scholar] [CrossRef]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.e9. [Google Scholar] [CrossRef]
- Colonna, M. The biology of TREM receptors. Nat. Rev. Immunol. 2023, 23, 580–594. [Google Scholar] [CrossRef]
- Li, R.; Wang, X.; He, P. The most prevalent rare coding variants of TREM2 conferring risk of Alzheimer’s disease: A systematic review and meta-analysis. Exp. Ther. Med. 2021, 21, 347. [Google Scholar] [CrossRef] [PubMed]
- Carmona, S.; Zahs, K.; Wu, E.; Dakin, K.; Bras, J.; Guerreiro, R. The role of TREM2 in Alzheimer’s disease and other neurodegenerative disorders. Lancet Neurol. 2018, 17, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Li, X.; Huang, T.; Jiang, L.L.; Tan, Z.; Zhang, M.; Cheng, I.H.; Wang, X.; Bu, G.; Zhang, Y.W.; et al. Intracellular trafficking of TREM2 is regulated by presenilin 1. Exp. Mol. Med. 2017, 49, e405. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Ji, I.J.; An, H.J.; Kang, M.J.; Kang, S.W.; Kim, D.H.; Yoon, S.Y. Disease-associated mutations of TREM2 alter the processing of N-linked oligosaccharides in the Golgi apparatus. Traffic 2015, 16, 510–518. [Google Scholar] [CrossRef]
- Joshi, P.; Riffel, F.; Satoh, K.; Enomoto, M.; Qamar, S.; Scheiblich, H.; Villacampa, N.; Kumar, S.; Theil, S.; Parhizkar, S.; et al. Differential interaction with TREM2 modulates microglial uptake of modified Aβ species. Glia 2021, 69, 2917–2932. [Google Scholar] [CrossRef]
- Sudom, A.; Talreja, S.; Danao, J.; Bragg, E.; Kegel, R.; Min, X.; Richardson, J.; Zhang, Z.; Sharkov, N.; Marcora, E.; et al. Molecular basis for the loss-of-function effects of the Alzheimer’s disease–associated R47H variant of the immune receptor TREM2. J. Biol. Chem. 2018, 293, 12634–12646. [Google Scholar] [CrossRef]
- Kleinberger, G.; Yamanishi, Y.; Suárez-Calvet, M.; Czirr, E.; Lohmann, E.; Cuyvers, E.; Struyfs, H.; Pettkus, N.; Wenninger-Weinzierl, A.; Mazaheri, F.; et al. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci. Transl. Med. 2014, 6, 243ra86. [Google Scholar] [CrossRef]
- Kleinberger, G.; Brendel, M.; Mracsko, E.; Wefers, B.; Groeneweg, L.; Xiang, X.; Focke, C.; Deußing, M.; Suárez-Calvet, M.; Mazaheri, F.; et al. The FTD-like syndrome causing TREM2 T66M mutation impairs microglia function, brain perfusion, and glucose metabolism. EMBO J. 2017, 36, 1837–1853. [Google Scholar] [CrossRef]
- Shi, Q.; Gutierrez, R.A.; Bhat, M.A. Microglia, Trem2, and Neurodegeneration. Neuroscientist 2025, 31, 159–176. [Google Scholar] [CrossRef]
- Gao CJiang, J.; Tan, Y.; Chen, S. Microglia in neurodegenerative diseases: Mechanism and potential therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 359. [Google Scholar] [CrossRef] [PubMed]
- Dash, R.; Choi, H.J.; Moon, I.S. Mechanistic insights into the deleterious roles of Nasu-Hakola disease associated TREM2 variants. Sci. Rep. 2020, 10, 3663. [Google Scholar] [CrossRef] [PubMed]
- Sirkis, D.W.; Aparicio, R.E.; Schekman, R. Neurodegeneration-associated mutant TREM2 proteins abortively cycle between the ER and ER-Golgi intermediate compartment. Mol. Biol. Cell 2017, 28, 2723–2733. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, L.I. Triggering Receptor Expressed on Myeloid Cells 2 (TREM2) Mutations: A Potential Common Cause of Alzheimer’s Disease and Musculoskeletal Disorders. FASEB J. 2019, 33, 587–598. [Google Scholar] [CrossRef]
- Schlepckow, K.; Kleinberger, G.; Fukumori, A.; Feederle, R.; Lichtenthaler, S.F.; Steiner, H.; Haass, C. An Alzheimer-associated TREM2 variant occurs at the ADAM cleavage site and affects shedding and phagocytic function. EMBO Mol. Med. 2017, 9, 1356–1365. [Google Scholar] [CrossRef]
- Qiao, W.; Chen, Y.; Zhong, J.; Madden, B.J.; Charlesworth, C.M.; Martens, Y.A.; Liu, C.C.; Knight, J.; Ikezu, T.C.; Kurti, A.; et al. Trem2 H157Y increases soluble TREM2 production and reduces amyloid pathology. Mol. Neurodegener. 2023, 18, 8. [Google Scholar] [CrossRef]
- Miyashita, A.; Wen, Y.; Kitamura, N.; Matsubara, E.; Kawarabayashi, T.; Shoji, M.; Tomita, N.; Furukawa, K.; Arai, H.; Asada, T.; et al. Lack of genetic association between TREM2 and late-onset Alzheimer’s disease in a Japanese population. J. Alzheimer’s Dis. JAD 2014, 41, 1031–1038. [Google Scholar] [CrossRef]
- Feuerbach, D.; Schindler, P.; Barske, C.; Joller, S.; Beng-Louka, E.; Worringer, K.A.; Kommineni, S.; Kaykas, A.; Ho, D.J.; Ye, C.; et al. ADAM17 is the main sheddase for the generation of human triggering receptor expressed in myeloid cells (hTREM2) ectodomain and cleaves TREM2 after Histidine 157. Neurosci Lett. 2017, 660, 109–114. [Google Scholar] [CrossRef]
- Thornton, P.; Sevalle, J.; Deery, M.J.; Fraser, G.; Zhou, Y.; Ståhl, S.; Franssen, E.H.; Dodd, R.B.; Qamar, S.; Gomez Perez-Nievas, B.; et al. TREM2 shedding by cleavage at the H157-S158 bond is accelerated for the Alzheimer’s disease-associated H157Y variant. EMBO Mol. Med. 2017, 9, 1366–1378. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.; Mez, J.; Vardarajan, B.N.; Staley, L.; Chung, J.; Zhang, X.; Farrell, J.J.; Rynkiewicz, M.J.; Cannon-Albright, L.A.; Teerlink, C.C.; et al. Association of Rare Coding Mutations with Alzheimer Disease and Other Dementias Among Adults of European Ancestry. JAMA Netw. Open 2019, 2, e191350. [Google Scholar] [CrossRef] [PubMed]
- Paloneva, J.; Autti, T.; Hakola, P.; Haltia, M.J. Polycystic Lipomembranous Osteodysplasia with Sclerosing Leukoencephalopathy (PLOSL). In GeneReviews (®); University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Ji, M.J.; Jung, S.; Seo, H.E.; Kim, S.Y.; Kim, W.R.; Kim, S.; Lee, J.S.; Noh, Y. Heterozygous TREM2 Mutation in Semantic Variant of Primary Progressive Aphasia. J. Clin. Neurol. 2020, 16, 352–354. [Google Scholar] [CrossRef] [PubMed]
- Haddad, G. Unraveling the Role of TREM2 and CD33 in Alzheimer’s Disease. Am. J. Stud. Res. 2024, 2, 3. [Google Scholar] [CrossRef]
- Suárez-Calvet, M.; Morenas-Rodríguez, E.; Kleinberger, G.; Schlepckow, K.; Araque Caballero, M.Á.; Franzmeier, N.; Capell, A.; Fellerer, K.; Nuscher, B.; Eren, E.; et al. Early increase of CSF sTREM2 in Alzheimer’s disease is associated with tau related-neurodegeneration but not with amyloid-β pathology. Mol. Neurodegener. 2019, 14, 1. [Google Scholar] [CrossRef]
- Tian, Y.; Xiao, X.; Liu, W.; Cheng, S.; Qian, N.; Wang, L.; Liu, Y.; Ai, R.; Zhu, X. TREM2 improves microglia function and synaptic development in autism spectrum disorders by regulating P38 MAPK signaling pathway. Mol. Brain 2024, 17, 12. [Google Scholar] [CrossRef]
- George, J. TREM2 as an evolving therapeutic target in Alzheimer’s disease. Neural Regen. Res. 2023, 18, 2680–2681. [Google Scholar] [CrossRef]
- Fassler, M.; Rappaport, M.S.; Cuño, C.B.; George, J. Engagement of TREM2 by a novel monoclonal antibody induces activation of microglia and improves cognitive function in Alzheimer’s disease models. J. Neuroinflamm. 2021, 18, 19. [Google Scholar] [CrossRef]
- Zhang, L.; Xiang, X.; Li, Y.; Bu, G.; Chen, X.F. TREM2 and sTREM2 in Alzheimer’s disease: From mechanisms to therapies. Mol. Neurodegener. 2025, 20, 43. [Google Scholar] [CrossRef]
- Mirescu, C. Characterization of the first TREM2 small molecule agonist, VG-3927, for clinical development in Alzheimer’s disease. Alzheimers Dement. 2025, 20 (Suppl. S6), e084622. [Google Scholar] [CrossRef]
- Vigil’s TREM2-Targeted Alzheimer’s Treatment Shows Early Promise, Moves on to Phase II. Available online: https://www.biospace.com/drug-development/vigils-trem2-targeted-alzheimers-treatment-shows-early-promise-moves-on-to-phase-ii (accessed on 1 July 2025).
- Ma, Y.N.; Hu, X.; Karako, K.; Song, P.; Tang, W.; Xia, Y. The potential and challenges of TREM2-targeted therapy in Alzheimer’s disease: Insights from the INVOKE-2 study. Front. Aging Neurosci. 2025, 17, 1576020. [Google Scholar] [CrossRef]
- Colonna, M.; Holtzman, D.M. Rethinking TREM2 as a target for Alzheimer’s disease after the INVOKE-2 trial failure. Nat. Med. 2025. [Google Scholar] [CrossRef]
- Serradas, M.L.; Ding, Y.; Martorell, P.V.; Kulińska, I.; Castro-Gomez, S. Therapeutic Targets in Innate Immunity to Tackle Alzheimer’s Disease. Cells 2024, 13, 1426. [Google Scholar] [CrossRef]
- van Lengerich, B.; Zhan, L.; Xia, D.; Chan, D.; Joy, D.; Park, J.I.; Tatarakis, D.; Calvert, M.; Hummel, S.; Lianoglou, S.; et al. A TREM2-activating antibody with a blood-brain barrier transport vehicle enhances microglial metabolism in Alzheimer’s disease models. Nat. Neurosci. 2023, 26, 416–429. [Google Scholar] [CrossRef]
- Schlepckow, K.; Monroe, K.M.; Kleinberger, G.; Cantuti-Castelvetri, L.; Parhizkar, S.; Xia, D.; Willem, M.; Werner, G.; Pettkus, N.; Brunner, B.; et al. Enhancing protective microglial activities with a dual function TREM2 antibody to the stalk region. EMBO Mol. Med. 2020, 12, e11227. [Google Scholar] [CrossRef]
- Zhao, P.; Xu, Y.; Jiang, L.; Fan, X.; Li, L.; Li, X.; Arase, H.; Zhao, Y.; Cao, W.; Zheng, H.; et al. A tetravalent TREM2 agonistic antibody reduced amyloid pathology in a mouse model of Alzheimer’s disease. Sci. Transl. Med. 2022, 14, eabq0095. [Google Scholar] [CrossRef]
- Masoudi, N.; Willen, J.; Daniels, C.; Jenkins, B.A.; Furber, E.C.; Kothiya, M.; Banjoko, M.B.; Gowda, R.; Hendricks, J.; Fang, Y.Y.; et al. Microglial-targeted gene therapy: Developing a disease modifying treatment for ALSP associated with CSF1R Mutations (ALSP-CSF1R) (P11-4.012). Neurology 2024, 102, 3061. [Google Scholar] [CrossRef]
- Yoo, Y.; Neumayer, G.; Shibuya, Y.; Mader, M.M.; Wernig, M. A cell therapy approach to restore microglial Trem2 function in a mouse model of Alzheimer’s disease. Cell Stem Cell. 2023, 30, 1043–1053.e6. [Google Scholar] [CrossRef]
- Deming, Y.; Li, Z.; Benitez, B.A.; Cruchaga, C. Triggering receptor expressed on myeloid cells 2 (TREM2): A potential therapeutic target for Alzheimer disease? Expert. Opin. Ther. Targets 2018, 22, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Wang, M.; Ni, C.; Yang, C.; Fu, C.; Zhang, X.; Chen, X.; Wu, X.; Hou, J.; Wang, L. TREM2 promotes the formation of a tumor-supportive microenvironment in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2025, 44, 20. [Google Scholar] [CrossRef]

| Name of Candidate | Type | Preclinical Development Results | Results in Clinical Trials | References |
|---|---|---|---|---|
| VG-3927 | Small Molecule Agonist | iPSC: increased anti-inflammatory activation hTREM2-5xFAD mice: reduced amyloid aggregates | Phase 1: reduced sTREM2 levels (50%) May be safe, further research needed, since there may be potential side effects | [163,164] |
| AL002-humanized IgG antibody | Monoclonal antibody | Increases microglial activation, proliferation and survival | Phase 2: failed to slow down AD despite microglial activation | [165,166] |
| CGX101: IgG4 antibody | Monoclonal antibody | In vitro and 5xFAD mice: reduced amyloid burden and cognitive decline Reduced p-Tau in mice | Currently in preclinical development phase | [161,168] |
| Ab-T1 | monoclonal antibody | Increased TREM2 expression, amyloid and apoptotic neuron uptake, reduced cognitive decline | Currently in preclinical development phase, but reduced sTREM2 levels in CSF from patients | [161] |
| 4D9 | Monoclonal antibody | Mouse models: protecting the microglia, enhanced TREM2 signaling, improved brain metabolism | Currently in preclinical development phase | [169] |
| Ab18 | Monoclonal antibody | Rodent models: increased amyloid clearance, improved synaptic marker intensity, reduced Tau phosphorylation | Currently in preclinical development phase | [170] |
| PR009 | Gene therapy | Stimulated TREM2 expression, maintain microglial balance | Currently in preclinical development phase | [172] |
| Transplantation of Trem2+/+ circulation-derived myeloid cells | Cell/Gene therapy | 5xFAD mice: restored microglial functions, DAM expression, reduced plaque load | Currently in preclinical development phase | [173] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, H.; Kim, D.; Yang, Y.; Bagyinszky, E.; An, S.S.A. TREM2 in Neurodegenerative Disorders: Mutation Spectrum, Pathophysiology, and Therapeutic Targeting. Int. J. Mol. Sci. 2025, 26, 7057. https://doi.org/10.3390/ijms26157057
Yang H, Kim D, Yang Y, Bagyinszky E, An SSA. TREM2 in Neurodegenerative Disorders: Mutation Spectrum, Pathophysiology, and Therapeutic Targeting. International Journal of Molecular Sciences. 2025; 26(15):7057. https://doi.org/10.3390/ijms26157057
Chicago/Turabian StyleYang, Hyewon, Danyeong Kim, YoungSoon Yang, Eva Bagyinszky, and Seong Soo A. An. 2025. "TREM2 in Neurodegenerative Disorders: Mutation Spectrum, Pathophysiology, and Therapeutic Targeting" International Journal of Molecular Sciences 26, no. 15: 7057. https://doi.org/10.3390/ijms26157057
APA StyleYang, H., Kim, D., Yang, Y., Bagyinszky, E., & An, S. S. A. (2025). TREM2 in Neurodegenerative Disorders: Mutation Spectrum, Pathophysiology, and Therapeutic Targeting. International Journal of Molecular Sciences, 26(15), 7057. https://doi.org/10.3390/ijms26157057